Co-reporter:Lei Wang
New Biotechnology 2017 Volume 38, Part A(Volume 38, Part A) pp:
Publication Date(Web):25 September 2017
DOI:10.1016/j.nbt.2016.10.003
The genetic code can be expanded to include unnatural amino acids (Uaas) by engineering orthogonal components involved in protein translation. To be compatible with live cells, side chains of Uaas have been limited to either chemically inert or bio-orthogonal (i.e., nonreactive toward biomolecules) functionalities. To introduce bioreactivity into live systems, the genetic code has recently been engineered to encode a new class of Uaas, the bioreactive Uaas. These Uaas, after being incorporated into proteins, specifically react with target natural amino acid residues via proximity-enabled bioreactivity, enabling the selective formation of new covalent linkages within and between proteins both in vitro and in live systems. The new covalent bonding ability has been harnessed within proteins to enhance photostability, increase thermostability, staple proteins recombinantly, and build optical nano-switches, and between proteins to pinpoint ligand-receptor interaction, target native receptors irreversibly, and generate covalent macromolecular inhibitors. These diverse bioreactivities, inaccessible to natural proteins, thus open doors to novel protein engineering and provide new avenues for biological studies, biotherapeutics and synthetic biology.
Co-reporter:Tomonori Kobayashi, Christian Hoppmann, Bing Yang, and Lei Wang
Journal of the American Chemical Society 2016 Volume 138(Issue 45) pp:14832-14835
Publication Date(Web):October 31, 2016
DOI:10.1021/jacs.6b08656
Chemical reactivity is essential for functional modification of biomolecules with small molecules and the development of covalent drugs. The reactivity between a chemical functional group of a small molecule and that of a large biomolecule cannot be reliably predicted from the reactivity of the corresponding functional groups separately installed on two small molecules, because the proximity effect on reactivity resulting from the binding of the small molecule to the biomolecule is challenging to achieve by mixing two small molecules. Here we present a new strategy to determine the chemical reactivity of two functional groups in the context of close proximity afforded by proteins. The functional groups to be tested were separately installed at the interface of two interacting proteins in the format of amino acid side chains via the expansion of the genetic code. Reaction of the two functional groups resulted in covalent cross-linking of interacting proteins, readily detectable by gel electrophoresis. Using this strategy, we evolved new synthetases to genetically encode Nε-fluoroacetyllysine (FAcK), an isosteric fluorine analogue of acetyllysine. We demonstrated that fluoroacetamide installed on FAcK, previously thought inert to biological functional groups, actually reacted with the thiol group of cysteine when in proximity. This strategy should be valuable for accurately evaluating chemical reactivity of small molecules toward large biomolecules, which will help avoid undesired side reactions of drugs and expand the repertoire of functional groups to covalently target biomolecules.
Co-reporter:Bradley L Pentelute, Lei Wang
Current Opinion in Chemical Biology 2016 Volume 34() pp:v-vi
Publication Date(Web):October 2016
DOI:10.1016/j.cbpa.2016.09.017
Co-reporter:Christian Hoppmann and Lei Wang
Chemical Communications 2016 vol. 52(Issue 29) pp:5140-5143
Publication Date(Web):21 Mar 2016
DOI:10.1039/C6CC01226D
Although small molecule covalent inhibitors have been widely explored, macromolecular covalent inhibitors are more difficult to design and implement. Here we present a strategy to enable a peptide to bind to its target protein covalently via proximity-enabled bioreactivity, improving its activity of inhibiting the p53–Mdm4 interaction by 10-fold.
Co-reporter:Haiyan Ren, Bing Yang, Cheng Ma, Ying S. Hu, Peng George Wang, and Lei Wang
ACS Chemical Biology 2016 Volume 11(Issue 10) pp:2679
Publication Date(Web):September 7, 2016
DOI:10.1021/acschembio.6b00579
Photobleaching of fluorescent proteins (FPs) is a major limitation to their use in advanced microscopy, and improving photostability remains highly challenging due to limited understanding of its molecular mechanism. Here we discovered a new mechanism to increase FP photostability. Cysteine oxidation, implicated in only photobleaching before, was found to drastically enhance FP photostability to the contrary. We generated a far-red FP mStable by introducing a cysteine proximal to the chromophore. Upon illumination, this cysteine was oxidized to sulfinic and sulfonic acids, enabling mStable more photostable than its ancestor mKate2 by 12-fold and surpassing other far-red FPs. mStable outperformed in laser scanning confocal imaging and super-resolution structured illumination microscopy. Moreover, photosensitization to oxidize a cysteine similarly introduced in another FP mPlum also increased its photostability by 23-fold. This postfolding cysteine sulfoxidation cannot be simply substituted by the isosteric aspartic acid, representing a unique mechanism valuable for engineering better photostability into FPs.
Co-reporter:Christian Hoppmann; Innokentiy Maslennikov; Senyon Choe
Journal of the American Chemical Society 2015 Volume 137(Issue 35) pp:11218-11221
Publication Date(Web):August 24, 2015
DOI:10.1021/jacs.5b06234
Optical modulation of proteins provides superior spatiotemporal resolution for understanding biological processes, and photoswitches built on light-sensitive proteins have been significantly advancing neuronal and cellular studies. Small molecule photoswitches could complement protein-based switches by mitigating potential interference and affording high specificity for modulation sites. However, genetic encodability and responsiveness to nonultraviolet light, two desired properties possessed by protein photoswitches, are challenging to be engineered into small molecule photoswitches. Here we developed a small molecule photoswitch that can be genetically installed onto proteins in situ and controlled by visible light. A pentafluoro azobenzene-based photoswitchable click amino acid (F-PSCaa) was designed to isomerize in response to visible light. After genetic incorporation into proteins via the expansion of the genetic code, F-PSCaa reacts with a nearby cysteine within the protein generating an azo bridge in situ. The resultant bridge is switchable by visible light and allows conformation and binding of CaM to be regulated by such light. This photoswitch should prove valuable in optobiology for its minimal interference, site flexibility, genetic encodability, and response to the more biocompatible visible light.
Co-reporter:Christian Hoppmann and Lei Wang
Chemical Communications 2016 - vol. 52(Issue 29) pp:NaN5143-5143
Publication Date(Web):2016/03/21
DOI:10.1039/C6CC01226D
Although small molecule covalent inhibitors have been widely explored, macromolecular covalent inhibitors are more difficult to design and implement. Here we present a strategy to enable a peptide to bind to its target protein covalently via proximity-enabled bioreactivity, improving its activity of inhibiting the p53–Mdm4 interaction by 10-fold.
.jpg)
![N-(6-hydrazinyl-6-oxohexyl)-6-[5-(2-oxo-1,3,3a,4,6,6a-hexahydrothieno[3,4-d]imidazol-4-yl)pentanoylamino]hexanamide](http://img.cochemist.com/ccimg/211300/211237-33-1.png)
![N-(6-hydrazinyl-6-oxohexyl)-6-[5-(2-oxo-1,3,3a,4,6,6a-hexahydrothieno[3,4-d]imidazol-4-yl)pentanoylamino]hexanamide](http://img.cochemist.com/ccimg/211300/211237-33-1_b.png)





































