Co-reporter:Dixie Bungard, Jacob S. Copple, Jing Yan, Jimmy J. Chhun, ... Matthew H.J. Cordes
Structure 2017 Volume 25, Issue 11(Volume 25, Issue 11) pp:
Publication Date(Web):7 November 2017
DOI:10.1016/j.str.2017.09.006
•The young, functional de novo protein Bsc4 has a rudimentary ability to fold•Bsc4 forms compact oligomers with high β sheet content and a hydrophobic core•Bsc4 lacks a specific quaternary state and binds dyes suggestive of amyloid oligomers•Young de novo proteins can have some structural order and native-like propertiesThe de novo evolution of protein-coding genes from noncoding DNA is emerging as a source of molecular innovation in biology. Studies of random sequence libraries, however, suggest that young de novo proteins will not fold into compact, specific structures typical of native globular proteins. Here we show that Bsc4, a functional, natural de novo protein encoded by a gene that evolved recently from noncoding DNA in the yeast S. cerevisiae, folds to a partially specific three-dimensional structure. Bsc4 forms soluble, compact oligomers with high β sheet content and a hydrophobic core, and undergoes cooperative, reversible denaturation. Bsc4 lacks a specific quaternary state, however, existing instead as a continuous distribution of oligomer sizes, and binds dyes indicative of amyloid oligomers or molten globules. The combination of native-like and non-native-like properties suggests a rudimentary fold that could potentially act as a functional intermediate in the emergence of new folded proteins de novo.Download high-res image (184KB)Download full-size image
Co-reporter:Christian G. Roessler;Sue A. Roberts;Branwen M. Hall;Wendy M. Ingram;Matthew H. J. Cordes;William R. Montfort;William J. Anderson
PNAS 2008 Volume 105 (Issue 7 ) pp:2343-2348
Publication Date(Web):2008-02-19
DOI:10.1073/pnas.0711589105
Proteins that share common ancestry may differ in structure and function because of divergent evolution of their amino acid
sequences. For a typical diverse protein superfamily, the properties of a few scattered members are known from experiment.
A satisfying picture of functional and structural evolution in relation to sequence changes, however, may require characterization
of a larger, well chosen subset. Here, we employ a “stepping-stone” method, based on transitive homology, to target sequences
intermediate between two related proteins with known divergent properties. We apply the approach to the question of how new
protein folds can evolve from preexisting folds and, in particular, to an evolutionary change in secondary structure and oligomeric
state in the Cro family of bacteriophage transcription factors, initially identified by sequence-structure comparison of distant
homologs from phages P22 and λ. We report crystal structures of two Cro proteins, Xfaso 1 and Pfl 6, with sequences intermediate
between those of P22 and λ. The domains show 40% sequence identity but differ by switching of α-helix to β-sheet in a C-terminal
region spanning ≈25 residues. Sedimentation analysis also suggests a correlation between helix-to-sheet conversion and strengthened
dimerization.
Co-reporter:Matthew H.J. Cordes, Katie L. Stewart
Structure (8 February 2012) Volume 20(Issue 2) pp:199-200
Publication Date(Web):8 February 2012
DOI:10.1016/j.str.2012.01.015
Fold switching may play a role in the evolution of new protein folds and functions. He et al., in this issue of Structure, use protein design to illustrate that the same drastic change in a protein fold can occur via multiple different mutational pathways.
Co-reporter:Branwen M. Hall, Sue A. Roberts, Annie Heroux, Matthew H.J. Cordes
Journal of Molecular Biology (18 January 2008) Volume 375(Issue 3) pp:802-811
Publication Date(Web):18 January 2008
DOI:10.1016/j.jmb.2007.10.082
Previously reported crystal structures of free and DNA-bound dimers of λ Cro differ strongly (about 4 Å backbone rmsd), suggesting both flexibility of the dimer interface and induced-fit protein structure changes caused by sequence-specific DNA binding. Here, we present two crystal structures, in space groups P3221 and C2 at 1.35 and 1.40 Å resolution, respectively, of a variant of λ Cro with three mutations in its recognition helix (Q27P/A29S/K32Q, or PSQ for short). One dimer structure (P3221; PSQ form 1) resembles the DNA-bound wild-type Cro dimer (1.0 Å backbone rmsd), while the other (C2; PSQ form 2) resembles neither unbound (3.6 Å) nor bound (2.4 Å) wild-type Cro. Both PSQ form 2 and unbound wild-type dimer crystals have a similar interdimer β-sheet interaction between the β1 strands at the edges of the dimer. In the former, an infinite, open β-structure along one crystal axis results, while in the latter, a closed tetrameric barrel is formed. Neither the DNA-bound wild-type structure nor PSQ form 1 contains these interdimer interactions. We propose that β-sheet superstructures resulting from crystal contact interactions distort Cro dimers from their preferred solution conformation, which actually resembles the DNA-bound structure. These results highlight the remarkable flexibility of λ Cro but also suggest that sequence-specific DNA binding may not induce large changes in the protein structure.
