Co-reporter:Michael Seitz, King Do, Andrew J. Ingram, Evan G. Moore, Gilles Muller and Kenneth N. Raymond
Inorganic Chemistry September 7, 2009 Volume 48(Issue 17) pp:8469-8479
Publication Date(Web):July 29, 2009
DOI:10.1021/ic901079s
The modular syntheses of three new octadentate, enantiopure ligands are reported, one with the bidentate chelating unit 2-hydroxyisophthalamide (IAM) and two with bidentate 1-hydroxy-2-pyridinone (1,2-HOPO) units. A new design principle is introduced for the chiral, non-racemic hexamines which constitute the central backbones for the presented class of ligands. The terbium(III) complex of the IAM ligand, as well as the europium(III) complexes of the 1,2-HOPO ligands, are synthesized and characterized by various techniques (NMR, UV, CD, luminescence spectroscopy). All species exhibit excellent stability and moderate to high luminescence efficiency (quantum yields ΦEu = 0.05−0.08 and ΦTb = 0.30−0.57) in aqueous solution at physiological pH. Special focus is put onto the properties of the complexes in regard to circularly polarized luminescence (CPL). The maximum luminescence dissymmetry factors (glum) in aqueous solution are high with ∣glum∣max = 0.08−0.40. Together with the very favorable general properties (good stability, high quantum yields, long lifetimes), the presented lanthanide complexes can be considered as good candidates for analytical probes based on CPL in biologically relevant environments.
Co-reporter:Cynthia M. Hong, David M. Kaphan, Robert G. Bergman, Kenneth N. Raymond, and F. Dean Toste
Journal of the American Chemical Society June 14, 2017 Volume 139(Issue 23) pp:8013-8013
Publication Date(Web):June 5, 2017
DOI:10.1021/jacs.7b03812
This study offers a detailed mechanistic investigation of host–guest encapsulation behavior in a new enzyme–mimetic metal–ligand host and provides the first observation of a conformational selection mechanism (as opposed to induced fit) in a supramolecular system. The Ga4L4 host described features a C3-symmetric ligand motif with meta-substituted phenyl spacers, which enables the host to initially self-assemble into an S4-symmetric structure and then subsequently isomerize to a T-symmetric tetrahedron for better accommodation of a sufficiently large guest. Selective inversion recovery 1H NMR studies provide structural insights into the self-exchange behaviors of the host and the guest individually in this dynamic system. Kinetic analysis of the encapsulation–isomerization event revealed that increasing the concentration of guest inhibits the rate of host–guest relaxation, a key distinguishing feature of conformational selection. A comprehensive study of this simple enzyme mimic provides insight into analogous behavior in biophysics and enzymology and aims to inform the design of efficient self-assembled microenvironment catalysts.
Co-reporter:Mark D. Levin; David M. Kaphan; Cynthia M. Hong; Robert G. Bergman; Kenneth N. Raymond;F. Dean Toste
Journal of the American Chemical Society 2016 Volume 138(Issue 30) pp:9682-9693
Publication Date(Web):July 26, 2016
DOI:10.1021/jacs.6b05442
The scope and mechanism of the microenvironment-catalyzed C(sp3)−C(sp3) reductive elimination from transition metal complexes [Au(III), Pt(IV)] is explored. Experiments detailing the effect of structural perturbation of neutral and anionic spectator ligands, reactive alkyl ligands, solvent, and catalyst structure are disclosed. Indirect evidence for a coordinatively unsaturated encapsulated cationic intermediate is garnered via observation of several inactive donor-arrested inclusion complexes, including a crystallographically characterized encapsulated Au(III) cation. Finally, based on stoichiometric experiments under catalytically relevant conditions, a detailed mechanism is outlined for the dual supramolecular and platinum-catalyzed C–C coupling between methyl iodide and tetramethyltin. Determination of major platinum species present under catalytic conditions and subsequent investigation of their chemistry reveals an unexpected interplay between cis–trans isomerism and the supramolecular catalyst in a Pt(II)/Pt(IV) cycle, as well as several off-cycle reactions.
Co-reporter:Carmelo Sgarlata and Kenneth N. Raymond
Analytical Chemistry 2016 Volume 88(Issue 13) pp:6923
Publication Date(Web):May 31, 2016
DOI:10.1021/acs.analchem.6b01684
The entropic and enthalpic driving forces for encapsulation versus sequential exterior guest binding to the [Ga4L6]12– supramolecular host in solution are very different, which significantly complicates the determination of these thermodynamic parameters. The simultaneous use of complementary techniques, such as NMR, UV–vis, and isothermal titration calorimetry, enables the disentanglement of such multiple host–guest interactions. Indeed, data collected by each technique measure different components of the host–guest equilibria and together provide a complete picture of the solution thermodynamics. Unfortunately, commercially available programs do not allow for global analysis of different physical observables. We thus resorted to a novel procedure for the simultaneous refinement of multiple parameters (ΔG°, ΔH°, and ΔS°) by treating different observables through a weighted nonlinear least-squares analysis of a constrained model. The refinement procedure is discussed for the multiple binding of the Et4N+ guest, but it is broadly applicable to the deconvolution of other intricate host–guest equilibria.
Co-reporter:Tiffany A. Pham, Alison B. Altman, S. Chantal E. Stieber, Corwin H. Booth, Stosh A. Kozimor, Wayne W. Lukens, Daniel T. Olive, Tolek Tyliszczak, Jian Wang, Stefan G. Minasian, and Kenneth N. Raymond
Inorganic Chemistry 2016 Volume 55(Issue 20) pp:9989-10002
Publication Date(Web):June 24, 2016
DOI:10.1021/acs.inorgchem.6b00684
A tetravalent cerium macrocyclic complex (CeLK4) was prepared with an octadentate terephthalamide ligand comprised of hard catecholate donors and characterized in the solution state by spectrophotometric titrations and electrochemistry and in the crystal by X-ray diffraction. The solution-state studies showed that L exhibits a remarkably high affinity toward Ce4+, with log β110 = 61(2) and ΔG = −348 kJ/mol, compared with log β110 = 32.02(2) for the analogous Pr3+ complex. In addition, L exhibits an unusual preference for forming CeL4– relative to formation of the analogous actinide complex, ThL4–, which has β110 = 53.7(5). The extreme stabilization of tetravalent cerium relative to its trivalent state is also evidenced by the shift of 1.91 V in the redox potential of the Ce3+/Ce4+ couple of the complex (measured at −0.454 V vs SHE). The unprecedented behavior prompted an electronic structure analysis using L3- and M5,4-edge X-ray absorption near-edge structure (XANES) spectroscopies and configuration interaction calculations, which showed that 4f-orbital bonding in CeLK4 has partial covalent character due to ligand-to-metal charge transfer (LMCT) in the ground state. The experimental results are presented in the context of earlier measurements on tetravalent cerium compounds, indicating that the amount of LMCT for CeLK4 is similar to that observed for [Et4N]2[CeCl6] and CeO2 and significantly less than that for the organometallic sandwich compound cerocene, (C8H8)2Ce. A simple model to rationalize changes in 4f orbital bonding for tri- and tetravalent lanthanide and actinide compounds is also provided.
Co-reporter:Lena J. Daumann, David S. Tatum, Christopher M. Andolina, Joseph I. Pacold, Anthony D’Aléo, Ga-lai Law, Jide Xu, and Kenneth N. Raymond
Inorganic Chemistry 2016 Volume 55(Issue 1) pp:114-124
Publication Date(Web):December 17, 2015
DOI:10.1021/acs.inorgchem.5b01927
A series of 10 tetradentate 1-hydroxy-pyridin-2-one (1,2-HOPO) ligands and corresponding eight-coordinated photoluminescent Eu(III) and Sm(III) complexes were prepared. Generally, the ligands differ by the linear (nLI) aliphatic linker length, from 2 to 8 methylene units between the bidentate 1,2-HOPO chelator units. The photoluminescent quantum yields (Φtot) were found to vary with the linker length, and the same trend was observed for the Eu(III) and Sm(III) complexes. The 2LI and 5LI bridged complexes are the brightest (Φtotxε). The change in ligand wrapping pattern between 2LI and 5LI complexes observed by X-ray diffraction (XRD) is further supported by density functional theory (DFT) calculations. The bimodal Φtot trends of the Eu(III) and Sm(III) complexes are rationalized by the change in ligand wrapping pattern as the bridge (nLI) is increased in length.
Co-reporter:Lena J. Daumann, Philipp Werther, Michael J. Ziegler, Kenneth N. Raymond
Journal of Inorganic Biochemistry 2016 Volume 162() pp:263-273
Publication Date(Web):September 2016
DOI:10.1016/j.jinorgbio.2016.01.006
•2-Hydroxisophthalamide (IAM) and 1,2-hydroxypyridonate (HOPO) mixed ligands are prepared•Photophysical properties of Tb and Eu complexes are reported.•When bound to a primary amine the above chromophores are better in sensitizing Tb and Eu.Following the success of the siderophore-inspired 1,2-hydroxypyridonate (HOPO) and 2-hydroxisophthalamide (IAM) chromophores in Eu(III) and Tb(III) luminescence, we designed three new ligands bearing both chromophores. Syntheses of the octadentate ligands 3,4,3-LI-IAM-1,2-HOPO and 3,4,3-LI-1,2-HOPO-IAM, where the chromophores are attached to different positions in the (LI = linear) spermine backbone, are reported in addition to a tetradentate ligand based on 1,5-diaminopentane. The Gd(III) complexes were prepared and revealed localized triplet states typical for the IAM and HOPO chromophores. Photophysical characterization of the Eu(III) and Tb(III) complexes revealed that the chromophores need to reside at a primary amine of the spermine backbone to be efficient in lanthanide excitation. These systems help us to understand the antenna effect in siderophore inspired chromophores and could be potential targets for sensing and biological imaging applications.Three antenna ligands bearing 1,2-hydroxypyridonate (HOPO) and 2-hydroxisophthalamide (IAM) chromophores are reported. Photophysical characterization of the Gd(III), Eu(III) and Tb(III) complexes revealed localized triplet states for the chromophores and that they need to reside on a primary amine of the spermine backbone to be efficient in lanthanide excitation.
Co-reporter:Sylvie L. Pailloux;Sean Nguyen;Stephanie Zhou;Marisa E. Hom;Michelle N. Keyser;Danil Smiles
Journal of Heterocyclic Chemistry 2016 Volume 53( Issue 4) pp:1065-1073
Publication Date(Web):
DOI:10.1002/jhet.2372
A hydroxypyridinone building block, bifunctionalized with thiazoline, has been prepared from orthogonally protected 2-(3-(benzyloxy)-4-(ethoxycarbonyl)-6-methyl-2-oxopyridin-1(2H)-yl) acetic acid. The reactivity of the dithiazolide has been explored with two primary amines, leading to the synthesis and characterization of four new hexadentate ligands. Their complexes with selected hard trivalent ions pertinent to potential molecular imaging applications have been surveyed.
Co-reporter:Casey J. Brown, F. Dean Toste, Robert G. Bergman, and Kenneth N. Raymond
Chemical Reviews 2015 Volume 115(Issue 9) pp:3012
Publication Date(Web):April 21, 2015
DOI:10.1021/cr4001226
Co-reporter:Kenneth N. Raymond, Benjamin E. Allred, and Allyson K. Sia
Accounts of Chemical Research 2015 Volume 48(Issue 9) pp:2496
Publication Date(Web):September 2, 2015
DOI:10.1021/acs.accounts.5b00301
This Account focuses on the coordination chemistry of the microbial iron chelators called siderophores. The initial research (early 1970s) focused on simple analogs of siderophores, which included hydroxamate, catecholate, or hydroxycarboxylate ligands. The subsequent work increasingly focused on the transport of siderophores and their microbial iron transport. Since these are pseudo-octahedral complexes often composed of bidentate ligands, there is chirality at the metal center that in principle is independent of the ligand chirality. It has been shown in many cases that chiral recognition of the complex occurs. Many techniques have been used to elucidate the iron uptake processes in both Gram-positive (single membrane) and Gram-negative (double membrane) bacteria. These have included the use of radioactive labels (of ligand, metal, or both), kinetically inert metal complexes, and Mössbauer spectroscopy. In general, siderophore recognition and transport involves receptors that recognize the metal chelate portion of the iron–siderophore complex. A second, to date less commonly found, mechanism called the siderophore shuttle involves the receptor binding an apo-siderophore.Since one of the primary ways that microbes compete with each other for iron stores is the strength of their competing siderophore complexes, it became important early on to characterize the solution thermodynamics of these species. Since the acidity of siderophores varies significantly, just the stability constant does not give a direct measure of the relative competitive strength of binding. For this reason, the pM value is compared. The pM, like pH, is a measure of the negative log of the free metal ion concentration, typically calculated at pH 7.4, and standard total concentrations of metal and ligand. The characterization of the electronic structure of ferric siderophores has done much to help explain the high stability of these complexes.A new chapter in siderophore science has emerged with the characterization of what are now called siderocalins. Initially found as a protein of the human innate immune system, these proteins bind both ferric and apo-siderophores to inactivate the siderophore transport system and hence deny iron to an invading pathogenic microbe. Siderocalins also can play a role in iron transport of the host, particularly in the early stages of fetal development. Finally, it is speculated that the molecular targets of siderocalins in different species differ based on the siderophore structures of the most important bacterial pathogens of those species.
Co-reporter:David M. Kaphan; F. Dean Toste; Robert G. Bergman
Journal of the American Chemical Society 2015 Volume 137(Issue 29) pp:9202-9205
Publication Date(Web):July 15, 2015
DOI:10.1021/jacs.5b01261
Supramolecular assembly 1 catalyzes a bimolecular aza-Prins cyclization featuring an unexpected transannular 1,5-hydride transfer. This reaction pathway, which is promoted by constrictive binding within the supramolecular cavity of 1, is kinetically disfavored in the absence of 1, as evidenced by the orthogonal reactivity observed in bulk solution. Mechanistic investigation through kinetic analysis and isotopic labeling studies indicates that the rate-limiting step of the transformation is the encapsulation of a transient iminium ion and supports the proposed 1,5-hydride transfer mechanism. This represents a rare example of such an extreme divergence of product selectivity observed within a catalytic metal–ligand supramolecular enzyme mimic.
Co-reporter:Derek M. Dalton; Scott R. Ellis; Eva M. Nichols; Richard A. Mathies; F. Dean Toste; Robert G. Bergman
Journal of the American Chemical Society 2015 Volume 137(Issue 32) pp:10128-10131
Publication Date(Web):August 10, 2015
DOI:10.1021/jacs.5b06317
The K12Ga4L6 supramolecular cage is photoactive and enables an unprecedented photoreaction not observed in bulk solution. Ga4L612– cages photosensitize the 1,3-rearrangement of encapsulated cinnamylammonium cation guests from the linear isomer to the higher energy branched isomer when irradiated with UVA light. The rearrangement requires light and guest encapsulation to occur. The Ga4L612– cage-mediated reaction mechanism was investigated by UV/vis absorption, fluorescence, ultrafast transient absorption, and electrochemical experiments. The results support a photoinduced electron transfer mechanism for the 1,3-rearrangement, in which the Ga4L612– cage absorbs photons and transfers an electron to the encapsulated cinnamylammonium ion, which undergoes C–N bond cleavage, followed by back electron transfer to the cage and recombination of the guest fragments to form the higher energy isomer.
Co-reporter:Lena J. Daumann; David S. Tatum; Benjamin E. R. Snyder; Chengbao Ni; Ga-lai Law; Edward I. Solomon
Journal of the American Chemical Society 2015 Volume 137(Issue 8) pp:2816-2819
Publication Date(Web):January 21, 2015
DOI:10.1021/ja5116524
We report the preparation and new insight into photophysical properties of luminescent hydroxypyridonate complexes [MIIIL]− (M = Eu or Sm) of the versatile 3,4,3-LI(1,2-HOPO) ligand (L). We report the crystal structure of this ligand with EuIII as well as insights into the coordination behavior and geometry in solution by using magnetic circular dichroism. In addition TD-DFT calculations were used to examine the excited states of the two different chromophores present in the 3,4,3-LI(1,2-HOPO) ligand. We find that the EuIII and SmIII complexes of this ligand undergo a transformation after in situ preparation to yield complexes with higher quantum yield (QY) over time. It is proposed that the lower QY in the in situ complexes is not only due to water quenching but could also be due to a lower degree of f-orbital overlap (in a kinetic isomer) as indicated by magnetic circular dichroism measurements.
Co-reporter:Anthony D’Aléo; Evan G. Moore; Jide Xu; Lena J. Daumann
Inorganic Chemistry 2015 Volume 54(Issue 14) pp:6807-6820
Publication Date(Web):July 7, 2015
DOI:10.1021/acs.inorgchem.5b00748
The synthesis of a series of octadentate ligands containing the 1-hydroxypyridin-2-one (1,2-HOPO) group in complex with europium(III) is reported. Within this series, the central bridge connecting two diethylenetriamine units linked to two 1,2-HOPO chromophores at the extremities (5-LIN-1,2-HOPO) is varied from a short ethylene chain (H(2,2)-1,2-HOPO) to a long pentaethylene oxide chain (H(17O5,2)-1,2-HOPO). The thermodynamic stability of the europium complexes has been studied and reveals these complexes may be effective for biological measurements. Extension of the central bridge results in exclusion of the inner-sphere water molecule observed for [Eu(H(2,2)-1,2-HOPO)]− going from a nonacoordinated to an octacoordinated Eu(III) ion. With the longer chain length ligands, the complexes display increased luminescence properties in aqueous medium with an optimum of 20% luminescence quantum yield for the [Eu(H(17O5,2)-1,2-HOPO)]− complex. The luminescence properties for [Eu(H(14O4,2)-1,2-HOPO)]− and [Eu(H(17O5,2)-1,2-HOPO)]− are better than that of the model bis-tetradentate [Eu(5LINMe-1,2-HOPO)2]− complex, suggesting a different geometry around the metal center despite the geometric freedom allowed by the longer central chain in the H(mOn,2) scaffold. These differences are also evidenced by examining the luminescence spectra at room temperature and at 77 K and by calculating the luminescence kinetic parameters of the europium complexes.
Co-reporter:William M. Hart-Cooper;Charles L. Perrin;Carmelo Sgarlata;F. Dean Toste;Robert G. Bergman
PNAS 2015 Volume 112 (Issue 50 ) pp:15303-15307
Publication Date(Web):2015-12-15
DOI:10.1073/pnas.1515639112
The mechanism of proton exchange in a metal–ligand enzyme active site mimic (compound 1) is described through amide hydrogen–deuterium
exchange kinetics. The type and ratio of cationic guest to host in solution affect the rate of isotope exchange, suggesting
that the rate of exchange is driven by a host whose cavity is occupied by water. Rate constants for acid-, base-, and water-mediated
