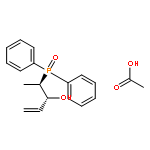
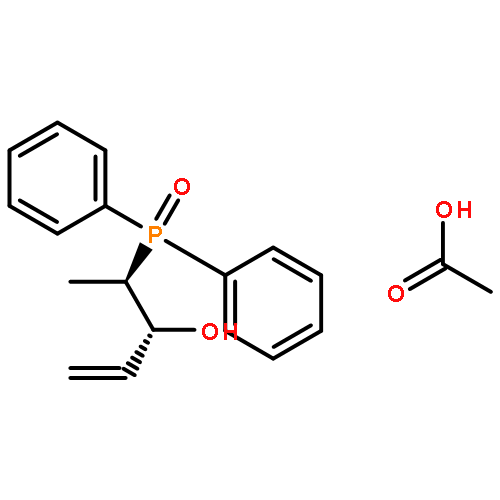
Co-reporter:Henri Brunner, Hikaru Kitamura, and Takashi Tsuno
Organometallics July 10, 2017 Volume 36(Issue 13) pp:2424-2424
Publication Date(Web):June 22, 2017
DOI:10.1021/acs.organomet.7b00311
The complexes [CpFe{Ph2P(CH2)nPPh2}NCMe]PF6, [CpFe(PPh2Me)2NCMe]PF6, and [CpFe{Ph2P(CH2)nPPh2}PPh2(OR)]PF6, R = Me, Et, and iPr, with chelate ring sizes between 4 and 7 were synthesized and characterized by spectroscopy and X-ray analysis. In these complexes, the monodentate ligands acetonitrile and PPh2(OR) tend to dissociate. The kinetics of the ligand exchanges MeCN/P(OMe)3 and PPh2(OR)/P(OMe)3 was measured. In the acetonitrile series, the first-order reaction of the five-membered chelate complex [CpFe(dppe)NCMe]PF6 had a half-life of 549 min in CDCl3 at 293 K. The other chelate complexes [CpFe(P-P)NCMe]PF6 and the nonchelate analogue [CpFe(PPh2Me)2NCMe]PF6 reacted faster by factors of 20–50. The PPh2(OR) series revealed a dramatic difference between the complexes [CpFe(P-P)PPh2(OR)]PF6 with five- and six-membered chelate rings. The PPh2(OR)/P(OMe)3 exchange in the dppp complex (six-membered chelate ring) was 500 times faster than in the dppe complex (five-membered chelate ring). This is due to the increase of the P–Fe–P angle in the dppp chelate ring, which diminishes the binding pocket of the PPh2(OR) ligand. In the nonchelate complex [CpFe(PPh2Me)2NCMe]PF6, a novel and unexpected bimolecular PPh2Me/PPh2(OMe) substitution was observed.
Co-reporter:Henri Brunner
Dalton Transactions 2017 vol. 46(Issue 15) pp:5103-5109
Publication Date(Web):2017/04/10
DOI:10.1039/C7DT00474E
In half-sandwich compounds of the type [Cp*ML1L2PPh3] the PPh3 propeller is stabilized by attractive CH/π interactions in which Co–H bonds specifically interact with the Ci and Co atoms of neighbouring phenyl rings, as in the T-shaped benzene dimer (i/o = ipso/ortho). This stabilization was not taken into account in a recent conformational analysis based on van der Waals energy calculations and minimization of steric compression (Dalton Trans., 2015, 44, 5451–5466). It is shown that in all 116 structures discussed in this analysis the CoH–Ci/o distances fall below the sum of the van der Waals radii, establishing attractive CH/π interactions, although the short contacts could easily be avoided by phenyl rotation to relieve steric strain. In 53 of the described structures there are acyl substituents which form conformation-determining Co–H⋯O(acyl) hydrogen bonds that are not taken into account in the recent analysis. The steric-only model is not an adequate description of M–PPh3 complexes.