![4-Amino-1-[(5S)-5-(hydroxymethyl)tetrahydro-2-furanyl]-2(1H)-pyri midinone](http://img.cochemist.com/ccimg/83900/83869-56-1.png)
![4-Amino-1-[(5S)-5-(hydroxymethyl)tetrahydro-2-furanyl]-2(1H)-pyri midinone](http://img.cochemist.com/ccimg/83900/83869-56-1_b.png)









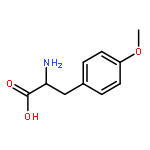






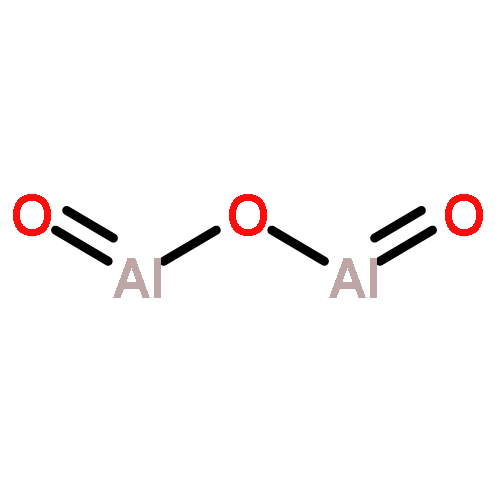
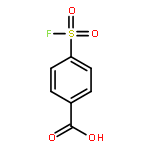





![(3AR,4R,5R,6AS)-4-FORMYL-2-OXOHEXAHYDRO-2H-CYCLOPENTA[B]FURAN-5-Y<WBR />L 4-BIPHENYLCARBOXYLATE](http://img.cochemist.com/ccimg/100/72-89-9.png)
![(3AR,4R,5R,6AS)-4-FORMYL-2-OXOHEXAHYDRO-2H-CYCLOPENTA[B]FURAN-5-Y<WBR />L 4-BIPHENYLCARBOXYLATE](http://img.cochemist.com/ccimg/100/72-89-9_b.png)