proton exchange vary by orders of magnitude depending on the guest, and differ by up to 200 million-fold relative to an alanine
polypeptide. These results suggest that the unusual microenvironment of the cavity of 1 can dramatically alter the reactivity
of associated water by magnitudes comparable to that of enzymes.
Co-reporter:David M. Kaphan;Mark D. Levin;Robert G. Bergman;F. Dean Toste
Science 2015 Vol 350(6265) pp:1235-1238
Publication Date(Web):04 Dec 2015
DOI:10.1126/science.aad3087
Faster elimination inside a cavity
Metals are adept at shuffling molecular bonds. They pry apart two atoms and then pair each one with a different partner. Sometimes the atoms get stuck on the metal, though, and the newly partnered products aren't released. Kaphan et al. designed a strategy for accelerating this elimination process (see the Perspective by Yan and Fujita). A hollow supramolecular capsule captured a gold or platinum complex and induced rapid bond formation between the carbon atoms in methyl groups bound to the metal. Generalization of this strategy could open the door to a wide range of chemical transformations that are currently held up by slow eliminations.
Science, this issue p. 1235; see also p. 1165
Co-reporter:Chen Zhao ; F. Dean Toste ; Kenneth N. Raymond ;Robert G. Bergman
Journal of the American Chemical Society 2014 Volume 136(Issue 41) pp:14409-14412
Publication Date(Web):September 29, 2014
DOI:10.1021/ja508799p
While the reactive pocket of many enzymes has been shown to modify reactions of substrates by changing their chemical properties, examples of reactions whose stereochemical course is completely reversed are exceedingly rare. We report herein a class of water-soluble host assemblies that is capable of catalyzing the substitution reaction at a secondary benzylic carbon center to give products with overall stereochemical retention, while reaction of the same substrates in bulk solution gives products with stereochemical inversion. Such ability of a biomimetic synthetic host assembly to reverse the stereochemical outcome of a nucleophilic substitution reaction is unprecedented in the field of supramolecular host–guest catalysis.
Co-reporter:Tiffany A. Pham ; Jide Xu
Journal of the American Chemical Society 2014 Volume 136(Issue 25) pp:9106-9115
Publication Date(Web):May 28, 2014
DOI:10.1021/ja503456r
A novel macrocyclic octadentate ligand incorporating terephthalamide binding units has been synthesized and evaluated for the chelation of Th4+. The thorium complex was structurally characterized by X-ray diffraction and in solution with kinetic studies and spectrophotometric titrations. Dye displacement kinetic studies show that the ligand is a much more rapid chelator of Th4+ than prevailing ligands (1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid and diethylenetriaminepentaacetic acid). Furthermore, the resulting complex was found to have a remarkably high thermodynamic stability, with a formation constant of 1054. These data support potential radiotherapeutic applications.
Co-reporter:Christopher J. Chang and Kenneth N. Raymond
Inorganic Chemistry 2014 Volume 53(Issue 4) pp:1791-1793
Publication Date(Web):February 7, 2014
DOI:10.1021/ic500099n
Co-reporter:Tatsuya Fukushima, Benjamin E. Allred, and Kenneth N. Raymond
ACS Chemical Biology 2014 Volume 9(Issue 9) pp:2092
Publication Date(Web):July 9, 2014
DOI:10.1021/cb500319n
Iron is an essential element for all organisms, and microorganisms produce small molecule iron-chelators, siderophores, to efficiently acquire Fe(III). Gram-positive bacteria possess lipoprotein siderophore-binding proteins (SBPs) on the membrane. Some of the SBPs bind both apo-siderophores (iron-free) and Fe-siderophore (iron-chelated) and only import Fe-siderophores. When the SBP initially binds an apo-siderophore, the SBP uses the Gram-positive siderophore-shuttle mechanism (the SBPs exchange Fe(III) from a Fe-siderophore to the apo-siderophore bound to the protein) and/or displacement mechanism (the apo-siderophore bound to the SBP is released and a Fe-siderophore is then bound to the protein) to import the Fe-siderophore. Previously, we reported that the Bacillus cereus SBP, YxeB, exchanges Fe(III) from a ferrioxamine B (FO) to a desferrioxamine B (DFO) bound to YxeB using the siderophore-shuttle mechanism although the iron exchange was indirectly elucidated. Synthetic Cr-DFO (inert metal FO analog) and Ga-DFO (nonreducible FO analog) are bound to YxeB and imported via YxeB and the corresponding permeases and ATPase. YxeB exchanges Fe(III) from FO and Ga(III) from Ga-DFO to DFO bound to the protein, indicating that the metal-exchange occurs without metal reduction. YxeB also binds DFO derivatives including acetylated DFO (apo-siderophore) and acetylated FO (AcFO, Fe-siderophore). The iron from AcFO is transferred to DFO when bound to YxeB, giving direct evidence of iron exchange. Moreover, YxeB also uses the displacement mechanism when ferrichrome (Fch) is added to the DFO:YxeB complex. Uptake by the displacement mechanism is a minor pathway compared to the shuttle mechanism.
Co-reporter:Dr. Courtney J. Hastings; Robert G. Bergman; Kenneth N. Raymond
Chemistry - A European Journal 2014 Volume 20( Issue 14) pp:3966-3973
Publication Date(Web):
DOI:10.1002/chem.201303885
Abstract
The self-assembled supramolecular host [Ga4L6]12− (1; L=N,N-bis(2,3-dihydroxybenzoyl)-1,5-diaminonaphthalene) catalyzes the Nazarov cyclization of 1,3-pentadienols with extremely high levels of efficiency. The catalyzed reaction proceeds at a rate over a million times faster than that of the background reaction, an increase comparable to those observed in some enzymatic systems. A detailed study was conducted to elucidate the reaction mechanism of both the catalyzed and uncatalyzed Nazarov cyclization of pentadienols. Kinetic analysis and 18O-exchange experiments implicate a mechanism, in which encapsulation, protonation, and water loss from substrate are reversible, followed by irreversible electrocyclization. Although electrocyclization is rate determining in the uncatalyzed reaction, the barrier for water loss and for electrocyclization are nearly equal in the assembly-catalyzed reaction. Analysis of the energetics of the catalyzed and uncatalyzed reaction revealed that transition-state stabilization contributes significantly to the dramatically enhanced rate of the catalyzed reaction.
Co-reporter:Jeffrey S. Mugridge ; Achim Zahl ; Rudi van Eldik ; Robert G. Bergman
Journal of the American Chemical Society 2013 Volume 135(Issue 11) pp:4299-4306
Publication Date(Web):February 7, 2013
DOI:10.1021/ja309949q
The supramolecular host assembly [Ga4L6]12- [1; L = 1,5-bis(2,3-dihydroxybenzamido)naphthalene] contains a flexible, hydrophobic interior cavity that can encapsulate cationic guest molecules and catalyze a variety of chemical transformations. The Ar–CH2 bond rotational barrier for encapsulated ortho-substituted benzyl phosphonium guest molecules is sensitive to the size and shape of the host interior space. Here we examine how changes in bulk solvent (water, methanol, or DMF) or applied pressure (up to 150 MPa) affect the rotational dynamics of encapsulated benzyl phosphonium guests, as a way to probe changes in host cavity size or flexibility. When host 1 is dissolved in organic solvents with large solvent internal pressures (∂U/∂V)T, we find that the free energy barrier to Ar–CH2 bond rotation increases by 1–2 kcal/mol, compared with that in aqueous solution. Likewise, when external pressure is applied to the host–guest complex in solution, the bond rotational rates for the encapsulated guests decrease. The magnitude of these rate changes and the volumes of activation obtained using either solvent internal pressure or applied external pressure are very similar. NOE distance measurements reveal shorter average host–guest distances (∼0.3 Å) in organic versus aqueous solution. These experiments demonstrate that increasing solvent internal pressure or applied external pressure reduces the host cavity size or flexibility, resulting in more restricted motions for encapsulated guest molecules. Changing bulk solvent or external pressure might therefore be used to tune the physical properties or reactivity of guest molecules encapsulated in a flexible supramolecular host.
Co-reporter:Chen Zhao ; Qing-Fu Sun ; William M. Hart-Cooper ; Antonio G. DiPasquale ; F. Dean Toste ; Robert G. Bergman
Journal of the American Chemical Society 2013 Volume 135(Issue 50) pp:18802-18805
Publication Date(Web):November 27, 2013
DOI:10.1021/ja411631v
The synthesis of a novel supramolecular tetrahedral assembly of K12Ga4L6 stoichiometry is reported. The newly designed chiral ligand exhibits high diastereoselective control during cluster formation, leading exclusively to a single diastereomer of the desired host. This new assembly also exhibits high stability toward oxidation or a low pH environment and is a more robust and efficient catalyst for asymmetric organic transformations of neutral substrates.
Co-reporter:Benjamin E. Allred, Colin Correnti, Matthew C. Clifton, Roland K. Strong, and Kenneth N. Raymond
ACS Chemical Biology 2013 Volume 8(Issue 9) pp:1882
Publication Date(Web):June 11, 2013
DOI:10.1021/cb4002552
The human protein siderocalin (Scn) inhibits bacterial iron acquisition by binding catechol siderophores. Several pathogenic bacteria respond by making stealth siderophores that are not recognized by Scn. Fluvibactin and vibriobactin, respectively of Vibrio fluvialis and Vibrio cholerae, include an oxazoline adjacent to a catechol. This chelating unit binds iron either in a catecholate or a phenolate-oxazoline coordination mode. The latter has been suggested to make vibriobactin a stealth siderophore without directly identifying the coordination mode in relation to Scn binding. We use Scn binding assays with the two siderophores and two oxazoline-substituted analogs and the crystal structure of Fe-fluvibactin:Scn to show that the oxazoline does not prevent Scn binding; hence, vibriobactin is not a stealth siderophore. We show that the phenolate-oxazoline coordination mode is present at physiological pH and is not bound by Scn. However, Scn binding shifts the coordination to the catecholate mode and thereby inactivates this siderophore.
Co-reporter:Allyson K. Sia;Tatsuya Fukushima;Ulla N. Andersen;Rita Nichiporuk;Benjamin E. Allred
PNAS 2013 Volume 110 (Issue 34 ) pp:13821-13826
Publication Date(Web):2013-08-20
DOI:10.1073/pnas.1304235110
Small molecule iron-chelators, siderophores, are very important in facilitating the acquisition of Fe(III), an essential element
for pathogenic bacteria. Many Gram-negative outer-membrane transporters and Gram-positive lipoprotein siderophore-binding
proteins have been characterized, and the binding ability of outer-membrane transporters and siderophore-binding proteins
for Fe-siderophores has been determined. However, there is little information regarding the binding ability of these proteins
for apo-siderophores, the iron-free chelators. Here we report that Bacillus cereus YxeB facilitates iron-exchange from Fe-siderophore to apo-siderophore bound to the protein, the first Gram-positive siderophore-shuttle
system. YxeB binds ferrioxamine B (FO, Fe-siderophore)/desferrioxamine B (DFO, apo-siderophore) in vitro. Disc-diffusion assays
and growth assays using the yxeB mutant reveal that YxeB is responsible for importing the FO. Cr-DFO (a FO analog) is bound by YxeB in vitro and B. cereus imports or binds Cr-DFO in vivo. In vivo uptake assays using Cr-DFO and FO and growth assays using DFO and Cr-DFO show that
B. cereus selectively imports and uses FO when DFO is present. Moreover, in vitro competition assays using Cr-DFO and FO clearly demonstrate
that YxeB binds only FO, not Cr-DFO, when DFO is bound to the protein. Iron-exchange from FO to DFO bound to YxeB must occur
when DFO is initially bound by YxeB. Because the metal exchange rate is generally first order in replacement ligand concentration,
protein binding of the apo-siderophore acts to dramatically enhance the iron exchange rate, a key component of the Gram-positive
siderophore-shuttle mechanism.
Co-reporter:Ga-Lai Law ; Christopher M. Andolina ; Jide Xu ; Vinh Luu ; Philip X. Rutkowski ; Gilles Muller ; David K. Shuh ; John K. Gibson
Journal of the American Chemical Society 2012 Volume 134(Issue 37) pp:15545-15549
Publication Date(Web):August 25, 2012
DOI:10.1021/ja306354n
A key distinction between the lanthanide (4f) and the actinide (5f) transition elements is the increased role of f-orbital covalent bonding in the latter. Circularly polarized luminescence (CPL) is an uncommon but powerful spectroscopy which probes the electronic structure of chiral, luminescent complexes or molecules. While there are many examples of CPL spectra for the lanthanides, this report is the first for an actinide. Two chiral, octadentate chelating ligands based on orthoamide phenol (IAM) were used to complex curium(III). While the radioactivity kept the amount of material limited to micromole amounts, spectra of the highly luminescent complexes showed significant emission peak shifts between the different complexes, consistent with ligand field effects previously observed in luminescence spectra.
Co-reporter:William M. Hart-Cooper ; Kristen N. Clary ; F. Dean Toste ; Robert G. Bergman
Journal of the American Chemical Society 2012 Volume 134(Issue 43) pp:17873-17876
Publication Date(Web):October 15, 2012
DOI:10.1021/ja308254k
A polyanionic supramolecular assembly (1) is shown to catalytically cyclize the monoterpene citronellal and two homologues. In contrast to cyclization in acidic aqueous solution, the hydrophobic interior of 1 prevents the capture of reactive intermediates by water. This effect was also observed in the gold-catalyzed cycloisomerization of an enyne. Due to the steric confinement of the catalyst’s interior, Prins cyclizations in 1 proceed cleanly both for substrates containing and lacking gem-dimethyl substitution. Encapsulation in 1 consequently imposes a degree of mechanistic control that, similar to enzyme catalysis, is not observed in bulk aqueous solution.
Co-reporter:Piper J. Klemm;William C. Floyd III;Christopher M. Andolina;Jean M. J. Fréchet
European Journal of Inorganic Chemistry 2012 Volume 2012( Issue 12) pp:2108-2114
Publication Date(Web):
DOI:10.1002/ejic.201101167
Abstract
Magnetic resonance imaging (MRI) contrast agents represent a worldwide billion-dollar market annually. While T1 relaxivity enhancement contrast agents receive greater attention and a significantly larger market share, the commercial potential for T2 relaxivity enhancing contrast agents remains a viable diagnostic option because of their increased relaxivity at high field strengths. Improvement of the contrast and biocompatibility of T2 MRI probes may enable new diagnostic prospects for MRI. Paramagnetic lanthanides have the potential to decrease T1 and T2 proton relaxation times, but are not commercially used in MRI diagnostics as T2 agents. In this article, oxygen donor chelates (hydroxypyridinone, HOPO, and terephthalamide, TAM) of various lanthanides are demonstrated as biocompatible macromolecular dendrimer conjugates for the development of T2 MRI probes. These conjugates have relaxivities of up to 374 mM–1 s–1 per dendrimer, high bioavailability, and low in vitro toxicity.
Co-reporter:Tatsuya Fukushima;Rita Nichiporuk;Allyson K. Sia;Zhongrui Zhou;Ulla N. Andersen;Benjamin E. Allred
PNAS 2012 Volume 109 (Issue 42 ) pp:16829-16834
Publication Date(Web):2012-10-16
DOI:10.1073/pnas.1210131109
Citrate is a common biomolecule that chelates Fe(III). Many bacteria and plants use ferric citrate to fulfill their nutritional
requirement for iron. Only the Escherichia coli ferric citrate outer-membrane transport protein FecA has been characterized; little is known about other ferric citrate-binding
proteins. Here we report a unique siderophore-binding protein from the Gram-positive pathogenic bacterium Bacillus cereus that binds multinuclear ferric citrate complexes. We have demonstrated that B. cereus ATCC 14579 takes up 55Fe radiolabeled ferric citrate and that a protein, BC_3466 [renamed FctC (ferric citrate-binding protein C)], binds ferric
citrate. The dissociation constant (Kd) of FctC at pH 7.4 with ferric citrate (molar ratio 1:50) is 2.6 nM. This is the tightest binding observed of any B. cereus siderophore-binding protein. Nano electrospray ionization–mass spectrometry (nano ESI-MS) analysis of FctC and ferric citrate
complexes or citrate alone show that FctC binds diferric di-citrate, and triferric tricitrate, but does not bind ferric di-citrate,
ferric monocitrate, or citrate alone. Significantly, the protein selectively binds triferric tricitrate even though this species
is naturally present at very low equilibrium concentrations.
Co-reporter:Christopher M. Andolina, Piper J. Klemm, William C. Floyd III, Jean M. J. Fréchet, and Kenneth N. Raymond
Macromolecules 2012 Volume 45(Issue 22) pp:8982-8990
Publication Date(Web):November 12, 2012
DOI:10.1021/ma302206g
Advances in clinical diagnostic instrumentation have enabled some imaging modalities to be run concurrently. For diagnostic purposes, multimodal imaging can allow for rapid location and accurate identification of a patient’s illness. The paramagnetic and near-infrared (NIR) properties of Dy(III) and Yb(III) are interesting candidates for the development of bimodal NIR and magnetic resonance imaging (MRI) contrast agents. To enhance their intrinsic bimodal properties, these lanthanides were chelated using the hexadentate-all-oxygen-donor-ligand TREN-bis(1-Me)-3,2-HOPO-TAM-NX (NX, where X = 1, 2, or 3) and subsequently conjugated to the esteramide dendrimer (EA) to improve bioavailability, solubility, and relaxivity. Of these new complexes synthesized and evaluated, DyN1-EA had the largest ionic T1 relaxivity, 7.60 mM–1 s–1, while YbN3-EA had the largest ionic T2 relaxivity with a NIR quantum yield of 0.17% when evaluated in mouse serum. This is the first Yb(III) bimodal NIR/T2 MRI contrast agent of its kind evaluated.
Co-reporter:Dr. Ga-Lai Law;Tiffany A. Pham;Dr. Jide Xu ; Kenneth N. Raymond
Angewandte Chemie International Edition 2012 Volume 51( Issue 10) pp:2371-2374
Publication Date(Web):
DOI:10.1002/anie.201106748
Co-reporter:Dr. Ga-Lai Law;Tiffany A. Pham;Dr. Jide Xu ; Kenneth N. Raymond
Angewandte Chemie 2012 Volume 124( Issue 10) pp:2421-2424
Publication Date(Web):
DOI:10.1002/ange.201106748
Co-reporter:Z. Jane Wang ; Casey J. Brown ; Robert G. Bergman ; Kenneth N. Raymond ;F. Dean Toste
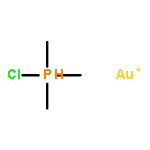
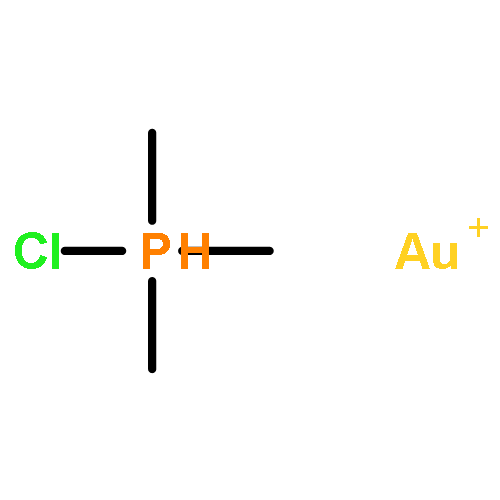
Journal of the American Chemical Society 2011 Volume 133(Issue 19) pp:7358-7360
Publication Date(Web):April 25, 2011
DOI:10.1021/ja202055v
Gold(I)–phosphine complexes are readily encapsulated by a tetrahedral supramolecular host (Ga4L6). We have investigated the catalytic activity of the resulting complexes for the intramolecular hydroalkoxylation of allenes. The catalytic activity of Me3PAuBr was increased 8-fold by encapsulation, as determined by initial rate kinetics, and we observed up to 67 catalytic turnovers by Me3PAu+ encapsulated in Ga4L6.
Co-reporter:Jeffrey S. Mugridge ; Robert G. Bergman
Journal of the American Chemical Society 2011 Volume 134(Issue 4) pp:2057-2066
Publication Date(Web):December 6, 2011
DOI:10.1021/ja2067324