Co-reporter:Henri Brunner, Takashi Tsuno
Inorganica Chimica Acta 2016 Volume 446() pp:132-142
Publication Date(Web):1 May 2016
DOI:10.1016/j.ica.2016.02.039
•The architecture of the triphenylphosphine propeller in transition metal complexes is controlled by weak CH/π interactions comparable to the archetypal T-shaped benzene dimer and not by steric-only interactions as claimed in the literature.•ortho-CH bonds bind to ipso- and ortho-carbon atoms of adjacent phenyl rings.•In the trans-[MCl2(PPh3)2] complexes it is the Cl/π interaction which determines the orientation of the PPh3 propeller within the molecule.The architecture of the triphenylphosphine propeller in Cr-PPh3 complexes and in the compounds trans-[MCl2(PPh3)2], M = Ni, Pd, and Pt, is analyzed on the basis of a CSD search. The three phenyl rings interact with each other by formation of weak CH/π bonds comparable to the archetypal T-shaped benzene dimer. ortho-CH bonds from inside the propeller bind to ipso- and ortho-carbon atoms of adjacent phenyl rings. In the broad energy minimum A/B there is a discontinuity in the transition from A to B. Binding switches from inside to outside ortho-carbon atoms. ortho-CH bonds from outside the propeller establish similar weak bonds with unsaturated ligands (and the metal atom) which control the arrangement of the PPh3 propeller within the molecule. In the trans-[MCl2(PPh3)2] complexes it is the Cl/π interaction which determines the orientation of the PPh3 propeller in the molecule.The architecture of the triphenylphosphine propeller in Cr-PPh3 complexes and trans-[MCl2(PPh3)2], M = Ni, Pd, and Pt, compounds is analyzed on the basis of a CSD search. The three phenyl rings interact with each other by formation of weak CH/π bonds. ortho-CH bonds bind to ipso- and ortho-carbon atoms of adjacent phenyl rings. In the trans-[MCl2(PPh3)2] complexes it is the Cl/π interaction which determines the orientation of the PPh3 propeller within the molecule.
Co-reporter:Henri Brunner and Takashi Tsuno
Organometallics 2015 Volume 34(Issue 7) pp:1287-1293
Publication Date(Web):March 18, 2015
DOI:10.1021/acs.organomet.5b00022
In the solid state and also in solution CpM–L–E–Ph compounds adopt preferential conformations, in which C–H bonds of the Cp ligand bind to the π system of the phenyl substituent. This holds for CpMo(CO)2–amidinato and −thioamidato as well as for CpM–P(OPh)3 complexes. In the solid state this is demonstrated by short contacts CH–Csp2, retrieved in a CSD search, comparable to those in the archetypal T-shaped benzene dimer. In solution the CH/π stabilization shows up in a typical high-field shift of the CpH signal, due to the anisotropy beam of the phenyl substituent, and in the thermodynamic stability of major and minor diastereomers in diastereomer equilibria. A special motif is the unsymmetrical bonding of two CpH bonds to the phenyl substituent, reminiscent of the new tilted T-shaped benzene dimer.
Co-reporter:Henri Brunner, Takashi Tsuno, and Michael Bodensteiner
Organometallics 2014 Volume 33(Issue 9) pp:2257-2265
Publication Date(Web):April 23, 2014
DOI:10.1021/om500175u
The Cambridge Structural Database comprises 89 structures with M–Prosphos chelate rings. On this basis a compilation analysis of the conformation of the M–Prophos chelate ring has been carried out. Typical conformational features, such as the puckering of the chelate ring and the arrangement of the phenyl rings, important for the transmission of the chiral information in enantioselective catalysts, are attributed to CH/π interactions within the M–Prophos system, although intramolecular interactions, in particular with Cp and Cp* ligands, also play a role. Distances below the van der Waals radii indicate weak bonding, provided the overlap angles are large enough. Normally, intermolecular interactions and packing effects are not structure-determining. Otherwise, recurrent motifs would not be as dominant, as established in the present analysis. The most important contribution comes from the interaction of the methyl group at the asymmetric center in the equatorial position of the five-membered chelate ring with the axial phenyl ring of the adjacent PPh2 group. The distances from the methyl group to the ipso and ortho carbon atoms of the axial phenyl ring are much shorter than those to Ci and Co of the equatorial phenyl ring. In addition, the overlap angles are better. In an active process the methyl group attracts one of the two sides of the phenyl rings to establish an effective CH/π interaction with one of the ortho carbon atoms. Within the PPh2 groups the interactions of the o-CH bond of one phenyl ring with the ipso and ortho carbon atoms of the other phenyl ring vary from strong to weak. Weak attractions are also observed between the hydrogen atoms of the CHMeCH2 backbone and the neighboring phenyl rings. The fact that the methyl group at the asymmetric center overwhelmingly occupies the equatorial position in the chelate ring is attributed to the missing phenyl–phenyl stabilization, when the methyl group switches into the exceptional axial position. Decreasing CH/π intensity, based on distance and overlap angle, is visualized by broad, normal, thin, and dashed arrows.