Co-reporter:Daniel M. Lajoie, Matthew H.J. Cordes
Toxicon (15 December 2015) Volume 108() pp:176-180
Publication Date(Web):15 December 2015
DOI:10.1016/j.toxicon.2015.10.008
•Phospholipase D toxins from pathogenic bacteria and fungi were characterized.•Both toxins convert phospholipid substrates to cyclic phosphate products.•The same cyclic products have been observed for distantly related spider toxins.•Cyclic phospholipids may contribute to pathogenicity of these diverse organisms.•These toxins most likely evolved by divergent evolution with horizontal gene transfer.Phospholipase D (PLD) toxins from sicariid spiders, which cause disease in mammals, were recently found to convert their primary substrates, sphingomyelin and lysophosphatidylcholine, to cyclic phospholipids. Here we show that two PLD toxins from pathogenic actinobacteria and ascomycete fungi, which share distant homology with the spider toxins, also generate cyclic phospholipids. This shared function supports divergent evolution of the PLD toxins from a common ancestor and suggests the importance of cyclic phospholipids in pathogenicity.
Co-reporter:Branwen M. Hall, Erin E. Vaughn, Adrian R. Begaye, Matthew H.J. Cordes
Journal of Molecular Biology (11 November 2011) Volume 413(Issue 5) pp:914-928
Publication Date(Web):11 November 2011
DOI:10.1016/j.jmb.2011.08.056
Cro proteins from different lambdoid bacteriophages are extremely variable in their target consensus DNA sequences and constitute an excellent model for evolution of transcription factor specificity. We experimentally tested a bioinformatically derived evolutionary code relating switches between pairs of amino acids at three recognition helix sites in Cro proteins to switches between pairs of nucleotide bases in the cognate consensus DNA half-sites. We generated all eight possible code variants of bacteriophage λ Cro and used electrophoretic mobility shift assays to compare binding of each variant to its own putative cognate site and to the wild-type cognate site; we also tested the wild-type protein against all eight DNA sites. Each code variant showed stronger binding to its putative cognate site than to the wild-type site, except some variants containing proline at position 27; each also bound its cognate site better than wild-type Cro bound the same site. Most code variants, however, displayed poorer affinity and specificity than wild-type λ Cro. Fluorescence anisotropy assays on λ Cro and the triple code variant (PSQ) against the two cognate sites confirmed the switch in specificity and showed larger apparent effects on binding affinity and specificity. Bacterial one-hybrid assays of λ Cro and PSQ against libraries of sequences with a single randomized half-site showed the expected switches in specificity at two of three coded positions and no clear switches in specificity at noncoded positions. With a few caveats, these results confirm that the proposed Cro evolutionary code can be used to reengineer Cro specificity.Download high-res image (114KB)Download full-size imageResearch Highlights► The evolutionary code partly explains diversity of Cro protein functional specificity. ► The code governs specificity at three positions in DNA binding site. ► Application to λ Cro gives reengineered DNA binding site preference. ► Evaluated by gel shift, fluorescence anisotropy and bacterial one-hybrid assays.

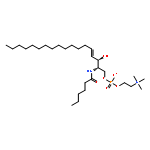
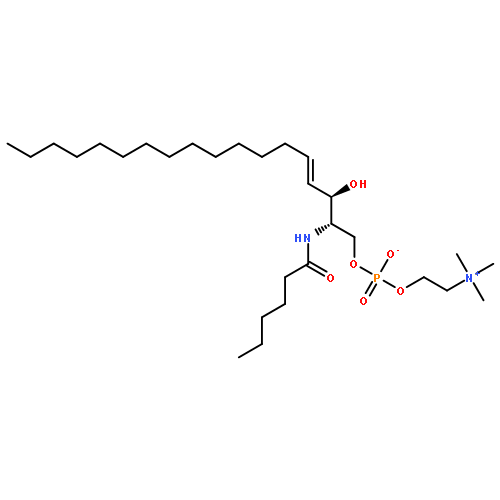
![[(E,2S,3R)-2-(dodecanoylamino)-3-hydroxyoctadec-4-enyl] 2-(trimethylazaniumyl)ethyl phosphate](http://img.cochemist.com/ccimg/475000/474923-21-2.png)
![[(E,2S,3R)-2-(dodecanoylamino)-3-hydroxyoctadec-4-enyl] 2-(trimethylazaniumyl)ethyl phosphate](http://img.cochemist.com/ccimg/475000/474923-21-2_b.png)