The self-assembled supramolecular complex [Ga4L6]12- (1; L = 1,5-bis[2,3-dihydroxybenzamido]naphthalene) can act as a molecular host in aqueous solution and bind cationic guest molecules to its highly charged exterior surface or within its hydrophobic interior cavity. The distinct internal cavity of host 1 modifies the physical properties and reactivity of bound guest molecules and can be used to catalyze a variety of chemical transformations. Noncovalent host–guest interactions in large part control guest binding, molecular recognition and the chemical reactivity of bound guests. Herein we examine equilibrium isotope effects (EIEs) on both exterior and interior guest binding to host 1 and use these effects to probe the details of noncovalent host–guest interactions. For both interior and exterior binding of a benzylphosphonium guest in aqueous solution, protiated guests are found to bind more strongly to host 1 (KH/KD > 1) and the preferred association of protiated guests is driven by enthalpy and opposed by entropy. Deuteration of guest methyl and benzyl C–H bonds results in a larger EIE than deuteration of guest aromatic C–H bonds. The observed EIEs can be well explained by considering changes in guest vibrational force constants and zero-point energies. DFT calculations further confirm the origins of these EIEs and suggest that changes in low-frequency guest C–H/D vibrational motions (bends, wags, etc.) are primarily responsible for the observed EIEs.
Co-reporter:Jide Xu ; Todd M. Corneillie ; Evan G. Moore ; Ga-Lai Law ; Nathaniel G. Butlin
Journal of the American Chemical Society 2011 Volume 133(Issue 49) pp:19900-19910
Publication Date(Web):October 19, 2011
DOI:10.1021/ja2079898
The synthesis, structure, and photophysical properties of several Tb(III) complexes with octadentate, macrotricyclic ligands that feature a bicapped topology and 2-hydroxyisophthalamide (IAM) chelating units are reported. These Tb(III) complexes exhibit highly efficient emission (Φtotal ≥ 50%), large extinction coefficients (εmax ≥ 20,000 M–1 cm–1), and long luminescence lifetimes (τH2O ≥ 2.45 ms) at dilute concentrations in standard biological buffers. The structure of the methyl-protected ligand was determined by single-crystal X-ray diffraction and confirms the macrotricyclic structure of the parent ligand; the amide groups of the methyl-protected cage compound generate an anion binding cavity that complexes a chloride anion. Once the ligand is deprotected, a conformational change generates a similar cavity, formed by the phenolate and ortho amide oxygen groups that strongly bind lanthanide ions. The Tb(III) complexes thus formed display long-term stability, with little if any change in their spectral properties (including lifetime, quantum yield, and emission spectrum) over time or in different chemical environments. Procedures to prepare functionalized derivatives with terminal amine, carboxylate, and N-hydroxysuccinimide groups suitable for derivatization and protein bioconjugation have also been developed. These bifunctional ligands have been covalently attached to a number of different proteins, and the terbium complexes’ exceptional photophysical properties are retained. These compounds establish a new aqueous stability and quantum yield standard for long-lifetime lanthanide reporters.
Co-reporter:Géza Szigethy and Kenneth N. Raymond
Journal of the American Chemical Society 2011 Volume 133(Issue 20) pp:7942-7956
Publication Date(Web):May 4, 2011
DOI:10.1021/ja201511u
Several linear, hexa- and tetradentate ligands incorporating a combination of 2,3-dihydroxy-terephthalamide (TAM) and hydroxypyridinone-amide (HOPO) moieties have been developed as uranyl chelating agents. Crystallographic analysis of several {UO2[TAM(HOPO)2]}2– complexes revealed a variable and crowded coordination geometry about the uranyl center. The TAM moiety dominates the bonding in hexadenate complexes, with linker rigidity dictating the equality of equatorial U–O bonding. Hexadentate TAM(HOPO)2 ligands demonstrated slow binding kinetics with uranyl affinities on average 6 orders of magnitude greater than those of similarly linked bis-HOPO ligands. Study of tetradentate TAM(HOPO) ligands revealed that the high uranyl affinity stems primarily from the presence of the TAM moiety and only marginally from increased ligand denticity. Uranyl affinities of TAM(HOPO)2 ligands were within experimental error, with TAM(o-phen-1,2-HOPO)2 exhibiting the most consistent uranyl affinity at variable pH.
Co-reporter:William C. Floyd ; III; Piper J. Klemm ; Danil E. Smiles ; Ayano C. Kohlgruber ; Valérie C. Pierre ; Justin L. Mynar ; Jean M. J. Fréchet
Journal of the American Chemical Society 2011 Volume 133(Issue 8) pp:2390-2393
Publication Date(Web):February 4, 2011
DOI:10.1021/ja110582e
One essential requirement for more sensitive gadolinium-based MRI contrast agents is to slow the molecular tumbling of the gadolinium(III) ion, which increases the gadolinium’s relaxivity (i.e., its ability to speed up the NMR relaxation of nearby water molecules). One route to this is through conjugation to high-molecular-weight polymers such as dendrimers. In this work, amine-functionalized TREN-bis(1,2-HOPO)-TAM-ethylamine and TREN-bis(1-Me-3,2-HOPO)-TAM-ethylamine ligands have been synthesized and attached to biocompatible 40 kDa esteramide (EA)- and poly-l-lysine (PLL)-based dendrimers capable of binding up to eight gadolinium complexes. These conjugates have T1 relaxivities of up to 38.14 ± 0.02 mM−1 s−1 per gadolinium at 37 °C, corresponding to relaxivities of up to 228 mM−1 s−1 per dendrimer molecule. This relaxivity expressed on a “per Gd” basis is several times that of the small-molecule complexes and an order of magnitude higher than that of current commercial agents. Because of their high performance and low toxicity, these macromolecules may constitute an attractive complement to currently available gadolinium(III)-based contrast agents.
Co-reporter:Jeffrey S. Mugridge ; Robert G. Bergman
Journal of the American Chemical Society 2011 Volume 133(Issue 29) pp:11205-11212
Publication Date(Web):June 29, 2011
DOI:10.1021/ja202254x
The self-assembled supramolecular host [Ga4L6]12- (1; L = 1,5-bis[2,3-dihydroxybenzamido]naphthalene) can encapsulate cationic guest molecules within its hydrophobic cavity and catalyze the chemical transformations of bound guests. The cavity of host 1 is lined with aromatic naphthalene groups, which create a magnetically shielded interior environment, resulting in upfield shifted (1–3 ppm) NMR resonances for encapsulated guest molecules. Using gauge independent atomic orbital (GIAO) DFT computations, we show that 1H NMR chemical shifts for guests encapsulated in 1 can be efficiently and accurately calculated and that valuable structural information is obtained by comparing calculated and experimental chemical shifts. The 1H NMR chemical shift calculations are used to map the magnetic environment of the interior of 1, discriminate between different host–guest geometries, and explain the unexpected downfield chemical shift observed for a particular guest molecule interacting with host 1.
Co-reporter:Casey J. Brown ; Gregory M. Miller ; Miles W. Johnson ; Robert G. Bergman
Journal of the American Chemical Society 2011 Volume 133(Issue 31) pp:11964-11966
Publication Date(Web):July 7, 2011
DOI:10.1021/ja205257x
The design of a supramolecular catalyst capable of high-turnover catalysis is reported. A ruthenium(II) catalyst is incorporated into a water-soluble supramolecular assembly, imparting the ability to catalyze allyl alcohol isomerization. The catalyst is protected from decomposition by sequestration inside the host but retains its catalytic activity with scope governed by confinement within the host. This host–guest complex is a uniquely active supramolecular catalyst, capable of >1000 turnovers.
Co-reporter:Praveena D. Garimella ; Ankona Datta ; Dante W. Romanini ; Kenneth N. Raymond ;Matthew B. Francis
Journal of the American Chemical Society 2011 Volume 133(Issue 37) pp:14704-14709
Publication Date(Web):July 29, 2011
DOI:10.1021/ja204516p
MRI contrast agents providing very high relaxivity values can be obtained through the attachment of multiple gadolinium(III) complexes to the interior surfaces of genome-free viral capsids. In previous studies, the contrast enhancement was predicted to depend on the rigidity of the linker attaching the MRI agents to the protein surface. To test this hypothesis, a new set of Gd-hydroxypyridonate based MRI agents was prepared and attached to genetically introduced cysteine residues through flexible and rigid linkers. Greater contrast enhancements were seen for MRI agents that were attached via rigid linkers, validating the design concept and outlining a path for future improvements of nanoscale MRI contrast agents.
Co-reporter:Chengbao Ni, David K. Shuh and Kenneth N. Raymond
Chemical Communications 2011 vol. 47(Issue 22) pp:6392-6394
Publication Date(Web):09 May 2011
DOI:10.1039/C1CC11329A
Uranyl complexes of a bis(methylterephthalamide) ligand (LH4) have been synthesized and characterized by X-ray crystallography. The structure is an unexpected [Me4N]8[L(UO2)]4 tetramer, formed via coordination of the two MeTAM units of L to two uranyl moieties. Addition of KOH to the tetramer gave the corresponding monomeric uranyl methoxide species [Me4N]K2[LUO2(OMe)].
Co-reporter:Trisha M. Hoette, Matthew C. Clifton, Anna M. Zawadzka, Meg A. Holmes, Roland K. Strong, and Kenneth N. Raymond
ACS Chemical Biology 2011 Volume 6(Issue 12) pp:1327
Publication Date(Web):October 6, 2011
DOI:10.1021/cb200331g
The innate immune system antibacterial protein Siderocalin (Scn) binds ferric carboxymycobactin (CMB) and also several catecholate siderophores. Although the recognition of catecholates by Scn has been thoroughly investigated, the binding interactions of Scn with the full spectrum of CMB isoforms have not been studied. Here we show that Scn uses different binding modes for the limited subset of bound CMB isoforms, resulting in a range of binding affinities that are much weaker than other siderophore targets of Scn. Understanding the binding interaction between Scn and CMBs provides clues for the influence of Scn on mycobacterial iron acquisition.
Co-reporter:Joseph Jankolovits;Dr. Christopher M. Andolina;Dr. Jeff W. Kampf; Kenneth N. Raymond; Vincent L. Pecoraro
Angewandte Chemie 2011 Volume 123( Issue 41) pp:9834-9838
Publication Date(Web):
DOI:10.1002/ange.201103851
Co-reporter:Dr. Géza Szigethy;Dr. Kenneth N. Raymond
Chemistry - A European Journal 2011 Volume 17( Issue 6) pp:1818-1827
Publication Date(Web):
DOI:10.1002/chem.201002372
Abstract
Seven water-soluble, tetradentate bis(3-hydroxy-N-methyl-pyridin-2-one) (bis-Me-3,2-HOPO) ligands were synthesized that vary only in linker geometry and rigidity. Solution-phase thermodynamic measurements were conducted between pH 1.6 and pH 9.0 to determine the effects of these variations on proton and uranyl cation affinity. Proton affinity decreases by introduction of the solubilizing triethylene glycol group as compared to unsubstituted reference ligands. Uranyl affinity was found to follow no discernable trends with incremental geometric modification. The butyl-linked 4 li-Me-3,2-HOPO ligand exhibited the highest uranyl affinity, consistent with prior in vivo decorporation results. Of the rigidly-linked ligands, the o-phenylene linker imparted the best uranyl affinity to the bis-Me-3,2-HOPO ligand platform.
Co-reporter:Joseph Jankolovits;Dr. Christopher M. Andolina;Dr. Jeff W. Kampf; Kenneth N. Raymond; Vincent L. Pecoraro
Angewandte Chemie International Edition 2011 Volume 50( Issue 41) pp:9660-9664
Publication Date(Web):
DOI:10.1002/anie.201103851
Co-reporter:Courtney J. Hastings;Mikael P. Backlund; Robert G. Bergman; Kenneth N. Raymond
Angewandte Chemie International Edition 2011 Volume 50( Issue 45) pp:10570-10573
Publication Date(Web):
DOI:10.1002/anie.201105325
Co-reporter:Jeffrey S. Mugridge ; Robert G. Bergman
Journal of the American Chemical Society 2010 Volume 132(Issue 4) pp:1182-1183
Publication Date(Web):January 5, 2010
DOI:10.1021/ja905170x
The self-assembled supramolecular host [Ga4L6]12− can bind cationic guest molecules to both the interior and exterior of the host assembly through noncovalent interactions. Very small equilibrium isotope effects (EIEs) have been precisely measured for the association of benzyltrimethylphosphonium isotopologues to the host exterior surface by adapting an NMR titration method originally developed by the Perrin group for measuring isotope effects on acidity constants. Deuteration of the phosphonium methyl groups was found to have a larger EIE than deuteration at the ring and benzyl positions, suggesting subtle differences in the noncovalent interactions between the host exterior and different guest C−H/D bonds. The application of this method to the measurement of EIEs on noncovalent host−guest interactions demonstrates the generality of this NMR technique in precisely measuring relative equilibrium constants.
Co-reporter:Courtney J. Hastings ; Michael D. Pluth ; Robert G. Bergman
Journal of the American Chemical Society 2010 Volume 132(Issue 20) pp:6938-6940
Publication Date(Web):May 5, 2010
DOI:10.1021/ja102633e
The water-soluble, self-assembled, tetrahedral assembly K12Ga4L6 (L = 1,5-biscatecholamidenaphthalene) catalyzes the Nazarov cyclization of 1,3-pentadienols with extremely high levels of efficiency. The catalyzed reaction proceeds over a million times faster than the background reaction, an increase comparable to those observed in some enzymatic systems. This catalysis operates under aqueous conditions at mild temperatures and pH, and the reaction is halted by the addition of an appropriate inhibitor. This unprecedented rate enhancement is attributed to both the stabilization of protonated reaction intermediates and the effect of constrictive binding on the bound guest.
Co-reporter:Jeffrey S. Mugridge ; Géza Szigethy ; Robert G. Bergman
Journal of the American Chemical Society 2010 Volume 132(Issue 45) pp:16256-16264
Publication Date(Web):October 26, 2010
DOI:10.1021/ja107656g
The supramolecular host assembly [Ga4L6]12− (1; L = 1,5-bis[2,3-dihydroxybenzamido]naphthalene) encapsulates cationic guest molecules within its hydrophobic cavity and catalyzes a variety of chemical transformations within its confined interior space. Despite the well-defined structure, the host ligand framework and interior cavity are very flexible and 1 can accommodate a wide range of guest shapes and sizes. These observations raise questions about the steric effects of confinement within 1 and how encapsulation fundamentally changes the motions of guest molecules. Here we examine the motional dynamics (guest bond rotation and tumbling) of encapsulated guest molecules to probe the steric consequences of encapsulation within host 1. Encapsulation is found to increase the Ph−CH2 bond rotational barrier for ortho-substituted benzyl phosphonium guest molecules by 3 to 6 kcal/mol, and the barrier is found to depend on both guest size and shape. The tumbling dynamics of guests encapsulated in 1 were also investigated, and here it was found that longer, more prolate-shaped guest molecules tumble more slowly in the host cavity than larger but more spherical guest molecules. The prolate guests reduce the host symmetry from T to C1 in solution at low temperatures, and the distortion of the host framework that is in part responsible for this symmetry reduction is observed directly in the solid state. Analysis of guest motional dynamics is a powerful method for interrogating host structure and fundamental host−guest interactions.
Co-reporter:Evan G. Moore ; Jide Xu ; Sheel C. Dodani ; Christoph J. Jocher ; Anthony D’Aléo ; Michael Seitz
Inorganic Chemistry 2010 Volume 49(Issue 9) pp:4156-4166
Publication Date(Web):April 5, 2010
DOI:10.1021/ic902219t
The synthesis, X-ray structure, solution stability, and photophysical properties of several trivalent lanthanide complexes of Yb(III) and Nd(III) using both tetradentate and octadentate ligand design strategies and incorporating the 1-methyl-3-hydroxy-pyridin-2-one (Me-3,2-HOPO) chelate group are reported. Both the Yb(III) and Nd(III) complexes have emission bands in the Near Infra-Red (NIR) region, and this luminescence is retained in aqueous solution (ΦtotYb ∼ 0.09−0.22%). Furthermore, the complexes demonstrate very high stability (pYb ∼ 18.8−21.9) in aqueous solution, making them good candidates for further development as probes for NIR imaging. Analysis of the low temperature (77 K) photophysical measurements for a model Gd(III) complex were used to gain an insight into the electronic structure, and were found to agree well with corresponding time-dependent density functional theory (TD-DFT) calculations at the B3LYP/6-311G++(d,p) level of theory for a simplified model monovalent sodium complex.
Co-reporter:Géza Szigethy
Inorganic Chemistry 2010 Volume 49(Issue 14) pp:6755-6765
Publication Date(Web):June 24, 2010
DOI:10.1021/ic1007878
A series of bis(3-hydroxy-N-methyl-pyridin-2-one) ligands was synthesized, and their respective uranyl complexes were characterized by single crystal X-ray diffraction analyses. These structures were inspected for high-energy conformations and evaluated using a series of metrics to measure co-planarity of chelating moieties with each other and the uranyl coordination plane, as well as to measure coordinative crowding about the uranyl dication. Both very short (ethyl, 3,4-thiophene and o-phenylene) and very long (α,α′-m-xylene and 1,8-fluorene) linkers provide optimal ligand geometries about the uranyl cation, resulting in planar, unstrained molecular arrangements. The planarity of the rigid linkers also suggests there is a degree of pre-organization for a planar coordination mode that is ideal for uranyl-selective ligand design. Comparison of intramolecular Namide−Ophenolate distances and 1H NMR chemical shifts of amide protons supports earlier results that short linkers provide the optimal geometry for intramolecular hydrogen bonding.
Co-reporter:Evan G. Moore ; Jide Xu ; Christoph J. Jocher ; Todd M. Corneillie
Inorganic Chemistry 2010 Volume 49(Issue 21) pp:9928-9939
Publication Date(Web):September 27, 2010
DOI:10.1021/ic101133w
The synthesis, stability, and photophysical properties of several Eu(III) complexes featuring the 1-hydroxypyridin-2-one (1,2-HOPO) chelate group in tetradentate and octadentate ligands are reported. These complexes pair highly efficient emission with exceptional stabilities (pEu ∼ 20.7−21.8) in aqueous solution at pH 7.4. Further analysis of their solution behavior has shown the observed luminescence intensity is significantly diminished below about pH ∼ 6 because of an apparent quenching mechanism involving protonation of the amine backbones. Nonetheless, under biologically relevant conditions, these complexes are promising candidates for applications in Homogeneous Time-Resolved Fluorescence (HTRF) assays and synthetic methodology to prepare derivatives with either a terminal amine or a carboxylate group suitable for bioconjugation has been developed. Lastly, we have demonstrated the use of these compounds as the energy donor in a Luminescence Resonance Energy Transfer (LRET) biological assay format.
Co-reporter:Ama P. S. Samuel;Jamie L. Lunkley;Gilles Muller
European Journal of Inorganic Chemistry 2010 Volume 2010( Issue 21) pp:3343-3347
Publication Date(Web):
DOI:10.1002/ejic.201000309
Abstract