Co-reporter:Henri Brunner, Takashi Tsuno, Gábor Balázs, and Michael Bodensteiner
The Journal of Organic Chemistry 2014 Volume 79(Issue 23) pp:11454-11462
Publication Date(Web):November 5, 2014
DOI:10.1021/jo5020454
In 1,2-Me,Ph substitution patterns of organic compounds the methyl group attracts one of the phenyl sides to establish a CH/π bond with one of the ortho carbon atoms (the Co side), leading to a characteristic tilting of the phenyl ring around its Ci–Cp axis. This phenyl rotation shortens the CMe–Co distances to bonding contacts between the methyl hydrogen atoms and the ortho carbon atom Co well below the van der Waals distance of 3.70 Å. On the other hand, it elongates the CMe–Co′ distances outside of the reach of any CH/π interaction (>3.70 Å). Our study is based on a search in the Cambridge Structural Database for substructures Me–C═C–Ph, Me–C–C–Ph, and Me–C–N–Ph with 1,2-Me,Ph substitution patterns. In the 1,2-Me,Ph substitution motif the torsion angle CMe–C–C–Ci determines the length of the CMe–Ci and CMe–Co distances. For aromatic compounds these torsion angles are close to 0°, but in five- and six-membered ring compounds and in open-chain compounds the torsion angles vary considerably. Universally, for torsion angles up to 80° CH/π bonds were found, whereas the long CMe–Ci and CMe–Co distances for torsion angles >80° do not allow a CH/π interaction. The results of the present CSD analysis are supported by calculations.
Co-reporter:Henri Brunner, Takaki Kurosawa, Manfred Muschiol, Takashi Tsuno, Gábor Balázs, and Michael Bodensteiner
Organometallics 2013 Volume 32(Issue 17) pp:4904-4911
Publication Date(Web):August 21, 2013
DOI:10.1021/om400631m
The compounds (RFe,RC)-/(SFe,RC)-[CpFe(Prophos)PPh(OMe)2]PF6 and (RFe,RC)-/(SFe,RC)-[CpFe(Prophos)PPh2(OR)]PF6 (R = Me, Et, iPr, tBu) were synthesized, starting from (RFe,RC)-/(SFe,RC)-[CpFe(Prophos)NCMe]PF6, and characterized, including X-ray analyses. Bubbling a stream of N2 through the solution speeded up the slow substitution reactions by removing the acetonitrile formed in the rate-determining cleavage of the Fe–NCMe bond. Due to their acceptor ligands P(OMe)3 and PPh(OMe)2 the complexes [CpFe(Prophos)P(OMe)3]PF6 and [CpFe(Prophos)PPh(OMe)2]PF6 are configurationally stable at the metal atom. In contrast, [CpFe(Prophos)PPh3]PF6 does not form, due to the large cone angle of the ligand PPh3. Ideally, the electronic and steric effects of the ligands PPh2(OR) (R = Me, Et, iPr, tBu) are such that they tend to dissociate from the congested complexes (RFe,RC)- and (SFe,RC)-[CpFe(Prophos)PPh2(OR)]PF6. In the series (RFe,RC)- and (SFe,RC)-[CpFe(Prophos)PPh2(OR)]PF6, (R = Me, Et, iPr, tBu) electron donation and the cone angle of the ligands increase. Thus, the rates of the ligand exchange with P(OMe)3 increased, initiated by the slow dissociation of the Fe–PPh2(OR) bond. For the RFe,RC/SFe,RC diastereomers of [CpFe(Prophos)PPh2(OR)]PF6 the half-lives of the first-order reactions were 125/350 h (R = Me), 75/275 h (R = Et), and 12/34 h (R = iPr) in CDCl3 at 60 °C.
Co-reporter:Henri Brunner;Michael Bodensteiner
Chirality 2013 Volume 25( Issue 10) pp:663-667
Publication Date(Web):
DOI:10.1002/chir.22199
ABSTRACT
Salicylidenimine palladium(II) complexes trans-Pd(O,N)2 adopt step and bowl arrangements. A stereochemical analysis subdivides 52 compounds into 41 step and 11 bowl types. Step complexes with chiral N-substituents and all the bowl complexes induce chiral distortions in the square planar system, resulting in Δ/Λ configuration of the Pd(O,N)2 unit. In complexes 1, 2, 3, 4, 5, 6 with enantiomerically pure N-substituents ligand chirality entails a specific square chirality and only one diastereomer assembles in the lattice. Dimeric Pd(O,N)2 complexes with bridging N-substituents in trans-arrangement are inherently chiral. For dimers 7, 8, 9, 10, 11 different chirality patterns for the Pd(O,N)2 square are observed. The crystals contain racemates of enantiomers. In complex 12 two independent molecules form a tight pair. The (RC) configuration of the ligand induces the same Δ chirality in the Pd(O,N)2 units of both molecules with varying square chirality due to the different crystallographic location of the independent molecules. In complexes 13 and 14 atrop isomerism induces specific configurations in the Pd(O,N)2 bowl systems. The square chirality is largest for complex 15 [(Diop)Rh(PPh3)Cl)], a catalyst for enantioselective hydrogenation. In the lattice of 15 two diastereomers with the same (RC,RC) configuration in the ligand Diop but opposite Δ and Λ square configurations co-crystallize, a rare phenomenon in stereochemistry. Chirality 25:663–667, 2013. © 2013 Wiley Periodicals, Inc.
Co-reporter:Takashi Tsuno, Haruka Iwabe, Henri Brunner
Inorganica Chimica Acta 2013 400() pp: 262-266
Publication Date(Web):
DOI:10.1016/j.ica.2013.02.023
Co-reporter:Takashi Tsuno, Daiki Kato, Henri Brunner, Hayato Ike
Inorganica Chimica Acta 2012 Volume 392() pp:331-334
Publication Date(Web):30 September 2012
DOI:10.1016/j.ica.2012.03.003
In the reaction of N,N′-methylenebis[(4S,5R)-4-methoxycarbonyl-5-methyloxazolidine] (1) with CuCl2 or RuCl3 in alcoholic solvents novel complexes, bis[(4S,5R)-4-methoxycarbonyl-5-methyl-1,3-oxazolidine]copper(II) dichloride (2) and bis[(4S,5R)-4-methoxycarbonyl-5-methyl-2-oxazoline]-(4S,5R)-4-methoxycarbonyl-5-methyl-1,3-oxazolidineruthenium(III) trichloride (3), respectively, were isolated and fully characterized by X-ray crystallography. In these reactions, which occurred under mild conditions, ligand 1 was cleaved at the bridging aminal position and degraded to (4S,5R)-4-methoxycarbonyl-5-methyl-1,3-oxazolidine 5. The trigonal bipyramidal Cu complex 3 contains two such oxazolidine ligands coordinated in different ways. One is bonded in a monodentate fashion via the nitrogen atom of the five-membered ring, the other binds as a bidentate ligand, additionally using the carbonyl oxygen of the ester group. In the octahedral Ru complex 4 there are one oxazolidine ligand 5 and two oxazoline ligands 6. Thus, in the synthesis of 4 the carbon atom between oxygen and nitrogen in the oxazolidine ring was partly oxidized from the half-aminal status to the carboxamide