Two luminescent terbium(III) complexes have been prepared from chiral ligands containing 2-hydroxyisophthalamide (IAM) antenna chromophores and their non-polarized and circularly-polarized luminescence properties have been studied. These tetradentate ligands, which form 2:1 ligand/TbIII complexes, utilize diaminocyclohexane (cyLI) and diphenylethylenediamine (dpenLI) backbones, which we reasoned would impart conformational rigidity and result in TbIII complexes that display both large luminescence quantum yield (Φ) values and strong circularly polarized luminescence (CPL) activities. Both TbIII complexes are highly emissive, with Φ values of 0.32 (dpenLI-Tb) and 0.60 (cyLI-Tb). Luminescence lifetime measurements in H2O and D2O indicate that while cyLI-Tb exists as a single species in solution, dpenLI-Tb exists as two species: a monohydrate complex with one H2O molecule directly bound to the TbIII ion and a complex with no water molecules in the inner coordination sphere. Both cyLI-Tb and dpenLI-Tb display increased CPL activity compared to previously reported TbIII complexes made with chiral IAM ligands. The CPL measurements also provide additional confirmation of the presence of a single emissive species in solution in the case of cyLI-Tb, and multiple emissive species in the case of dpenLI-Tb.
Co-reporter:JeffreyS. Mugridge;RobertG. Bergman ;KennethN. Raymond
Angewandte Chemie International Edition 2010 Volume 49( Issue 21) pp:3635-3637
Publication Date(Web):
DOI:10.1002/anie.200906569
Co-reporter:JeffreyS. Mugridge;RobertG. Bergman ;KennethN. Raymond
Angewandte Chemie International Edition 2010 Volume 49( Issue 21) pp:
Publication Date(Web):
DOI:10.1002/anie.201001707
Co-reporter:Evan G. Moore, Amanda P. S. Samuel and Kenneth N. Raymond
Accounts of Chemical Research 2009 Volume 42(Issue 4) pp:542
Publication Date(Web):March 26, 2009
DOI:10.1021/ar800211j
Ligand-sensitized, luminescent lanthanide(III) complexes are of considerable importance because their unique photophysical properties (microsecond to millisecond lifetimes, characteristic and narrow emission bands, and large Stokes shifts) make them well suited as labels in fluorescence-based bioassays. The long-lived emission of lanthanide(III) cations can be temporally resolved from scattered light and background fluorescence to vastly enhance measurement sensitivity. One challenge in this field is the design of sensitizing ligands that provide highly emissive complexes with sufficient stability and aqueous solubility for practical applications. In this Account, we give an overview of some of the general properties of the trivalent lanthanides and follow with a summary of advances made in our laboratory in the development of highly luminescent Tb(III) and Eu(III) complexes for applications in biotechnology. A focus of our research has been the optimization of these compounds as potential commercial agents for use in homogeneous time-resolved fluorescence (HTRF) technology. Our approach involves developing high-stability octadentate Tb(III) and Eu(III) complexes that rely on all-oxygen donor atoms and using multichromophore chelates to increase molar absorptivity; earlier examples utilized a single pendant chromophore (that is, a single “antenna”). Ligands based on 2-hydroxyisophthalamide (IAM) provide exceptionally emissive Tb(III) complexes with quantum yield values up to ∼60% that are stable at the nanomolar concentrations required for commercial assays. Through synthetic modification of the IAM chromophore and time-dependent density functional theory (TD-DFT) calculations, we have developed a method to predict absorption and emission properties of these chromophores as a tool to guide ligand design. Additionally, we have investigated chiral IAM ligands that yield Tb(III) complexes possessing both high quantum yield values and strong circularly polarized luminescence (CPL) activity. To efficiently sensitize Eu(III) emission, we have used the 1-hydroxypyridin-2-one (1,2-HOPO) chelate to create remarkable ligands that combine excellent photophysical properties and exceptional aqueous stabilities. A more complete understanding of this chromophore has been achieved by combining low-temperature phosphorescence measurements with the same TD-DFT approach used with the IAM system. Eu(III) complexes with strong CPL activity have also been obtained with chiral 1,2-HOPO ligands. We have also undertaken kinetic analysis of radiative and nonradiative decay pathways for a series of Eu(III) complexes; the importance of the metal ion symmetry on the ensuing photophysical properties is clear. Lastly, we describe a Tb(III)-IAM compound—now carried through to commercial availability—that offers improved performance in the common HTRF platform and has the potential to vastly improve sensitivity.
Co-reporter:Ankona Datta and Kenneth N. Raymond
Accounts of Chemical Research 2009 Volume 42(Issue 7) pp:938
Publication Date(Web):June 8, 2009
DOI:10.1021/ar800250h
Magnetic resonance imaging (MRI) is a particularly effective tool in medicine because of its high depth penetration (from 1 mm to 1 m) and ability to resolve different soft tissues. The MRI signal is generated by the relaxation of in vivo water molecule protons. MRI images can be improved by administering paramagnetic agents, which increase the relaxation rates of nearby water protons, thereby enhancing the MRI signal. The lanthanide cation Gd3+ is generally used because of its favorable electronic properties; high toxicity, however, necessitates strongly coordinating ligands to keep Gd3+ completely bound while in the patient. In this Account, we give a coordination chemistry overview of contrast agents (CAs) based on Gd−hydroxypyridinone (HOPO), which show improved MRI contrast and high thermodynamic stabilities. Tris-bidentate HOPO-based ligands developed in our laboratory were designed to complement the coordination preferences of Gd3+, especially its oxophilicity. The HOPO ligands provide a hexadentate coordination environment for Gd3+, in which all of the donor atoms are oxygen. Because Gd3+ favors eight or nine coordination, this design provides two to three open sites for inner-sphere water molecules. These water molecules rapidly exchange with bulk solution, hence affecting the relaxation rates of bulk water molecules. The parameters affecting the efficiency of these contrast agents have been tuned to improve contrast while still maintaining a high thermodynamic stability for Gd3+ binding. The Gd−HOPO-based contrast agents surpass current commercially available agents because of a higher number of inner-sphere water molecules, rapid exchange of inner-sphere water molecules via an associative mechanism, and a long electronic relaxation time. The contrast enhancement provided by these agents is at least twice that of commercial contrast agents, which are based on polyaminocarboxylate ligands. Advances in MRI technology have made significant contributions to the improvement of clinical diagnostics by allowing visualization of underlying pathology. However, understanding the mechanism of a disease at the molecular level requires improved imaging sensitivity. The ultimate goal is to visually distinguish between different disease targets or markers, such as enzymes, hormones, proteins, or small molecules, at biologically relevant concentrations (from micro- to nanomolar). Although MRI techniques can provide images of the organs and tissues in which these biomarkers are regulated, the high sensitivity required to visualize the biological targets within the tissues is currently lacking; contrast enhancements of 50-fold beyond current agents are required to achieve this goal. According to the theory of paramagnetic relaxation, the contrast enhancement can be further improved by slowing the tumbling rate of the MRI agent. Theoretically, this enhancement would be greater for contrast agents with an optimal rate of water exchange. The Gd−HOPO-based contrast agents have optimal water-exchange rates, whereas the commercial agents have slower non-optimal water-exchange rates; thus, the Gd−HOPO agents are ideal for attachment to macromolecules, which will slow down the tumbling rate and increase contrast. This strategy has been recently tested with the Gd−HOPO agents via covalent attachment to virus capsids, affording contrast enhancements 10-fold beyond commercial agents.
Co-reporter:Michael D. Pluth, Robert G. Bergman and Kenneth N. Raymond
Accounts of Chemical Research 2009 Volume 42(Issue 10) pp:1650
Publication Date(Web):July 10, 2009
DOI:10.1021/ar900118t
Synthetic supramolecular host assemblies can impart unique reactivity to encapsulated guest molecules. Synthetic host molecules have been developed to carry out complex reactions within their cavities, despite the fact that they lack the type of specifically tailored functional groups normally located in the analogous active sites of enzymes. Over the past decade, the Raymond group has developed a series of self-assembled supramolecules and the Bergman group has developed and studied a number of catalytic transformations. In this Account, we detail recent collaborative work between these two groups, focusing on chemical catalysis stemming from the encapsulation of protonated guests and expanding to acid catalysis in basic solution. We initially investigated the ability of a water-soluble, self-assembled supramolecular host molecule to encapsulate protonated guests in its hydrophobic core. Our study of encapsulated protonated amines revealed rich host−guest chemistry. We established that self-exchange (that is, in−out guest movement) rates of protonated amines were dependent on the steric bulk of the amine rather than its basicity. The host molecule has purely rotational tetrahedral (T) symmetry, so guests with geminal N-methyl groups (and their attendant mirror plane) were effectively desymmetrized; this allowed for the observation and quantification of the barriers for nitrogen inversion followed by bond rotation. Furthermore, small nitrogen heterocycles, such as N-alkylaziridines, N-alkylazetidines, and N-alkylpyrrolidines, were found to be encapsulated as proton-bound homodimers or homotrimers. We further investigated the thermodynamic stabilization of protonated amines, showing that encapsulation makes the amines more basic in the cavity. Encapsulation raises the effective basicity of protonated amines by up to 4.5 pKa units, a difference almost as large as that between the moderate and strong bases carbonate and hydroxide. The thermodynamic stabilization of protonated guests was translated into chemical catalysis by taking advantage of the potential for accelerating reactions that take place via positively charged transition states, which could be potentially stabilized by encapsulation. Orthoformates, generally stable in neutral or basic solution, were found to be suitable substrates for catalytic hydrolysis by the assembly. Orthoformates small enough to undergo encapsulation were readily hydrolyzed by the assembly in basic solution, with rate acceleration factors up to 3900 compared with those of the corresponding uncatalyzed reactions. Furthering the analogy to enzymes that obey Michaelis−Menten kinetics, we observed competitive inhibition with the inhibitor NPr4+, thereby confirming that the interior cavity of the assembly was the active site for catalysis. Mechanistic studies revealed that the assembly is required for catalysis and that the rate-limiting step of the reaction involves proton transfer from hydronium to the encapsulated substrate. Encapsulation in the assembly changes the orthoformate hydrolysis from an A-1 mechanism (in which decomposition of the protonated substrate is the rate-limiting step) to an A-SE2 mechanism (in which proton transfer is the rate-limiting step). The study of hydrolysis in the assembly was next extended to acetals, which were also catalytically hydrolyzed by the assembly in basic solution. Acetal hydrolysis changed from the A-1 mechanism in solution to an A-2 mechanism inside the assembly, where attack of water on the protonated substrate is rate limiting. This work provides rare examples of assembly-catalyzed reactions that proceed with substantial rate accelerations despite the absence of functional groups in the cavity and with mechanisms fully elucidated by quantitative kinetic studies.
Co-reporter:Rebecca J. Abergel ; Anna M. Zawadzka ; Trisha M. Hoette
Journal of the American Chemical Society 2009 Volume 131(Issue 35) pp:12682-12692
Publication Date(Web):August 11, 2009
DOI:10.1021/ja903051q
Bacillibactin and enterobactin are hexadentate catecholate siderophores produced by bacteria upon iron limitation to scavenge ferric ion and seem to be the ultimate siderophores of their two respective domains: Gram-positive and Gram-negative. Iron acquisition mediated by these trilactone-based ligands necessitates enzymatic hydrolysis of the scaffold for successful intracellular iron delivery. The esterases BesA and Fes hydrolyze bacillibactin and enterobactin, respectively, as well as the corresponding iron complexes. Bacillibactin binds iron through three 2,3-catecholamide moieties linked to a trithreonine scaffold via glycine spacers, whereas in enterobactin the iron-binding moieties are directly attached to a tri-l-serine backbone; although apparently minor, these structural differences result in markedly different iron coordination properties and iron transport behavior. Comparison of the solution thermodynamic and circular dichroism properties of bacillibactin, enterobactin and the synthetic analogs d-enterobactin, SERGlyCAM and d-SERGlyCAM has determined the role of each different feature in the siderophores’ molecular structures in ferric complex stability and metal chirality. While opposite metal chiralities in the different complexes did not affect transport and incorporation in Bacillus subtilis, ferric complexes formed with the various siderophores did not systematically promote growth of the bacteria. The bacillibactin esterase BesA is less specific than the enterobactin esterase Fes; BesA can hydrolyze the trilactones of both siderophores, while only the tri-l-serine trilactone is a substrate of Fes. Both enzymes are stereospecific and cannot cleave tri-d-serine lactones. These data provide a complete picture of the microbial iron transport mediated by these two siderophores, from initial recognition and transport to intracellular iron release.
Co-reporter:Carmelo Sgarlata ; Jeffrey S. Mugridge ; Michael D. Pluth ; Bryan E. F. Tiedemann ; Valeria Zito ; Giuseppe Arena
Journal of the American Chemical Society 2009 Volume 132(Issue 3) pp:1005-1009
Publication Date(Web):December 31, 2009
DOI:10.1021/ja9056739
NMR, UV−vis, and isothermal titration calorimetry (ITC) measurements probe different aspects of competing host−guest equilibria as simple alkylammonium guest molecules interact with both the exterior (ion-association) and interior (encapsulation) of the [Ga4L6]12− supramolecular assembly in water. Data obtained by each independent technique measure different components of the host−guest equilibria and only when analyzed together does a complete picture of the solution thermodynamics emerge. Striking differences between the internal and external guest binding are found. External binding is enthalpy driven and mainly due to attractive interactions between the guests and the exterior surface of the assembly while encapsulation is entropy driven as a result of desolvation and release of solvent molecules from the host cavity.
Co-reporter:Casey J. Brown ; Robert G. Bergman
Journal of the American Chemical Society 2009 Volume 131(Issue 48) pp:17530-17531
Publication Date(Web):November 12, 2009
DOI:10.1021/ja906386w
The chiral supramolecular catalyst Ga4L6 [L = 1,5-bis(2,3-dihydroxybenzoylamino)naphthalene] is a molecular tetrahedron that catalyzes the 3-aza-Cope rearrangement of allyl enammonium cations. This catalysis is accomplished by preorganizing the substrate in a reactive conformation within the host. This work demonstrates that through the use of enantiopure assembly, its chiral cavity is capable of catalyzing the 3-aza-Cope rearrangement enantioselectively, with yields of 21−74% and enantiomeric excesses from 6 to 64% at 50 °C. At lower temperatures, the enantioselectivity improved, reaching 78% ee at 5 °C. This is the highest enantioselectivity to date induced by the chiral cavity of a supramolecular assembly.
Co-reporter:Anthony D’Aléo, Evan G. Moore, Géza Szigethy, Jide Xu and Kenneth N. Raymond
Inorganic Chemistry 2009 Volume 48(Issue 19) pp:9316-9324
Publication Date(Web):September 1, 2009
DOI:10.1021/ic901161z
The efficiency of Eu3+ luminescence by energy transfer from an antenna ligand can be strongly dependent on the metal ion coordination geometry. The geometric component of the Eu(III) sensitization has been probed using series of tetradentate 1,2-HOPO derivatives that are connected by bridges of varying length and geometry. The ligands are N,N′-(1,2-phenylene)bis(1-hydroxy-6-oxo-1,6-dihydropyridine-2-carboxamide) for the ligand (L1), 1-hydroxy-N-(2-(1-hydroxy-6-oxo-1,6-dihydropyridine-2-carboxamido)benzyl)-6-oxo-1,6-dihydropyridine-2-carboxamide (L2) and N,N′-(1,2-phenylenebis(methylene))bis(1-hydroxy-6-oxo-1,6-dihydropyridine-2-carboxamide) (L3). Spectroscopic characterization of both the Gd(III) and the Eu(III) metal complexes, time-dependent density functional theory (TD-DFT) analysis of model compounds and evaluation of the kinetic parameters for the europium emission were completed. Some striking differences were observed in the luminescence quantum yield by altering the bridging unit. The [Eu(L2)2]− derivative shows efficient sensitization coupled with good metal centered emission. For [Eu(L3)2]−, the large quenching of the luminescence quantum yield compared to [Eu(L2)2]− is primarily a result of one inner sphere water molecule bound to the europium cation while for [Eu(L1)2]−, the low luminescence quantum yield can be attributed to inefficient sensitization of the europium ion.
Co-reporter:Rebecca J. Abergel ; Anthony D’Aléo ; Clara Ng Pak Leung ; David K. Shuh
Inorganic Chemistry 2009 Volume 48(Issue 23) pp:10868-10870
Publication Date(Web):November 10, 2009
DOI:10.1021/ic9013703
Although widely used in bioassays, the spectrofluorimetric method described here uses the antenna effect as a tool to probe the thermodynamic parameters of ligands that sensitize lanthanide luminescence. The Eu3+ coordination chemistry, solution thermodynamic stability, and photophysical properties of the spermine-based hydroxypyridonate octadentate chelator 3,4,3-LI(1,2-HOPO) are reported. The complex [EuIII(3,4,3-LI(1,2-HOPO))]− luminesces with a long lifetime (805 μs) and a quantum yield of 7.0% in aqueous solution, at pH 7.4. These remarkable optical properties were exploited to determine the high (and proton-independent) stability of the complex (log β110 = 20.2(2)) and to define the influence of the ligand scaffold on the stability and photophysical properties.
Co-reporter:Eric J. Werner, Julia Kozhukh, Mauro Botta, Evan G. Moore, Stefano Avedano, Silvio Aime and Kenneth N. Raymond
Inorganic Chemistry 2009 Volume 48(Issue 1) pp:277-286
Publication Date(Web):November 25, 2008
DOI:10.1021/ic801730u