status.Graphical abstractIn the reaction of N,N′-methylenebis[(4S,5R)-4-methoxycarbonyl-5-methyloxazolidine] (1) with CuCl2 or RuCl3 in alcoholic solvents novel complexes, bis[(4S,5R)-4-methoxycarbonyl-5-methyl-1,3-oxazolidine]copper(II) dichloride (2) and bis[(4S,5R)-4-methoxycarbonyl-5-methyl-2-oxazoline]-[(4S,5R)-4-methoxycarbonyl-5-methyl-1,3-oxazolidine]ruthenium(III) trichloride (3), respectively, were isolated and fully characterized by X-ray crystallography.Highlights► Novel complexes 3 and 4 were isolated and characterized by X-ray analysis. ► The trigonal bipyramidal Cu complex 3 contains two such oxazolidine ligands coordinated in different ways. ► In the octahedral Ru complex 4 there are one oxazolidine ligand 5 and two oxazoline ligands 6.
Co-reporter: Henri Brunner;Manfred Muschiol; Takashi Tsuno;Hyato Ike;Takaki Kurosawa;Kazuhito Koyama
Angewandte Chemie International Edition 2012 Volume 51( Issue 4) pp:1067-1070
Publication Date(Web):
DOI:10.1002/anie.201104960
Co-reporter:Henri Brunner, Takaki Kurosawa, Manfred Muschiol, Takashi Tsuno, and Hayato Ike
Organometallics 2012 Volume 31(Issue 8) pp:3395-3401
Publication Date(Web):April 6, 2012
DOI:10.1021/om3001789
The compounds (RFe,RC)-/(SFe,RC)-[CpFe(Prophos)NCMe]X (X = I, PF6), configurationally labile at the metal center, were used in the MeCN/ligand exchange reactions with cyclohexyl isocyanide (CyNC) and tert-butyl isocyanide (tBuNC). Kinetic measurements showed that the MeCN/CyNC exchange in diastereomerically pure (SFe,RC)-[CpFe(Prophos)NCMe]X proceeded via the slow SN1-type dissociation of the Fe–NCMe bond, already observed in the MeCN/phosphite exchange reactions. The product (RFe,RC)-/(SFe,RC)-[CpFe(Prophos)CNCy]X (X = I, PF6) was formed in diastereomer ratios between 40:60 and 60:40. However, specific for the MeCN/CyNC exchange in (SFe,RC)-[CpFe(Prophos)NCMe]PF6, in some of the samples a fast initial reaction interfered, initiated by traces of oxygen, which oxidized the cation in (SFe,RC)-[CpFe(Prophos)NCMe]PF6 to (SFe,RC)-[CpFe(Prophos)NCMe]2+. This dipositive cation started an electrocatalytic chain reaction, producing (RFe,RC)-/(SFe,RC)-[CpFe(Prophos)CNCy]PF6 with a high stereoselectivity of 2:98 in favor of (SFe,RC)-[CpFe(Prophos)CNCy]PF6. Deactivation processes terminated the chain reaction, depending on the varying amounts of (SFe,RC)-[CpFe(Prophos)NCMe]2+ present in the system. Larger amounts of oxygen or oxidants, such as I2 and AgPF6, caused immediate complete conversion to (RFe,RC)/(SFe,RC)-[CpFe(Prophos)CNR]PF6 in a diastereomer ratio of 2:98. In contrast to the hexafluorophosphate salt, addition of a crystal of iodine did not initiate the chain reaction in the iodide salt [CpFe(Prophos)NCMe]I, because I2 added to I– to form I3–, which did not oxidize the cation of [CpFe(Prophos)NCMe]I. Instead, there was slow conversion according to the dissociative pathway. The correlation between the configuration of (RFe,RC)- and (SFe,RC)-[CpFe(Prophos)CNCy]X and the conformation of the Fe-Prophos chelate ring on the one hand and the correlation with the P–P coupling constants of the Prophos ligand on the other hand was corroborated.