Four new Gd(III) complexes based on the 1,2-hydroxypyridinone chelator have been synthesized and evaluated as potential magentic resonance imaging contrast agents. Previously reported work examining Gd-TREN-1,2-HOPO (3; HOPO = hydroxypyridinone) suggests that the 1,2-HOPO unit binds strongly and selectively to Gd(III), encouraging further study of the stability and relaxivity properties of this class of compounds. Among the new complexes presented in this paper are the homopodal Gd-Ser-TREN-1,2-HOPO (Gd-5) and three heteropodal bis-1,2-HOPO-TAM complexes (Gd-6, Gd-7, and Gd-8; TAM = terephthalamide). Conditional stability constants were determined, and all pGd values are in the range of 18.5−19.7, comparable to other analogous HOPO complexes and currently used commercial contrast agents. Relaxivities for all complexes are about twice those of commercial agents, ranging from 7.8 to 10.5 mM−1 s−1 (20 MHz; 25 °C), and suggest two innersphere water molecules in fast exchange. Luminescent measurements were used to verify the number of coordinated waters for Gd-5, and VT 17O NMR experiments were employed for the highly soluble Gd-TREN-bis-1,2-HOPO-TAM-N3 (Gd-8) complex to measure a fast water exchange rate, 298kex = 1/τM, of 5.1 (±0.4) × 108 s−1 (298τM ∼ 2 ns).
Co-reporter:Michael D. Pluth, Darren W. Johnson, Géza Szigethy, Anna V. Davis, Simon J. Teat, Allen G. Oliver, Robert G. Bergman and Kenneth N. Raymond
Inorganic Chemistry 2009 Volume 48(Issue 1) pp:111-120
Publication Date(Web):November 20, 2008
DOI:10.1021/ic8012848
The molecular structure of the spontaneously assembled supramolecular cluster [M4L6]n− has been explored with different metals (M = GaIII, FeIII, TiIV) and different encapsulated guests (NEt4+, BnNMe3+, Cp2Co+, Cp*2Co+) by X-ray crystallography. While the identity of the metal ions at the vertices of the M4L6 structure is found to have little effect on the assembly structure, encapsulated guests significantly distort the size and shape of the interior cavity of the assembly. Cations on the exterior of the assembly are found to interact with the assembly through either π−π, cation−π, or CH−π interactions. In some cases, the exterior guests interact with only one assembly, but cations with the ability to form multiple π−π interactions are able to interact with adjacent assemblies in the crystal lattice. The solvent accessible cavity of the assembly is modeled using the rolling probe method and found to range from 253−434 Å3, depending on the encapsulated guest. On the basis of the volume of the guest and the volume of the cavity, the packing coefficient for each host−guest complex is found to range from 0.47−0.67.
Co-reporter:Amanda P. S. Samuel, Jide Xu and Kenneth N. Raymond
Inorganic Chemistry 2009 Volume 48(Issue 2) pp:687-698
Publication Date(Web):December 17, 2008
DOI:10.1021/ic801904s
A series of highly luminescent Tb(III) complexes of para-substituted 2-hydroxyisophthalamide ligands (5LI-IAM-X) has been prepared (X = H, CH3, (C═O)NHCH3, SO3−, NO2, OCH3, F, Cl, Br) to probe the effect of substituting the isophthalamide ring on ligand and Tb(III) emission in order to establish a method for predicting the effects of chromophore modification on Tb(III) luminescence. The energies of the ligand singlet and triplet excited states are found to increase linearly with the π-withdrawing ability of the substituent. The experimental results are supported by time-dependent density functional theory calculations performed on model systems, which predict ligand singlet and triplet energies within ∼5% of the experimental values. The quantum yield (Φ) values of the Tb(III) complexes increase with the triplet energy of the ligand, which is in part due to decreased non-radiative deactivation caused by thermal repopulation of the triplet. Together, the experimental and theoretical results serve as a predictive tool that can guide the synthesis of ligands used to sensitize lanthanide luminescence.
Co-reporter:Géza Szigethy and Kenneth N. Raymond
Inorganic Chemistry 2009 Volume 48(Issue 24) pp:11489-11491
Publication Date(Web):November 23, 2009
DOI:10.1021/ic901815b
Structural characterization of a mononuclear uranyl complex with a tetradentate, thiophene-linked bis(3-hydroxy-N-methylpyridin-2-one) ligand reveals the most planar coordination geometry yet observed with this ligand class. The introduction of ethylsulfanyl groups onto the thiophene linker disrupts this planar, conjugated ligand arrangement, resulting in the formation of dimeric (UO2)2L2 species in which each ligand spans two uranyl centers. Relative energy calculations reveal that this tendency toward dimer formation is the result of steric interference between ethylsulfanyl substitutents and linking amides.
Co-reporter:Anna M. Zawadzka, Rebecca J. Abergel, Rita Nichiporuk, Ulla N. Andersen and Kenneth N. Raymond
Biochemistry 2009 Volume 48(Issue 16) pp:
Publication Date(Web):March 2, 2009
DOI:10.1021/bi8018674
During growth under iron limitation, Bacillus cereus and Bacillus anthracis, two human pathogens from the Bacillus cereus group of Gram-positive bacteria, secrete two siderophores, bacillibactin (BB) and petrobactin (PB), for iron acquisition via membrane-associated substrate-binding proteins (SBPs) and other ABC transporter components. Since PB is associated with virulence traits in B. anthracis, the PB-mediated iron uptake system presents a potential target for antimicrobial therapies; its characterization in B. cereus is described here. Separate transporters for BB, PB, and several xenosiderophores are suggested by 55Fe-siderophore uptake studies. The PB precursor, 3,4-dihydroxybenzoic acid (3,4-DHB), and the photoproduct of FePB (FePBν) also mediate iron delivery into iron-deprived cells. Putative SBPs were recombinantly expressed, and their ligand specificity and binding affinity were assessed using fluorescence spectroscopy. The noncovalent complexes of the SBPs with their respective siderophores were characterized using ESI-MS. The differences between solution phase behavior and gas phase measurements are indicative of noncovalent interactions between the siderophores and the binding sites of their respective SBPs. These studies combined with bioinformatics sequence comparison identify SBPs from five putative transporters specific for BB and enterobactin (FeuA), 3,4-DHB and PB (FatB), PB (FpuA), schizokinen (YfiY), and desferrioxamine and ferrichrome (YxeB). The two PB receptors show different substrate ranges: FatB has the highest affinity for ferric 3,4-DHB, iron-free PB, FePB, and FePBν, whereas FpuA is specific to only apo- and ferric PB. The biochemical characterization of these SBPs provides the first identification of the transporter candidates that most likely play a role in the B. cereus group pathogenicity.
Co-reporter:Anthony D'Aléo;Jide Xu;King Do;Gilles Muller;KennethN. Raymond
Helvetica Chimica Acta 2009 Volume 92( Issue 11) pp:2439-2460
Publication Date(Web):
DOI:10.1002/hlca.200900161
Abstract
The synthesis of the cyclen derivative H4L1⋅2 HBr containing four 2-hydroxybenzamide groups is described. The spectroscopic properties of the LnIII conplexes of L1 (Ln=Gd, Tb, Yb, and Eu) reveal changes of the UV/VIS-absorption, circular-dichroism-absorption, luminescence, and circularly polarized luminescence spectra. It is shown that at least two metal-complex species are present in solution, whose relative amounts are pH dependent. At pH>8.0, an intense long-lived emission is observed (for [TbL1] and [YbL1]), while at pH<8.0, a weaker, shorter-lived species predominates. Unconventional LnIII emitters (Pr, Nd, Sm, Dy, and Tm) were sensitized in basic solution, both in the VIS and in the near-IR, to measure the emission of these ions.
Co-reporter:Michael D. Pluth;Dorothea Fiedler;Jeffrey S. Mugridge;Robert G. Bergman
PNAS 2009 Volume 106 (Issue 26 ) pp:10438-10443
Publication Date(Web):2009-06-30
DOI:10.1073/pnas.0809806106
Cyclic amines can be encapsulated in a water-soluble self-assembled supramolecular host upon protonation. The hydrogen-bonding
ability of the cyclic amines, as well as the reduced degrees of rotational freedom, allows for the formation of proton-bound
homodimers inside of the assembly that are otherwise not observable in aqueous solution. The generality of homodimer formation
was explored with small N-alkyl aziridines, azetidines, pyrrolidines, and piperidines. Proton-bound homodimer formation is observed for N-alkylaziridines (R = methyl, isopropyl, tert-butyl), N-alkylazetidines (R = isopropyl, tert-butyl), and N-methylpyrrolidine. At high concentration, formation of a proton-bound homotrimer is observed in the case of N-methylaziridine. The homodimers stay intact inside the assembly over a large concentration range, thereby suggesting cooperative
encapsulation. Both G3(MP2)B3 and G3B3 calculations of the proton-bound homodimers were used to investigate the enthalpy of
the hydrogen bond in the proton-bound homodimers and suggest that the enthalpic gain upon formation of the proton-bound homodimers
may drive guest encapsulation.
Co-reporter:Michael D. Pluth, Robert G. Bergman and Kenneth N. Raymond
The Journal of Organic Chemistry 2009 Volume 74(Issue 1) pp:58-63
Publication Date(Web):December 2, 2008
DOI:10.1021/jo802131v
A self-assembled supramolecular host catalyzes the hydrolysis of acetals in basic aqueous solution. The mechanism of hydrolysis is consistent with the Michaelis−Menten kinetic model. Further investigation of the rate-limiting step of the reaction revealed a negative entropy of activation (ΔS⧧ = −9 cal mol−1 K−1) and an inverse solvent isotope effect (k(H2O)/k(D2O) = 0.62). These data suggest that the mechanism of hydrolysis that takes place inside the assembly proceeds through an A-2 mechanism, in contrast to the A-1 mechanism operating in the uncatalyzed reaction. Comparison of the rates of acetal hydrolysis in the assembly with the rate of the reaction of unencapsulated substrates reveals rate accelerations of up to 980 over the background reaction for the substrate 1,1-diethoxyethane.
Co-reporter:Michael D. Pluth ; Bryan E. F. Tiedemann ; Herman van Halbeek ; Rudi Nunlist
Inorganic Chemistry 2008 Volume 47(Issue 5) pp:1411-1413
Publication Date(Web):February 9, 2008
DOI:10.1021/ic7020885
The diffusion coefficient of the self-assembled supramolecular cluster [Ga4L6]12− depends on the cationic counterions in solution. Diffusion coefficients were determined using the pulsed-gradient spin–echo 1H NMR method and fit using nonlinear least-squares refinement. Saturation studies revealed a small number of ion-association sites on the exterior of the assembly and the direct observation of ion association in water. The addition of excess alkali-metal cations displaces the ion-associated hydrophobic tetraalkylammonium cations. Comparisons between tetraethyl- and tetrapropylammonium show a preference for ion association with the more hydrophobic cation.
Co-reporter:Anthony D’Aléo ; ; Jide Xu ; ; Evan G. Moore ; ; Christoph J. Jocher ; ;
Inorganic Chemistry 2008 Volume 47(Issue 14) pp:6109-6111
Publication Date(Web):June 14, 2008
DOI:10.1021/ic8003189
The synthesis, crystal structure, solution stability, and photophysical properties of an aryl group bridging two 1-hydroxypyridin-2-one units complexed to Eu(III) are reported. The results show that this backbone unit increases the rigidity of the ensuing complex, and also the conjugation of the ligand. As a result of the latter, the singlet absorption energy is decreased, along with the energy of the lowest excited triplet state. The resulting efficiency of sensitization for the Eu(III) ion is influenced by these phenomena, yielding an overall quantum yield of 6.2% in aqueous solution. The kinetic parameters arising from the luminescence data reveal an enhanced nonradiative decay rate for this compound when compared to previously reported aliphatic bridges.
Co-reporter:Amanda P. S. Samuel ; Evan G. Moore ; Marco Melchior ; Jide Xu
Inorganic Chemistry 2008 Volume 47(Issue 17) pp:7535-7544
Publication Date(Web):August 1, 2008
DOI:10.1021/ic800328g
A series of octadentate ligands featuring the 2-hydroxyisophthalamide (IAM) antenna chromophore to sensitize Tb(III) and Eu(III) luminescence has been prepared and characterized. The length of the alkyl amine scaffold that links the four IAM moieties has been varied to investigate the effect of the ligand backbone on the stability and photophysical properties of the Ln(III) complexes. The amine backbones utilized in this study are N,N,N′,N′-tetrakis-(2-aminoethyl)-ethane-1,2-diamine [H(2,2)-], N,N,N′,N′-tetrakis-(2-aminoethyl)-propane-1,3-diamine [H(3,2)-], and N,N,N′,N′-tetrakis-(2-aminoethyl)-butane-1,4-diamine [H(4,2)-]. These ligands also incorporate methoxyethylene [MOE] groups on each of the IAM chromophores to increase their water solubility. The aqueous ligand protonation constants and Tb(III) and Eu(III) formation constants were determined from solution thermodynamic studies. The resulting values indicate that at physiological pH the Eu(III) and Tb(III) complexes of H(2,2)-IAM-MOE and H(4,2)-IAM-MOE are sufficiently stable to prevent dissociation at nanomolar concentrations. The photophysical measurements for the Tb(III) complexes gave overall quantum yield values of 0.56, 0.39, and 0.52 respectively for the complexes with H(2,2)-IAM-MOE, H(3,2)-IAM-MOE, and H(4,2)-IAM-MOE, while the corresponding Eu(III) complexes displayed significantly weaker luminescence, with quantum yield values of 0.0014, 0.0015, and 0.0058, respectively. Analysis of the steady state Eu(III) emission spectra provides insight into the solution symmetries of the complexes. The combined solubility, stability, and photophysical performance of the Tb(III) complexes in particular make them well suited to serve as the luminescent reporter group in high sensitivity time-resolved fluoroimmunoassays.
Co-reporter:Michael Seitz ; Evan G. Moore
Inorganic Chemistry 2008 Volume 47(Issue 19) pp:8665-8673
Publication Date(Web):August 27, 2008
DOI:10.1021/ic800269w
The neutral complexes of two ligands based on the 1-oxo-2-hydroxy-isoquinoline motif with group 13 metals (Al, Ga, In) show bright blue-violet luminescence in organic solvents. The corresponding transition can be attributed to ligand-centered singlet emission, characterized by a small Stokes shifts of only a few nanometers combined with lifetimes in the range between 1 and 3 ns. The fluorescence efficiency is high, with quantum yields of up to 37% in benzene solution. The crystal structure of one of the indium(III) complexes (trigonal space group R3̅, a = b = 13.0384(15) Å, c = 32.870(8) Å, α = β = 90°, γ = 120°, V = 4839.3(14) Å3, Z = 6) shows a six-coordinate geometry around the indium center, which is close to trigonal-prismatic, with a twist angle between the two trigonal faces of 20.7°. Time-dependent density functional theory calculations (Al and Ga, B3LYP/6-31G(d); In, B3LYP/LANL2DZ) of the fac and mer isomers with one of the two ligands indicate that there is no clear preference for either one of the isomeric forms of the metal complexes. In addition, the metal centers do not have a significant influence on the electronic structure nor, as a consequence, on the predominant intraligand optical transitions.
Co-reporter:Evan G. Moore ; Michael Seitz
Inorganic Chemistry 2008 Volume 47(Issue 19) pp:8571-8573
Publication Date(Web):August 26, 2008
DOI:10.1021/ic801060x
The synthesis, structure, and characterization of a [Yb(Tren-Me-3,2-HOPO)(H2O)2] complex are reported. As a result of its YbIII emission in the near-infrared region, sensitized by the Me-3,2-HOPO chromophore, this complex can be utilized for the first time to determine the hydration state, q, via the luminescence lifetimes and hence the solution structure of these Me-3,2-HOPO-type ligands, which have attracted significant interest in complex with GdIII as possible next-generation magnetic resonance imaging contrast agents.
Co-reporter:Evan G. Moore ; Jide Xu ; Christoph J. Jocher ; Ingrid Castro-Rodriguez
Inorganic Chemistry 2008 Volume 47(Issue 8) pp:3105-3118
Publication Date(Web):March 1, 2008
DOI:10.1021/ic702144n
The synthesis, X-ray structure, stability, and photophysical properties of several trivalent lanthanide complexes formed from two differing bis-bidentate ligands incorporating either alkyl or alkyl ether linkages and featuring the 1-hydroxy-2-pyridinone (1,2-HOPO) chelate group in complex with Eu(III), Sm(III), and Gd(III) are reported. The Eu(III) complexes are among some of the best examples, pairing highly efficient emission (ΦtotEu ∼ 21.5%) with high stability (pEu ∼ 18.6) in aqueous solution, and are excellent candidates for use in biological assays. A comparison of the observed behavior of the complexes with differing backbone linkages shows remarkable similarities, both in stability and photophysical properties. Low temperature photophysical measurements for a Gd(III) complex were also used to gain insight into the electronic structure and were found to agree with corresponding time-dependent density functional theory (TD-DFT) calculations for a model complex. A comparison of the high resolution Eu(III) emission spectra in solution and from single crystals also revealed a more symmetric coordination geometry about the metal ion in solution due to dynamic rotation of the observed solid state structure.
Co-reporter:Christoph J. Jocher ; Evan G. Moore ; Jason D. Pierce
Inorganic Chemistry 2008 Volume 47(Issue 18) pp:7951-7953
Publication Date(Web):August 16, 2008
DOI:10.1021/ic8010162
The synthesis, characterization, and photophysical properties are reported for several Ln(III) complexes of a tetradentate chelate, 5LIO-MAM, derived from the common flavor enhancer “maltol”. Eu(III), Yb(III), and Nd(III) form stable ML2 complexes in aqueous solution that emit in the red or near-infrared (NIR) upon excitation at ca. 330 nm. The synthesis, aqueous stability, and photophysical properties are reported for a novel tetradentate ligand derived from maltol, a commonly used flavor enhancer. In aqueous solution, this chelate forms stable complexes with Ln(III) cations, and sensitized emission was observed from Eu(III), Yb(III), and Nd(III). A comparison with recently reported and structurally analogous ligands reveals a slightly higher basicity but lower complex stability with Eu(III) [pEu = 14.7(1)]. A very poor metal-centered quantum yield with Eu(III) was observed (Φtot = 0.04%), which can be rationalized by the similar energy of the ligand triplet state and the Eu(III) 5D0 emissive level. Instead, sensitized emission from the Yb(III) and Nd(III) cations was observed, which emit in the NIR.
Co-reporter:Géza Szigethy;Jide Xu;Anne E. V. Gorden;Simon J. Teat;David K. Shuh
European Journal of Inorganic Chemistry 2008 Volume 2008( Issue 13) pp:2143-2147
Publication Date(Web):
DOI:10.1002/ejic.200800144
Abstract
As part of a study to characterize the detailed coordination behavior of PuIV, single-crystal X-ray diffraction structures have been determined for PuIV and CeIV complexes with the naturally occurring ligand maltol (3-hydroxy-2-methylpyran-4-one) and its derivative bromomaltol (5-bromo-3-hydroxy-2-methylpyran-4-one). Although CeIV is generally accepted as a structural analog for PuIV, and the maltol complexes of these two metals are isostructural, the corresponding bromomaltol complexes are strikingly different with respect to ligand orientation about the metal ion: All complexes exhibit trigonal dodecahedral coordination geometry but the CeIV–bromomaltol complex displays an uncommon ligand arrangement not found in the PuIV complex.(© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2008)
Co-reporter:Michael Seitz
European Journal of Organic Chemistry 2008 Volume 2008( Issue 16) pp:2697-2700
Publication Date(Web):
DOI:10.1002/ejoc.200800147
Abstract
2-Hydroxyisoquinolin-1-one (1,2-HOIQO) is a new member of the important class of aromatic cyclic hydroxamic acid ligands which are widely used in metal sequestering applications and metal chelating therapy. The first general approach for the introduction of substituents at the aromatic ring of the chelating moiety is presented. As a useful derivative, the highly water-soluble sulfonic acid has been synthesized by an efficient route that allows general access to 1,2-HOIQO 3-carboxylic acid amides, which are the most relevant for applications. (© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2008)
Co-reporter:Rebecca J. Abergel
JBIC Journal of Biological Inorganic Chemistry 2008 Volume 13( Issue 2) pp:229-240
Publication Date(Web):2008 February
DOI:10.1007/s00775-007-0314-y
The mechanism and effectiveness of iron removal from transferrin by three series of new potential therapeutic iron sequestering agents have been analyzed with regard to the structures of the chelators. All compounds are hexadentate ligands composed of a systematically varied combination of methyl-3,2-hydroxypyridinone (Me-3,2-HOPO) and 2,3-dihydroxyterephthalamide (TAM) binding units linked to a polyamine scaffold through amide linkers; each series is based on a specific backbone: tris(2-aminoethyl)amine, spermidine, or 5-LIO(TAM), where 5-LIO is 2-(2-aminoethoxy)ethylamine. Rates of iron removal from transferrin were determined spectrophotometrically for the ten ligands, which all efficiently acquire ferric ion from diferric transferrin with a hyperbolic dependence on ligand concentration (saturation kinetics). The effect of the two iron-binding subunits Me-3,2-HOPO and TAM and of the scaffold structures on iron removal ability is discussed. At the low concentrations corresponding to therapeutic dose, TAM-containing ligands exhibit the fastest rates of iron removal, which correlates with their high affinity for ferric ion and suggests the insertion of such binding units into future therapeutic chelating agents. In addition, urea polyacrylamide gel electrophoresis was used to measure the individual microscopic rates of iron removal from the three iron-bound transferrin species (diferric transferrin, N-terminal monoferric transferrin, C-terminal monoferric transferrin) by the representative chelators 5-LIO(Me-3,2-HOPO)2(TAM) and 5-LIO(TAMmeg)2(TAM), where TAMmeg is 2,3-dihydroxy-1-(methoxyethylcarbamoyl)terephthalamide. Both ligands show preferential removal from the C-terminal site of the iron-binding protein. However, cooperative effects between the two binding sites differ with the chelator. Replacement of hydroxypyridinone moieties by terephthalamide groups renders the N-terminal site more accessible to the ligand and may represent an advantage for iron chelation therapy.
Co-reporter:ShannonM. Biros;RobertM. Yeh ;KennethN. Raymond
Angewandte Chemie 2008 Volume 120( Issue 32) pp:6151-6153
Publication Date(Web):
DOI:10.1002/ange.200801226
Co-reporter:EricJ. Werner Dr.;Ankona Datta Dr.;ChristophJ. Jocher Dr.;KennethN. Raymond
Angewandte Chemie 2008 Volume 120( Issue 45) pp:8696-8709
Publication Date(Web):
DOI:10.1002/ange.200800212
Abstract
Um die klinischen Anwendungsmöglichkeiten der Kernspintomographie (MRI) zu verbessern, wird intensiv nach effizienteren Kontrastmitteln gesucht. Dazu müssen die Koordinationschemie von Gadolinium(III) und die Parameter, die seine Wirksamkeit als Protonenrelaxationsagens beeinflussen, berücksichtigt werden. Will man jeden Parameter optimieren, so sind auch Eigenschaften wie Löslichkeit und In-vivo-Toxizität zu beachten, was die Entwicklung sicherer und starker Relaxationsagentien zu einem anspruchsvollen Ziel macht. Dieser Kurzaufsatz stellt die neuesten Fortschritte auf diesem Gebiet vor, wobei ein besonderes Augenmerk auf Gadolinium(III)-Hydroxypyridinon-Chelatkomplexen liegt.
Co-reporter:ShannonM. Biros;RobertM. Yeh ;KennethN. Raymond
Angewandte Chemie International Edition 2008 Volume 47( Issue 32) pp:6062-6064
Publication Date(Web):
DOI:10.1002/anie.200801226
Co-reporter:EricJ. Werner Dr.;Ankona Datta Dr.;ChristophJ. Jocher Dr.;KennethN. Raymond
Angewandte Chemie International Edition 2008 Volume 47( Issue 45) pp:8568-8580
Publication Date(Web):
DOI:10.1002/anie.200800212
Abstract
The desire to improve and expand the scope of clinical magnetic resonance imaging (MRI) has prompted the search for contrast agents of higher efficiency. The development of better agents requires consideration of the fundamental coordination chemistry of the gadolinium(III) ion and the parameters that affect its efficacy as a proton relaxation agent. In optimizing each parameter, other practical issues, such as solubility and in vivo toxicity, must also be addressed, making the attainment of safe, high-relaxivity agents a challenging goal. This Minireview presents recent advances in the field, with an emphasis on gadolinium(III) hydroxypyridinone chelate complexes.
Co-reporter:EvanG. Moore Dr.;Géza Szigethy;Jide Xu Dr.;Lars-Olof Pålsson Dr.;Andrew Beeby ;KennethN. Raymond
Angewandte Chemie International Edition 2008 Volume 47( Issue 49) pp:9500-9503
Publication Date(Web):
DOI:10.1002/anie.200802337
Co-reporter:EvanG. Moore Dr.;Géza Szigethy;Jide Xu Dr.;Lars-Olof Pålsson Dr.;Andrew Beeby ;KennethN. Raymond
Angewandte Chemie 2008 Volume 120( Issue 49) pp:9642-9645
Publication Date(Web):
DOI:10.1002/ange.200802337
Co-reporter:Michael D. Pluth and Kenneth N. Raymond
Chemical Society Reviews 2007 vol. 36(Issue 2) pp:161-171
Publication Date(Web):20 Nov 2006
DOI:10.1039/B603168B
Synthetic chemists have provided a wide array of supramolecular assemblies able to encapsulate guest molecules. The scope of this tutorial review focuses on supramolecular host molecules capable of reversibly encapsulating polyatomic guests. Much work has been done to determine the mechanism of guest encapsulation and guest release. This review covers common methods of monitoring and characterizing guest exchange such as NMR, UV-VIS, mass spectrometry, electrochemistry, and calorimetry and also presents representative examples of guest exchange mechanisms. The guest exchange mechanisms of hemicarcerands, cucurbiturils, hydrogen-bonded assemblies, and metal–ligand assemblies are discussed. Special attention is given to systems which exhibit constrictive binding, a motif common in supramolecular guest exchange systems.
Co-reporter:Anne E. V Gorden Dr.;David K. Shuh Dr.;Bryan E. F. Tiedemann;Richard E. Wilson;Jide Xu Dr.
Chemistry - A European Journal 2007 Volume 13(Issue 2) pp:
Publication Date(Web):19 DEC 2006
DOI:10.1002/chem.200790005
Co-reporter:Robert G. Bergman;Michael D. Pluth
Science 2007 Volume 316(Issue 5821) pp:85-88
Publication Date(Web):06 Apr 2007
DOI:10.1126/science.1138748
Abstract
Although many enzymes can promote chemical reactions by tuning substrate properties purely through the electrostatic environment of a docking cavity, this strategy has proven challenging to mimic in synthetic host-guest systems. Here, we report a highly charged, water-soluble, metal-ligand assembly with a hydrophobic interior cavity that thermodynamically stabilizes protonated substrates and consequently catalyzes the normally acidic hydrolysis of orthoformates in basic solution, with rate accelerations of up to 890-fold. The catalysis reaction obeys Michaelis-Menten kinetics and exhibits competitive inhibition, and the substrate scope displays size selectivity, consistent with the constrained binding environment of the molecular host.
Co-reporter:Michael D. Pluth;Robert G. Bergman ;Kenneth N. Raymond
Angewandte Chemie International Edition 2007 Volume 46(Issue 45) pp:
Publication Date(Web):9 OCT 2007
DOI:10.1002/anie.200703371
Changing on the inside: Acetals are a commonly used protecting groups for aldehydes and ketones in organic synthesis because of their ease of installation and resistance to cleavage in neutral or basic solution. A self-assembled supramolecular assembly has been shown to catalyze the hydrolysis of acetals and ketals in basic solution (see scheme).
Co-reporter:Bryan E. F. Tiedemann;Kenneth N. Raymond Dr.
Angewandte Chemie International Edition 2007 Volume 46(Issue 26) pp:
Publication Date(Web):24 MAY 2007
DOI:10.1002/anie.200701002
A quick in and out: A supramolecular cluster encapsulates a cation guest with a linear alkyl side chain, which rapidly extends and retracts through apertures in the triangular faces of the tetrahedral host. This motion results in a dynamic second-order Jahn–Teller distortion (see picture).
Co-reporter:Bryan E. F. Tiedemann;Kenneth N. Raymond Dr.
Angewandte Chemie 2007 Volume 119(Issue 26) pp:
Publication Date(Web):24 MAY 2007
DOI:10.1002/ange.200701002
Ein schnelles Rein und Raus: Die lineare Alkylseitenkette eines in einen supramolekularen Cluster eingeschlossenen kationischen Gasts ragt mal durch Öffnungen in den Dreiecksflächen des tetraedrischen Wirts nach draußen und zieht sich dann wieder zurück. Diese Bewegung führt zu einer dynamischen Jahn-Teller-Verzerrung zweiter Ordnung (siehe Bild).
Co-reporter:Michael D. Pluth;Robert G. Bergman ;Kenneth N. Raymond
Angewandte Chemie 2007 Volume 119(Issue 45) pp:
Publication Date(Web):9 OCT 2007
DOI:10.1002/ange.200703371
Schutzlos ausgeliefert: Die Acetalgruppe ist eine häufig genutzte Schutzgruppe für Aldehyde und Ketone, da sie leicht eingeführt werden kann und in neutraler oder basischer Lösung stabil ist. Ein selbstorganisiertes supramolekulares Aggregat wird beschrieben, das die Hydrolyse von Acetalen und Ketalen unter basischen Bedingungen katalysiert (siehe Schema).
Co-reporter:Ulla N. Andersen, Georg Seeber, Dorothea Fiedler, Kenneth N. Raymond, Dayin Lin, Don Harris
Journal of the American Society for Mass Spectrometry 2006 Volume 17(Issue 3) pp:292-296
Publication Date(Web):March 2006
DOI:10.1016/j.jasms.2005.10.011
Self-assembled supramolecular host–guest complexes have been characterized by electrospray ionization mass spectrometry. The spectra obtained by use of a Q-TOF instrument equipped with a Z-spray ion source show primarily the 3- and 4- charge states of the assemblies. The assemblies have the general formula [guest ⊂ Ga4L6]11− where L represents the chelating bidentate catechol ligand 1,5-bis(2′,3′-dihydroxy-benzamido)naphthalene and guests are tetramethyl ammonium (Me4N+), tetraethyl ammonium (Et4N+), tetra-n-propyl ammonium (Pr4N+) and decamethylcobaltocenium (Cp*2Co+) cations. For the first time, the mass spectrum of the empty assembly [Ga4L6]12− is reported. This article also reports that provided the electrospray ion source is capable of preserving noncovalent interactions, it is possible to observe host–guest complexes containing both weak binding guests as well as sterically demanding guests in the mass spectra. The present data suggest that electrospray mass spectrometry is a powerful tool for characterization of supramolecular host–guest complexes.
Co-reporter:Dorothea Fiedler;Robert G. Bergman
Angewandte Chemie 2006 Volume 118(Issue 5) pp:
Publication Date(Web):21 DEC 2005
DOI:10.1002/ange.200501938
Im Käfig eingeschlossen: Ein supramolekulares Metall-Ligand-System wird genutzt, um zwei reaktive metallorganische Intermediate in wässriger Lösung zu stabilisieren. Während sich die beiden Organometallkomplexe in organischen Lösungsmitteln binnen Stunden und in Gegenwart von Wasser binnen Minuten zersetzen, sind sie nach Einschluss in den supramolekularen Käfig über Wochen inert. Trotz ihrer Stabilisierung sind die Gastmoleküle aber noch in der Lage, mit CO zu reagieren.
Co-reporter:Jide Xu Dr.
Angewandte Chemie 2006 Volume 118(Issue 39) pp:
Publication Date(Web):4 SEP 2006
DOI:10.1002/ange.200602060
Stränge und andere Zwänge: In supramolekularen Actinoidarchitekturen der Form viersträngiger Helices sind zwei überdacht-quadratisch-antiprismatisch koordinierte ThIV-Zentren durch vier verbrückende 4-Acylpyrazolon-Chelatstränge verbunden. Die Clusterbildung erfolgt nach dem Prinzip der „unvereinbaren Koordinationszahlen“.
Co-reporter:Jide Xu Dr.
Angewandte Chemie International Edition 2006 Volume 45(Issue 39) pp:
Publication Date(Web):4 SEP 2006
DOI:10.1002/anie.200602060
Let's twist again: Actinide quadruple-stranded helical supramolecular architectures were synthesized in which two capped square-antiprismatic ThIV centers are coordinated by four bis(bidentate) 4-acylpyrazolone chelating strands. This example shows a cluster formation caused by incommensurate coordination numbers.
Co-reporter:Dorothea Fiedler, Robert G. Bergman,Kenneth N. Raymond
Angewandte Chemie International Edition 2006 45(5) pp:745-748
Publication Date(Web):
DOI:10.1002/anie.200501938
Co-reporter:Bryan E. F. Tiedemann,Kenneth N. Raymond
Angewandte Chemie International Edition 2006 45(1) pp:83-86
Publication Date(Web):
DOI:10.1002/anie.200502209
Co-reporter:Bryan E. F. Tiedemann Dr.
Angewandte Chemie 2006 Volume 118(Issue 1) pp:
Publication Date(Web):22 NOV 2005
DOI:10.1002/ange.200502209
Ein Ende bleibt sichtbar: Ein Gast, der aus einem kationischen Sandwichkomplex, einer Alkylkette und einer anionischen Sulfonatgruppe besteht, wird partiell in einen [Ga4L6]12−-Wirtcluster eingelagert (siehe Schema). Die kationische Kopfgruppe wird schnell in den Wirthohlraum aufgenommen, die Sulfonateinheit dagegen nicht, weil die Alkylsulfonatkette aus der Öffnung einer dreieckigen Fläche des tetraedrischen Clusters herausragt.
Co-reporter:Anne E. V. Gorden Dr.;David K. Shuh Dr.;Bryan E. F. Tiedemann;Richard E. Wilson;Jide Xu Dr.
Chemistry - A European Journal 2005 Volume 11(Issue 9) pp:
Publication Date(Web):15 APR 2005
DOI:10.1002/chem.200590027
Co-reporter:Anne E. V. Gorden Dr.;David K. Shuh Dr.;Bryan E. F. Tiedemann;Richard E. Wilson;Jide Xu Dr.
Chemistry - A European Journal 2005 Volume 11(Issue 9) pp:
Publication Date(Web):3 MAR 2005
DOI:10.1002/chem.200401193
The first single-crystal X-ray diffraction analysis of a hydroxypyridonate plutonium(IV) complex is presented, that of the tetradentate ligand 5LIO(Me-3,2-HOPO) with PuIV. The [PuIV{5LIO(Me-3,2-HOPO)}2] complex crystallizes in the space group Pna21 with the asymmetric unit cell containing two unique eight-coordinate plutonium complexes and one perchlorate anion. According to shape measure analysis, the geometry of both Pu centers is closest to a bicapped trigonal prism (C2v symmetry, for Pu 1: S(C2v)=13.48°, S(D4d)=15.43°, S(D4d)=16.10°). The average bond length for the PuO(phenolic) is 2.31(4) Å, whereas the PuO(amide) distances are slightly longer, averaging 2.40(2) Å. The preparative chemistry of this compound and the implications of the structure are discussed.
Co-reporter:Jide Xu;Anne E. V. Gorden;Kenneth N. Raymond
European Journal of Organic Chemistry 2004 Volume 2004(Issue 15) pp:
Publication Date(Web):12 JUL 2004
DOI:10.1002/ejoc.200400080
The linear octadentate ligand 3,4,3-LICAM(C) (2) is one of the most effective chelating agents for PuIV that is not acutely toxic; however, at physiological pH, due to the weak acidity of the catechol hydroxy groups and the large proton dependence of the complexation reaction, only three of the four catecholate subunits are coordinated to PuIV. To overcome this disadvantage, a new topological class of octadentate ligands based on tetrapodal amine backbones and 2,3-dihydroxyterephthalamide (3) (TAM) binding units were designed and synthesized. The amide substituents provide a handle by which the functionality of the ligand can be readily modified, and this synthetic strategy and procedure can be extended to prepare a variety of new multidentate metal-coordination and extraction agents. A streamlined synthesis for the terephthalamides, both symmetric and asymmetric, was recently reported. In this work, the synthetic details of their incorporation into octadentate systems for use as coordination or extraction agents for PuIV or other metals in the +4 oxidation state are described. (© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2004)
Co-reporter:Julia L. Brumaghim;Martin Michels;Daniela Pagliero;Kenneth N. Raymond
European Journal of Organic Chemistry 2004 Volume 2004(Issue 24) pp:
Publication Date(Web):1 DEC 2004
DOI:10.1002/ejoc.200400533
Supramolecular assemblies with internal cavities are being developed as nanoscale reaction vessels to protect or modify the reactivity of guest species through encapsulation. Diazonium cations and the tropylium cation were examined for their ability to encapsulate in the tetrahedral [Ga4L6]12− supramolecular assembly. The 4-(diethylamino)benzenediazonium cation 1 readily formed a 1:1 host−guest complex with this assembly, and this encapsulation prevented 1 from reacting with 2,4-pentanedione in D2O. The tropylium cation also formed a 1:1 host−guest complex with the [Ga4L6]12− assembly, greatly slowing its decomposition in D2O. Encapsulation in the protected environment of this host cavity alters the reactivity of these guest molecules, giving them greater stability. (© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2004)
Co-reporter:Julia L. Brumaghim;Martin Michels;Kenneth N. Raymond
European Journal of Organic Chemistry 2004 Volume 2004(Issue 22) pp:
Publication Date(Web):2 NOV 2004
DOI:10.1002/ejoc.200400428
Encapsulation of guest molecules inside supramolecular host assemblies provides a way to stabilize reactive species in aqueous solution. The stabilization of reactive phosphonium/ketone adducts of the general formula [R1MeC(OH)PR3]+ by encapsulation as guest molecules within a [Ga4L6]12− tetrahedral metal−ligand assembly is reported; although these cations decompose in aqueous solution, encapsulation inside the hydrophobic cavity of the assembly lengthens their lifetimes considerably, in some cases up to weeks. By varying the phosphane (PMe3, PEt3, PPhMe2, and PPh2Me) and ketone (acetone, methyl ethyl ketone, 1,1,1-trifluoroacetone, and fluoroacetone) which form these adducts, as well as the pD of the solutions, it was determined that the pH of the solution as well as the size and shape of the guest cations play an important role in the stability of these host−guest complexes. Encapsulation of chiral guests in the chiral [Ga4L6]12− assembly results in the formation of diastereomers, as characterized by 1H, 19F, and 31P NMR spectroscopy. Although the [Ga4L6]12− assembly is formed from non-chiral ligands, the assembly itself has ΔΔΔΔ or ΛΛΛΛ chirality around the metal centers. Due to the chirality of this assembly, diastereomeric selectivity is observed upon initial guest encapsulation (typical diastereomeric excesses are 30−50%). This initial diastereomeric selectivity decreases over time to reach an equilibrium but does not become 1:1, indicating both kinetic and thermodynamic processes promote selective guest encapsulation. These experiments demonstrate further the applications of nanoscale reaction vessels, self-assembled by design from non-chiral ligands, in providing a chiral and hydrophobic environment for guest molecules in aqueous solution. (© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2004)
Co-reporter:Dorothea Fiedler;Robert G. Bergman
Angewandte Chemie 2004 Volume 116(Issue 48) pp:
Publication Date(Web):9 DEC 2004
DOI:10.1002/ange.200490164
Co-reporter:Dorothea Fiedler;Robert G. Bergman
Angewandte Chemie 2004 Volume 116(Issue 48) pp:
Publication Date(Web):18 NOV 2004
DOI:10.1002/ange.200461776
Katalytische Behälter: Ein supramolekulares Metall-Ligand-System [M4L6] dient als katalytischer Wirt für die unimolekulare Umlagerung von Enammonium-Kationen (siehe Schema). Der beschränkte Innenraum in der supramolekularen Struktur erzwingt eine reaktive Konformation des eingeschlossenen Substrats. Dies hat eine bis zu 850fach höhere Geschwindigkeit der Umlagerung zur Folge.