Co-reporter:Henri Brunner, Hayato Ike, Manfred Muschiol, Takashi Tsuno, Naohisa Umegaki, and Manfred Zabel
Organometallics 2011 Volume 30(Issue 3) pp:414-421
Publication Date(Web):January 20, 2011
DOI:10.1021/om100276t
The compounds [CpFe(Prophos)Cl] and [CpFe(Prophos)I] were prepared in photochemical reactions of [CpFe(CO)2Cl] and [CpFe(CO)2I] with (R)-Prophos. They consist of pairs of RFe,RC and SFe,RC diastereomers which only differ in the configuration at the metal atom. The diastereomerically pure compounds (SFe,RC)-[CpFe(Prophos)Cl] and (RFe,RC)-[CpFe(Prophos)I], which have the same relative configurations, were isolated. They epimerize via change of the Fe configuration and approach the equilibria (RFe,RC)-/(SFe,RC)-[CpFe(Prophos)Cl] = 5/95 and (RFe,RC)-/(SFe,RC)-[CpFe(Prophos)I] = 95/5 in first-order reactions with half-lives of 43 min at 20 °C and 50 min at 50 °C in C6D6, respectively. The reaction of (RFe,RC)-/(SFe,RC)-[CpFe(Prophos)I] = 95/5 with KCN afforded the cyano complex [CpFe(Prophos)CN] in the diastereomer ratio RFe,RC/SFe,RC = 50/50. Both diastereomers (RFe,RC)- and (SFe,RC)-[CpFe(Prophos)CN] could be isolated diastereomerically pure. The compounds (RFe,RC)- and (SFe,RC)-[CpFe(Prophos)CN] are configurationally stable at the metal center. There is no diastereomer interconversion, not even at higher temperatures. The carbonyl complexes [CpFe(Prophos)CO]I, [CpFe(Prophos)CO]PF6, and [IndFe(Prophos)CO]I were prepared in thermal reactions of [CpFe(CO)2I] and [IndFe(CO2)I] with (R)-Prophos or in an autoclave reaction of [CpFe(Prophos)I]/NH4PF6 with CO under pressure. All the carbonyl complexes are configurationally stable at the metal center. Seven diastereomers were characterized by X-ray crystallography. Including the two diastereomers (RFe,RC)-[CpRu(Prophos)Br] and (RFe,RC)-[CpRu(Prophos)I], a conformational analysis of the M-Prophos chelate ring was carried out, resulting in characteristic differences between major and minor diastereomers.
Co-reporter:Henri Brunner, Hayato Ike, Manfred Muschiol, Takashi Tsuno, Kazuhiro Koyama, Takaki Kurosawa, and Manfred Zabel
Organometallics 2011 Volume 30(Issue 13) pp:3666-3676
Publication Date(Web):June 14, 2011
DOI:10.1021/om2003893
The diastereomers (RFe,RC)/(SFe,RC)-[CpFe(Prophos)NCMe]X (X = I, PF6), 5:95, differing only in the metal configuration, were prepared from (RFe,RC)/(SFe,RC)-[CpFe(Prophos)I] (95:5) in acetonitrile in the absence or presence of NH4PF6. The diastereomers interconverted by change of the Fe configuration in first-order reactions in CDCl3 at 293 K with half-lives of 216 min (iodide) and 96 min (hexafluorophosphate) in an SN1-type dissociation of the MeCN ligand, followed by pyramidal inversion of the 16-electron intermediates (RFe,RC)- and (SFe,RC)-[CpFe(Prophos)]+ and recombination with MeCN. In the presence of phosphite ligands there was MeCN/ligand exchange, the kinetics of which was measured. The rates of the MeCN/phosphite exchange decreased with increasing cone angle for P(OCH2)3CMe (101°), P(OMe)3 (107°), and P(OPh)3 (128°). PPh3 (145°) did not enter the vacant coordination site in the intermediates (RFe,RC)- and (SFe,RC)-[CpFe(Prophos)]+. Phosphines such as Ph2PCH2CH2PPh2 and PBu3 bound only loosely to the intermediates. The phosphite complexes (RFe,RC)/(SFe,RC)-[CpFe(Prophos)P(OR)3]X were configurationally stable at the metal atom. In the MeCN/phosphite exchange reactions the diastereomeric ratio of the products was constant, explained by an equilibrium between the intermediates in an energy diagram in which the barrier for the unimolecular pyramidal inversion was lower than the barriers for the bimolecular reactions of the intermediates with the phosphite ligands. A correlation between the major diastereomers (SFe,RC)-[CpFe(Prophos)NCMe]X (X = I, PF6) and the diastereomers of the phosphite complexes with the same relative configuration and the favored envelope conformation of the Fe-Prophos chelate ring was corroborated, and a new correlation with the P–P coupling constants of the Prophos ligand was established, including all the compounds of a former study.