Co-reporter:Dennis H. Leung;Dorothea Fiedler;Robert G. Bergman
Angewandte Chemie 2004 Volume 116(Issue 8) pp:
Publication Date(Web):11 FEB 2004
DOI:10.1002/ange.200352772
Innenraumdesign: Eine Wirt-Gast-Verbindung aus der chiralen supramolekularen Tetraeder-Kapsel [Ga4L6]12− (L=verbrückender Ligand) und einem Halbsandwich-Iridiumkomplex aktiviert C-H-Bindungen von Aldehyden. Die Aktivierung findet innerhalb der Kapsel statt, ist hochgradig größen- und formspezifisch und verläuft mit passabler Diastereoselektivität (d.r. 55:45–70:30).
Co-reporter:Dorothea Fiedler;Robert G. Bergman
Angewandte Chemie International Edition 2004 Volume 43(Issue 48) pp:
Publication Date(Web):9 DEC 2004
DOI:10.1002/anie.200490169
Co-reporter:Dorothea Fiedler;Robert G. Bergman
Angewandte Chemie International Edition 2004 Volume 43(Issue 48) pp:
Publication Date(Web):19 NOV 2004
DOI:10.1002/anie.200461776
Catalytic containers: A supramolecular metal–ligand assembly [M4L6] is utilized as a catalytic host for the unimolecular carbon–carbon bond-forming rearrangement of enammonium cations (see scheme). The restricted reaction space of the supramolecular structure forces the substrate to adopt a reactive conformation upon binding to the interior. The assembly achieves up to 850-fold rate acceleration of the rearrangement.
Co-reporter:Dennis H. Leung;Dorothea Fiedler;Robert G. Bergman
Angewandte Chemie International Edition 2004 Volume 43(Issue 8) pp:
Publication Date(Web):11 FEB 2004
DOI:10.1002/anie.200352772
Interior design: A chiral supramolecular tetrahedral [Ga4L6]12− host (L=bis(bidentate) ligand) is shown to encapsulate a half-sandwich iridium complex. This host–guest assembly reacts with aldehydes through CH bond activation, which occurs within the host interior. This activation occurs with high size and shape specificity as well as modest diastereoselectivity (d.r. 55:45–70:30) owing to the well-defined shape of the host cavity.
Co-reporter:Marco Ziegler;Anna V. Davis;Darren W. Johnson
Angewandte Chemie 2003 Volume 115(Issue 6) pp:
Publication Date(Web):6 FEB 2003
DOI:10.1002/ange.200390151
„Es wollte sich einschleichen“: Liganden ohne Neigung zur Tetraederbildung (L2) schieben sich in ein enantiomerenreines tetraedrisches ΔΔΔΔ-[Ga4L16]12−-Aggregat mit labil gebundenen Metall- und Ligandenkomponenten (L1) ein, ohne seine supramolekulare Struktur zu beeinträchtigen (siehe Schema). Diese Erhaltung der Konfiguration der ursprünglichen Architektur ist ein Fall von Strukturgedächtnis.
Co-reporter:Kenneth N. Raymond;Emily A. Dertz;Sanggoo S. Kim;
Proceedings of the National Academy of Sciences 2003 100(7) pp:3584-3588
Publication Date(Web):March 24, 2003
DOI:10.1073/pnas.0630018100
Bacteria have aggressive acquisition processes for iron, an essential nutrient. Siderophores are small iron chelators that
facilitate cellular iron transport. The siderophore enterobactin is a triscatechol derivative of a cyclic triserine lactone.
Studies of the chemistry, regulation, synthesis, recognition, and transport of enterobactin make it perhaps the best understood
of the siderophore-mediated iron uptake systems, displaying a lot of function packed into this small molecule. However, recent
surprises include the isolation of corynebactin, a closely related trithreonine triscatechol derivative lactone first found
in Gram-positive bacteria, and the crystal structure of a ferric enterobactin complex of a protein identified as an antibacterial
component of the human innate immune system.
Co-reporter:Marco Ziegler;Anna V. Davis;Darren W. Johnson
Angewandte Chemie International Edition 2003 Volume 42(Issue 6) pp:
Publication Date(Web):6 FEB 2003
DOI:10.1002/anie.200390183
Non-tetrahedron-forming L2ligands insert into a resolved ΔΔΔΔ-[Ga4L16]12− tetrahedral assembly of labile metal and ligand components without compromising its supramolecular structure (see scheme). Preservation of the chirality of the initial architecture verifies its structural memory.
Co-reporter:Anna V. Davis;Robert M. Yeh
PNAS 2002 Volume 99 (Issue 8 ) pp:4793-4796
Publication Date(Web):2002-04-16
DOI:10.1073/pnas.052018299
Co-reporter:Carmen M. Barnés;Elizabeth C. Theil
PNAS 2002 Volume 99 (Issue 8 ) pp:5195-5200
Publication Date(Web):2002-04-16
DOI:10.1073/pnas.032089399
Ferritin concentrates iron as a hydrous ferric oxide in a protein cavity (8 nm in diameter) by using eight pores along the
threefold symmetry axes of the octahedral supramolecular structure. The role of ligand exchange in the entry of Fe(II) hexahydrate
into ferritin protein has been studied with [Cr(TREN)(H2O)(OH)]2+ [TREN = N(CH2CH2NH2)3], a model for Fe(H2O)62+ with only two exchangeable ligands. The results show that five different ferritin proteins, varying in pore structure, oxidation
sites, and nucleation sites, bind Cr(TREN) at functional protein sites, based on inhibition of iron mineralization and oxidation.
Properties of Cr(TREN)–ferritin adducts include an increased isoelectric point, a shift in the Cr(TREN) UV/vis spectrum consistent
with exchange of water for protein carboxylate or thiolate ligands, binding affinities of 50–250 μM, and a slow rate of dissociation
(k = 4 × 10−6 sec−1). The relationship of Cr(TREN) inhibition of iron oxidation and mineralization by Cr(TREN) to the known structures of the
various ferritins tested suggests that Cr(TREN) plugs the ferritin pores, obstructing Fe(II) entry in folded and unfolded
pores. Because only two exchangeable waters are sufficient for pore binding of Cr(TREN), the physiological Fe(II) donor must
bind to the pore with few exchangeable ligands. These results show the advantage of using stable model complexes to explore
properties of transient Fe–protein complexes during Fe mineralization in ferritin.
Co-reporter:Andreas J. Terpin;Marco Ziegler;Darren W. Johnson
Angewandte Chemie 2001 Volume 113(Issue 1) pp:
Publication Date(Web):4 JAN 2001
DOI:10.1002/1521-3757(20010105)113:1<161::AID-ANGE161>3.0.CO;2-6
Co-reporter:Marco Ziegler;J. J. Mira;Ulla N. Andersen;Darren W. Johnson ;Julie A. Leary
Angewandte Chemie 2001 Volume 113(Issue 4) pp:
Publication Date(Web):15 FEB 2001
DOI:10.1002/1521-3757(20010216)113:4<755::AID-ANGE7550>3.0.CO;2-0
Co-reporter:Seth M. Cohen;Stéphane Petoud Dr.
Chemistry - A European Journal 2001 Volume 7(Issue 1) pp:
Publication Date(Web):3 JAN 2001
DOI:10.1002/1521-3765(20010105)7:1<272::AID-CHEM272>3.0.CO;2-Y
The synthesis, characterization, and metal-binding studies of chelate-functionalized dendrimers is reported. Salicylate, catecholate, and hydroxypyridinonate bidentate chelators have been coupled to the surface of both poly(propyleneimine) (Astramol) and poly(amidoamine) (Starburst, PAMAM) dendrimers up to the fifth generation (64 endgroups). A general method has been developed for the facile and high quality chromatographic purification of poly(propyleneimine) and poly(amidoamine) dendrimer derivatives. One- and two-dimensional (TOCSY) 1H NMR experiments and electrospray ionization mass spectrometry (ESI-MS) have confirmed the exhaustive coupling of these chelators to the primary amine functionalities of the dendrimers. Spectrophotometric titrations were used to investigate the metal binding ability of these macrochelates. Spectral analysis shows that ferric iron binding to these ligands is localized to the chelating endgroups. The ability of these dendritic polymers to bind large numbers of metal ions may lead to applications as metal sequestering agents for waste remediation technologies.
Co-reporter:Marco Ziegler Dr.;Julia L. Brumaghim Dr.;Kenneth N. Raymond Dr.
Angewandte Chemie 2000 Volume 112(Issue 22) pp:
Publication Date(Web):14 NOV 2000
DOI:10.1002/1521-3757(20001117)112:22<4285::AID-ANGE4285>3.0.CO;2-D
Co-reporter:Tatjana N. Parac Dr.;Markus Scherer Dr. Dr.
Angewandte Chemie 2000 Volume 112(Issue 7) pp:
Publication Date(Web):4 APR 2000
DOI:10.1002/(SICI)1521-3757(20000403)112:7<1288::AID-ANGE1288>3.0.CO;2-J
Co-reporter:Jide Xu Dr.;Kenneth N. Raymond Dr.
Angewandte Chemie 2000 Volume 112(Issue 15) pp:
Publication Date(Web):2 AUG 2000
DOI:10.1002/1521-3757(20000804)112:15<2857::AID-ANGE2857>3.0.CO;2-2
Co-reporter:Alain Stintzi;Carmen Barnes;Jide Xu
PNAS 2000 Volume 97 (Issue 20 ) pp:10691-10696
Publication Date(Web):2000-09-26
DOI:10.1073/pnas.200318797
A mechanism of ion transport across membranes is reported.
Microbial transport of Fe3+ generally delivers iron, a
growth-limiting nutrient, to cells via highly specific
siderophore-mediated transport systems. In contrast, iron transport in
the fresh water bacterium Aeromonas hydrophila is found
to occur by means of an indiscriminant siderophore transport system
composed of a single multifunctional receptor. It is shown that
(i) the siderophore and Fe3+ enter the
bacterium together, (ii) a ligand exchange step occurs
in the course of the transport, and (iii) a redox
process is not involved in iron exchange. To the best of our knowledge,
there have been no other reports of a ligand exchange mechanism in
bacterial iron transport. The ligand exchange step occurs at the cell
surface and involves the exchange of iron from a ferric siderophore to
an iron-free siderophore already bound to the receptor. This ligand
exchange mechanism is also found in Escherichia coli and
seems likely to be widely distributed among microorganisms.
Co-reporter:Jide Xu;Tatjana N. Parac
Angewandte Chemie 1999 Volume 111(Issue 19) pp:
Publication Date(Web):24 SEP 1999
DOI:10.1002/(SICI)1521-3757(19991004)111:19<3055::AID-ANGE3055>3.0.CO;2-K
Sowohl eine Tripelhelix als auch einenmeso-Komplex bilden die GaIII- und AlIII-Komplexe mit einem zweifach zweizähnigen Bis-Hydroxypyridinon-Liganden H2L. Die beiden Formen liegen in Lösung im Gleichgewicht vor, wobei die Bildung der helicalen Struktur in Gegenwart von Wasser, das als Gastmolekül im Hohlraum der Helix ausreichend Platz findet, begünstigt ist (die Struktur des helicalen H2O⊂[Al2L3]-Komplexes ist gezeigt).
Co-reporter:Darren W. Johnson;Jide Xu;Rolf W. Saalfrank
Angewandte Chemie 1999 Volume 111(Issue 19) pp:
Publication Date(Web):24 SEP 1999
DOI:10.1002/(SICI)1521-3757(19991004)111:19<3058::AID-ANGE3058>3.0.CO;2-2
Ein annähernd trigonales Antiprisma mit Metall-Metall-Abständen im Nanometerbereich bilden die sechs Metallionen im kristallinen, homochiralen [Ga6(L2)6] (siehe Struktur). Dieser Metall-Ligand-„Zylinder” leitet sich von einem dreizählig-symmetrischen β-Diketonliganden ab und ist das erste Beispiel für eine neue Form von Metall-Ligand-Clustern.
Co-reporter:Markus Scherer;Dana L. Caulder;Darren W. Johnson
Angewandte Chemie 1999 Volume 111(Issue 11) pp:
Publication Date(Web):26 MAY 1999
DOI:10.1002/(SICI)1521-3757(19990601)111:11<1689::AID-ANGE1689>3.0.CO;2-W
Ein einzigartiges Ligandendesign ermöglicht die Bildung sowohl eines M2L3-Tripelhelicats als auch eines M4L6-Tetraeders (M = Ti, Ga; L = ein auf 2,6-Diaminoanthracen basierender Ligand). Obwohl das Tetraeder entropisch ungünstig ist, reicht eine starke Wirt-Gast-Wechselwirkung zu einem Me4N+-Ion aus, um das Gleichgewicht zum Tetraeder zu verschieben. Bemerkenswert ist, daß das Helicat quantitativ in das Tetraeder überführt werden kann, wenn man einfach Me4N+-Ionen zugibt (siehe Bild).
Co-reporter:Xiankai Sun;Darren W. Johnson;Dana L. Caulder;Ryan E. Powers;Edward H. Wong
Angewandte Chemie 1999 Volume 111(Issue 9) pp:
Publication Date(Web):28 APR 1999
DOI:10.1002/(SICI)1521-3757(19990503)111:9<1386::AID-ANGE1386>3.0.CO;2-I
Heterometall-Mesocate [M2Pd3Br6L6]4− (M = TiIV, SnIV; L = Dianion von 4-Diphenylphosphanyl-1,2-dihydroxybenzol) wurden synthetisiert, bei denen die Kombination zweier nicht zueinander passender Symmetrieelemente, die von verschiedenen, über einen starren, bifunktionellen Liganden verbundenen Metallionen herrühren, zur gezielten Bildung eines C3h-symmetrischen Clusters führt (siehe Bild).
Co-reporter:Jide Xu;Tatjana N. Parac
Angewandte Chemie International Edition 1999 Volume 38(Issue 19) pp:
Publication Date(Web):24 SEP 1999
DOI:10.1002/(SICI)1521-3773(19991004)38:19<2878::AID-ANIE2878>3.0.CO;2-W
Both a triple helix as well as ameso complex are formed by the GaIII and AlIII complexes with a bis-bidentate bis-hydroxypyridinone ligand H2L. The two forms are in equilibrium in solution, though formation of the helical structure in the presence of water, which as guest molecule finds sufficient space in the cavity of the helix, is favored (the structure of the helical H2O⊂[Al2L3] complex is shown).
Co-reporter:Darren W. Johnson;Jide Xu;Rolf W. Saalfrank
Angewandte Chemie International Edition 1999 Volume 38(Issue 19) pp:
Publication Date(Web):24 SEP 1999
DOI:10.1002/(SICI)1521-3773(19991004)38:19<2882::AID-ANIE2882>3.0.CO;2-T
A near trigonal antiprism with metal–metal distances in the nanometer regime is formed by the six metal ions in the crystalline, homochiral [Ga6(L2)6] (see structure). This metal–ligand “cylinder” is based on a threefold symmetric, β-diketone ligand, and represents a new geometry for metal–ligand clusters.
Co-reporter:Markus Scherer;Dana L. Caulder;Darren W. Johnson
Angewandte Chemie International Edition 1999 Volume 38(Issue 11) pp:
Publication Date(Web):26 MAY 1999
DOI:10.1002/(SICI)1521-3773(19990601)38:11<1587::AID-ANIE1587>3.0.CO;2-R
A unique ligand design allows the formation of both an M2L3 triple helicate and an M4L6 tetrahedron (M=Ti, Ga; L=ligand based on 2,6-diaminoanthracene). Although the tetrahedron is entropically disfavored, a strong host–guest interaction with Me4N+ is enough to drive the equilibrium towards the tetrahedron. Remarkably, the helicate can be quantitatively converted into the tetrahedron simply by addition of Me4N+ (shown schematically).
Co-reporter:Xiankai Sun;Darren W. Johnson;Dana L. Caulder;Ryan E. Powers;Edward H. Wong
Angewandte Chemie International Edition 1999 Volume 38(Issue 9) pp:
Publication Date(Web):12 MAY 1999
DOI:10.1002/(SICI)1521-3773(19990503)38:9<1303::AID-ANIE1303>3.0.CO;2-8
Mixed-metal mesocates [M2Pd3Br6L6]4− (M=TiIV, SnIV; L=4-diphenylphosphanyl-catecholate) have been synthesized, in which the two incommensurate symmetry elements generated by the different metal ions are linked by a rigid, bifunctional ligand to generate a C3h-symmetrical cluster (see picture).
Co-reporter:Dana L. Caulder;Ryan E. Powers;Tatjana N. Parac
Angewandte Chemie International Edition 1998 Volume 37(Issue 13‐14) pp:
Publication Date(Web):17 DEC 1998
DOI:10.1002/(SICI)1521-3773(19980803)37:13/14<1840::AID-ANIE1840>3.0.CO;2-D
A remarkable selectivity on the basis of size is observed for the encapsulation of Et4N+ in the presence of Me4N+ and Pr4N+ by a predesigned [Ga4L6]12− homochiral tetrahedral cluster (L=bis-bidentate ligand). Immediate and quantitative stepwise replacement of R4N+ counterions in the cluster cavity is observed by 1H NMR spectroscopy (see below). The encapsulation of Et4N+ is also observed in the solid state.
Co-reporter:Christian Brückner;Ryan E. Powers
Angewandte Chemie International Edition 1998 Volume 37(Issue 13‐14) pp:
Publication Date(Web):17 DEC 1998
DOI:10.1002/(SICI)1521-3773(19980803)37:13/14<1837::AID-ANIE1837>3.0.CO;2-A
General design principles for the formation of tetrahedral M4L4 clusters (L=ligand) are outlined and exemplified by the formation and structure of the [Ti4L4]8− species pictured on the right.
Co-reporter:Christian Brückner;Ryan E. Powers
Angewandte Chemie 1998 Volume 110(Issue 13‐14) pp:
Publication Date(Web):12 MAR 1999
DOI:10.1002/(SICI)1521-3757(19980703)110:13/14<1937::AID-ANGE1937>3.0.CO;2-G
Allgemeingültige Prinzipien für das Design von tetraedrischen M4L4-Clustern (L=Ligand) werden am Beispiel der Bildung und Struktur der rechts abgebildeten [Ti4L4]8−-Spezies näher erläutert.
Co-reporter:Dana L. Caulder;Ryan E. Powers;Tatjana N. Parac
Angewandte Chemie 1998 Volume 110(Issue 13‐14) pp:
Publication Date(Web):12 MAR 1999
DOI:10.1002/(SICI)1521-3757(19980703)110:13/14<1940::AID-ANGE1940>3.0.CO;2-J
Nicht zu groß und nicht zu klein sollte der Gast im Hohlraum sein: Der homochirale tetraedrische Cluster [Ga4L6]12− weist eine hohe Selektivität für den Einschluß von Et4N+ gegenüber Pr4N+ auf, das seinerseits eingeschlossenes Me4N+ verdrängt (L=zweifach zweizähniger Ligand); dieser sukzessive Austausch von R4N+-Ionen verläuft 1H-NMR-spektroskopischen Untersuchungen zufolge schnell und quantitativ (siehe unten). Der Einschluß von Et4N+ wurde auch im Festkörper nachgewiesen.
Co-reporter:Michael Seitz ; King Do ; Andrew J. Ingram ; Evan G. Moore ; Gilles Muller
Inorganic Chemistry () pp:
Publication Date(Web):July 29, 2009
DOI:10.1021/ic901079s
The modular syntheses of three new octadentate, enantiopure ligands are reported, one with the bidentate chelating unit 2-hydroxyisophthalamide (IAM) and two with bidentate 1-hydroxy-2-pyridinone (1,2-HOPO) units. A new design principle is introduced for the chiral, non-racemic hexamines which constitute the central backbones for the presented class of ligands. The terbium(III) complex of the IAM ligand, as well as the europium(III) complexes of the 1,2-HOPO ligands, are synthesized and characterized by various techniques (NMR, UV, CD, luminescence spectroscopy). All species exhibit excellent stability and moderate to high luminescence efficiency (quantum yields ΦEu = 0.05−0.08 and ΦTb = 0.30−0.57) in aqueous solution at physiological pH. Special focus is put onto the properties of the complexes in regard to circularly polarized luminescence (CPL). The maximum luminescence dissymmetry factors (glum) in aqueous solution are high with ∣glum∣max = 0.08−0.40. Together with the very favorable general properties (good stability, high quantum yields, long lifetimes), the presented lanthanide complexes can be considered as good candidates for analytical probes based on CPL in biologically relevant environments.
Co-reporter:Chengbao Ni, David K. Shuh and Kenneth N. Raymond
Chemical Communications 2011 - vol. 47(Issue 22) pp:NaN6394-6394
Publication Date(Web):2011/05/09
DOI:10.1039/C1CC11329A
Uranyl complexes of a bis(methylterephthalamide) ligand (LH4) have been synthesized and characterized by X-ray crystallography. The structure is an unexpected [Me4N]8[L(UO2)]4 tetramer, formed via coordination of the two MeTAM units of L to two uranyl moieties. Addition of KOH to the tetramer gave the corresponding monomeric uranyl methoxide species [Me4N]K2[LUO2(OMe)].
Co-reporter:Michael D. Pluth and Kenneth N. Raymond
Chemical Society Reviews 2007 - vol. 36(Issue 2) pp:NaN171-171
Publication Date(Web):2006/11/20
DOI:10.1039/B603168B
Synthetic chemists have provided a wide array of supramolecular assemblies able to encapsulate guest molecules. The scope of this tutorial review focuses on supramolecular host molecules capable of reversibly encapsulating polyatomic guests. Much work has been done to determine the mechanism of guest encapsulation and guest release. This review covers common methods of monitoring and characterizing guest exchange such as NMR, UV-VIS, mass spectrometry, electrochemistry, and calorimetry and also presents representative examples of guest exchange mechanisms. The guest exchange mechanisms of hemicarcerands, cucurbiturils, hydrogen-bonded assemblies, and metal–ligand assemblies are discussed. Special attention is given to systems which exhibit constrictive binding, a motif common in supramolecular guest exchange systems.
.jpg)
![1,3-Benzenedicarboxamide,4-methoxy-N,N'-dimethyl-5-[(2-thioxo-3-thiazolidinyl)carbonyl]-](http://img.cochemist.com/ccimg/872800/872705-77-6.png)
![1,3-Benzenedicarboxamide,4-methoxy-N,N'-dimethyl-5-[(2-thioxo-3-thiazolidinyl)carbonyl]-](http://img.cochemist.com/ccimg/872800/872705-77-6_b.png)

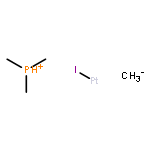
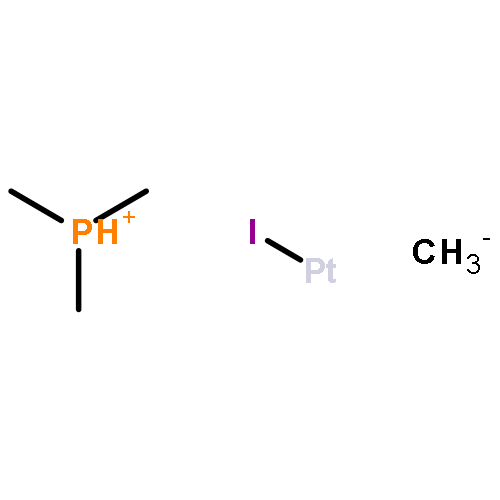

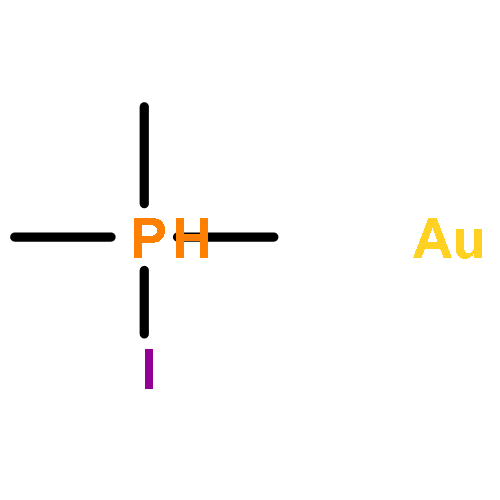

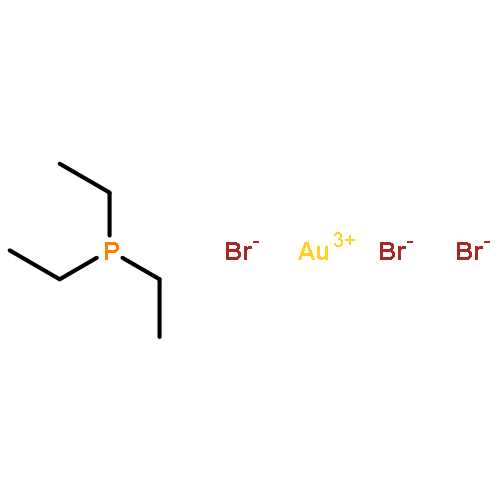




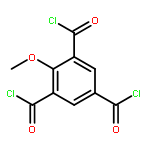
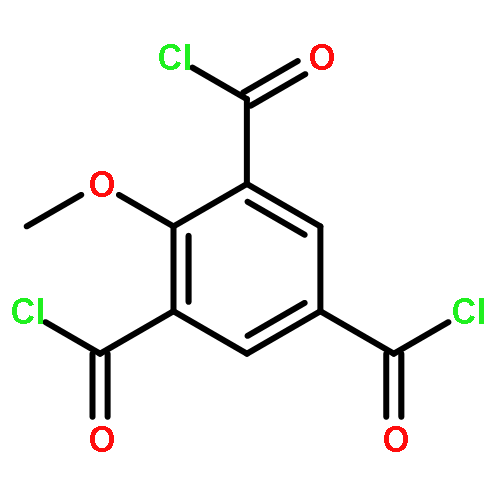


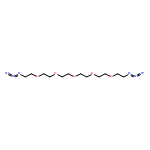
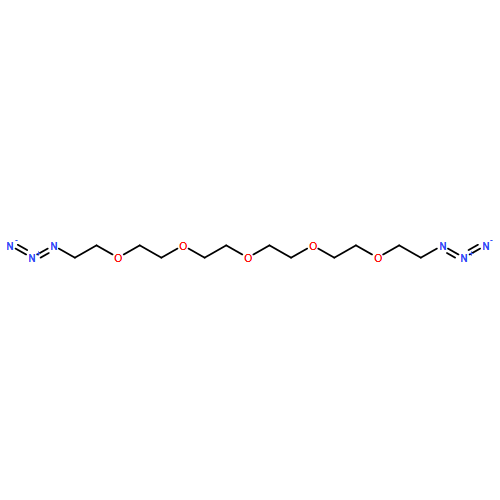




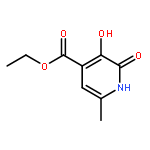



![2-Thiazolidinethione, 3,3'-[(2-methoxy-1,3-phenylene)dicarbonyl]bis-](http://img.cochemist.com/ccimg/247100/247039-29-8.png)
![2-Thiazolidinethione, 3,3'-[(2-methoxy-1,3-phenylene)dicarbonyl]bis-](http://img.cochemist.com/ccimg/247100/247039-29-8_b.png)
![4-Pyridinecarboxamide, N,N'-[[(2-aminoethyl)imino]di-2,1-ethanediyl]bis[1,2-dihydro-1-methyl-2-oxo-3-(phenylmethoxy)-](/data/chemimg/1774500/186834-28-6.png)
![4-Pyridinecarboxamide, N,N'-[[(2-aminoethyl)imino]di-2,1-ethanediyl]bis[1,2-dihydro-1-methyl-2-oxo-3-(phenylmethoxy)-](/data/chemimg/1774500/186834-28-6_b.png)

![Carbamic acid, [2-[bis(2-aminoethyl)amino]ethyl]-, 1,1-dimethylethylester](http://img.cochemist.com/ccimg/179200/179167-09-0.png)
![Carbamic acid, [2-[bis(2-aminoethyl)amino]ethyl]-, 1,1-dimethylethylester](http://img.cochemist.com/ccimg/179200/179167-09-0_b.png)







![Benzenamine, 5-methoxy-4-[2-[2-(2-methoxyethoxy)ethoxy]ethoxy]-2-nitro-](http://img.cochemist.com/ccimg/114700/114692-81-8.png)
![Benzenamine, 5-methoxy-4-[2-[2-(2-methoxyethoxy)ethoxy]ethoxy]-2-nitro-](http://img.cochemist.com/ccimg/114700/114692-81-8_b.png)






![4-Oxazolecarboxamide,N,N-bis[3-[(2,3-dihydroxybenzoyl)amino]propyl]-2-(2,3-dihydroxyphenyl)-4,5-dihydro-5-methyl-,(4S,5R)-](http://img.cochemist.com/ccimg/103200/103185-30-4.png)
![4-Oxazolecarboxamide,N,N-bis[3-[(2,3-dihydroxybenzoyl)amino]propyl]-2-(2,3-dihydroxyphenyl)-4,5-dihydro-5-methyl-,(4S,5R)-](http://img.cochemist.com/ccimg/103200/103185-30-4_b.png)








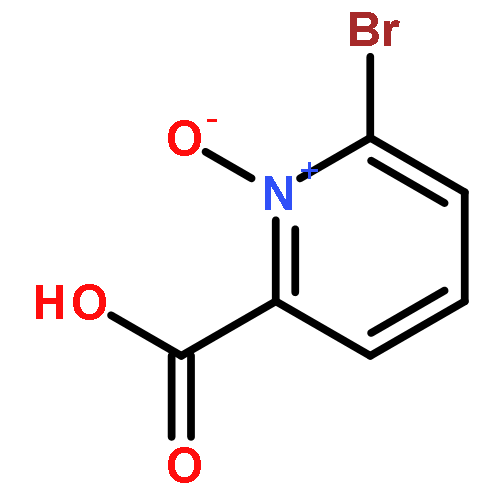





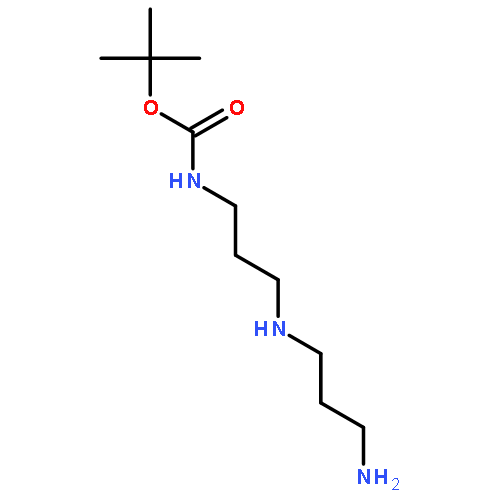
![2-Thiazolidinethione, 3-[2,3-bis(phenylmethoxy)benzoyl]-](http://img.cochemist.com/ccimg/81300/81254-86-6.png)
![2-Thiazolidinethione, 3-[2,3-bis(phenylmethoxy)benzoyl]-](http://img.cochemist.com/ccimg/81300/81254-86-6_b.png)


![L-Threonine, N-[2,3-bis(phenylmethoxy)benzoyl]-](http://img.cochemist.com/ccimg/74300/74272-79-0.png)
![L-Threonine, N-[2,3-bis(phenylmethoxy)benzoyl]-](http://img.cochemist.com/ccimg/74300/74272-79-0_b.png)









![2-METHYL-2-PROPANYL [1-(4-AMINOPHENYL)-3-PYRROLIDINYL]CARBAMATE](http://img.cochemist.com/ccimg/55500/55400-73-2.png)
![2-METHYL-2-PROPANYL [1-(4-AMINOPHENYL)-3-PYRROLIDINYL]CARBAMATE](http://img.cochemist.com/ccimg/55500/55400-73-2_b.png)


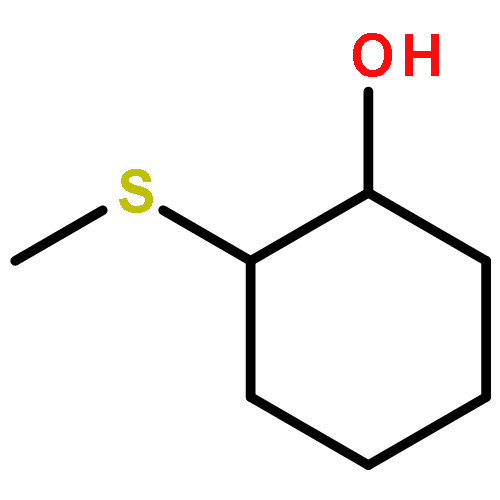
![4-[3-[ACETYL(HYDROXY)AMINO]PROPYLAMINO]-2-[2-[3-[ACETYL(HYDROXY)AMINO]PROPYLAMINO]-2-OXOETHYL]-2-HYDROXY-4-OXOBUTANOIC ACID](http://img.cochemist.com/ccimg/35500/35418-52-1.png)
![4-[3-[ACETYL(HYDROXY)AMINO]PROPYLAMINO]-2-[2-[3-[ACETYL(HYDROXY)AMINO]PROPYLAMINO]-2-OXOETHYL]-2-HYDROXY-4-OXOBUTANOIC ACID](http://img.cochemist.com/ccimg/35500/35418-52-1_b.png)

![Manganate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/12700/21393-64-6.png)
![Manganate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/12700/21393-64-6_b.png)




![Iron,[cyclo[glycylglycylglycyl-N5-(acetyl-kO)-N5-(hydroxy-kO)-L-ornithyl-N5-(acetyl-kO)-N5-(hydroxy-kO)-L-ornithyl-N5-(acetyl-kO)-N5-(hydroxy-kO)-L-ornithyl]ato(3-)]-, (OC-6-64)-](http://img.cochemist.com/ccimg/15700/15630-64-5.png)
![Iron,[cyclo[glycylglycylglycyl-N5-(acetyl-kO)-N5-(hydroxy-kO)-L-ornithyl-N5-(acetyl-kO)-N5-(hydroxy-kO)-L-ornithyl-N5-(acetyl-kO)-N5-(hydroxy-kO)-L-ornithyl]ato(3-)]-, (OC-6-64)-](http://img.cochemist.com/ccimg/15700/15630-64-5_b.png)


![Ferrate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/522800/14875-96-8.png)
![Ferrate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/522800/14875-96-8_b.png)












![[3-(hydroxymethyl)-2-methoxy-5-methylphenyl]methanol](http://img.cochemist.com/ccimg/6400/6327-85-1.png)
![[3-(hydroxymethyl)-2-methoxy-5-methylphenyl]methanol](http://img.cochemist.com/ccimg/6400/6327-85-1_b.png)














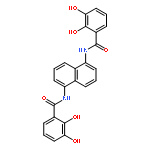
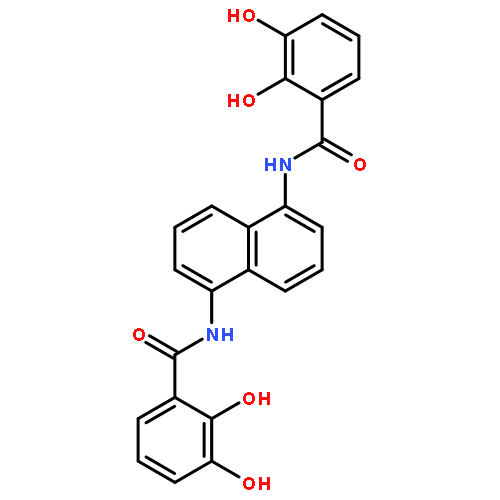





![4-Oxazolecarboxamide,N-[3-[(2,3-dihydroxybenzoyl)amino]propyl]-2-(2,3-dihydroxyphenyl)-N-[3-[[[(4S,5R)-2-(2,3-dihydroxyphenyl)-4,5-dihydro-5-methyl-4-oxazolyl]carbonyl]amino]propyl]-4,5-dihydro-5-methyl-,(4S,5R)-](http://img.cochemist.com/ccimg/88300/88217-23-6.png)
![4-Oxazolecarboxamide,N-[3-[(2,3-dihydroxybenzoyl)amino]propyl]-2-(2,3-dihydroxyphenyl)-N-[3-[[[(4S,5R)-2-(2,3-dihydroxyphenyl)-4,5-dihydro-5-methyl-4-oxazolyl]carbonyl]amino]propyl]-4,5-dihydro-5-methyl-,(4S,5R)-](http://img.cochemist.com/ccimg/88300/88217-23-6_b.png)







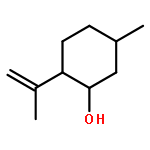

![Benzamide,N,N',N''-[(3S,7S,11S)-2,6,10-trioxo-1,5,9-trioxacyclododecane-3,7,11-triyl]tris[2,3-dihydroxy-](http://img.cochemist.com/ccimg/28400/28384-96-5.png)
![Benzamide,N,N',N''-[(3S,7S,11S)-2,6,10-trioxo-1,5,9-trioxacyclododecane-3,7,11-triyl]tris[2,3-dihydroxy-](http://img.cochemist.com/ccimg/28400/28384-96-5_b.png)

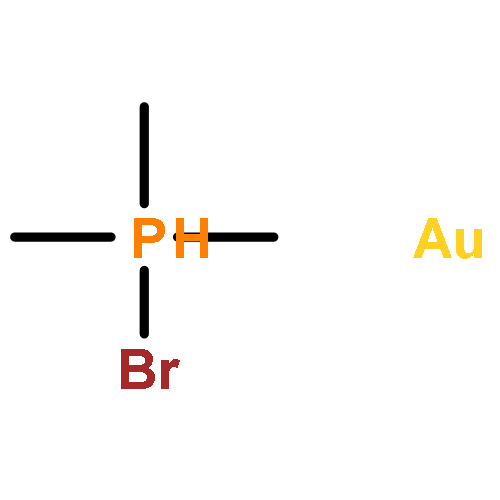


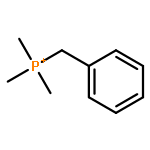

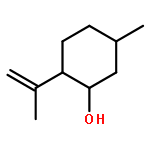





![N-[1-(1,3-BENZODIOXOL-5-YL)-2-PROPANYL]FORMAMIDE](http://img.cochemist.com/ccimg/7500/7440-51-9.png)
![N-[1-(1,3-BENZODIOXOL-5-YL)-2-PROPANYL]FORMAMIDE](http://img.cochemist.com/ccimg/7500/7440-51-9_b.png)


![2-Thiazolidinethione,3-[[1,2-dihydro-1-methyl-2-oxo-3-(phenylmethoxy)-4-pyridinyl]carbonyl]-](http://img.cochemist.com/ccimg/169300/169237-39-2.png)
![2-Thiazolidinethione,3-[[1,2-dihydro-1-methyl-2-oxo-3-(phenylmethoxy)-4-pyridinyl]carbonyl]-](http://img.cochemist.com/ccimg/169300/169237-39-2_b.png)