Co-reporter:Henri Brunner, Manfred Muschiol, Takashi Tsuno, Takemoto Takahashi and Manfred Zabel
Organometallics 2010 Volume 29(Issue 2) pp:428-435
Publication Date(Web):December 29, 2009
DOI:10.1021/om900830a
This paper reports the synthesis, isomer separation, and X-ray characterization of the compounds (SRu,SC)-/(RRu,SC)-[CpRu(Chairphos)Cl], Chairphos = (S)-1,3-bis(diphenylphosphanyl)butane, and cis-/trans-[CpRu(Dppm-Me)Cl], Dppm-Me = 1,1-bis(diphenylphosphanyl)ethane. The Cl/I exchange reactions proceeded with predominant retention of the metal configuration, accompanied by some inversion, except for trans-[CpRu(Dppm-Me)Cl], which was stereospecifically converted to trans-[CpRu(Dppm-Me)I]. Temperature-dependent kinetic measurements afforded rates and activation parameters of the Cl/I exchange and epimerization reactions that follow basilica-type energy profiles. Dissociation of Cl− from [CpRu(Chairphos)Cl] and [CpRu(Dppm-Me)Cl] gives pyramidal intermediates [CpRu(Chairphos)]+ and [CpRu(Dppm-Me)]+, which maintain the metal configuration. The 16-electron intermediates can react with excess I− to form the iodo complexes with retention of the metal configuration, or they can change the metal configuration by pyramidal inversion, leading to formation of iodo complexes with inverted metal configuration. The kinetic measurements show that the pyramidal inversion via planar transition states depends on the P−Ru−P′ angles. It increases with decreasing chelate ring size, because small P−Ru−P′ angles resist planarization in the transition, which requires larger P−Ru−P′ angles.
Co-reporter:Henri Brunner and Takashi Tsuno
Dalton Transactions 2017 - vol. 46(Issue 15) pp:NaN5109-5109
Publication Date(Web):2017/03/27
DOI:10.1039/C7DT00474E
In half-sandwich compounds of the type [Cp*ML1L2PPh3] the PPh3 propeller is stabilized by attractive CH/π interactions in which Co–H bonds specifically interact with the Ci and Co atoms of neighbouring phenyl rings, as in the T-shaped benzene dimer (i/o = ipso/ortho). This stabilization was not taken into account in a recent conformational analysis based on van der Waals energy calculations and minimization of steric compression (Dalton Trans., 2015, 44, 5451–5466). It is shown that in all 116 structures discussed in this analysis the CoH–Ci/o distances fall below the sum of the van der Waals radii, establishing attractive CH/π interactions, although the short contacts could easily be avoided by phenyl rotation to relieve steric strain. In 53 of the described structures there are acyl substituents which form conformation-determining Co–H⋯O(acyl) hydrogen bonds that are not taken into account in the recent analysis. The steric-only model is not an adequate description of M–PPh3 complexes.