Co-reporter:Ge Meng, Meilin Zheng, Mei Wang, Jing Tong, Weijuan Ge, Jiehe Zhang, Aqun Zheng, Jingya Li, Lixin Gao, Jia Li
European Journal of Medicinal Chemistry 2016 Volume 122() pp:756-769
Publication Date(Web):21 October 2016
DOI:10.1016/j.ejmech.2016.05.060
•17 new compounds with two heterocycles were designed, synthesized as PTP1B inhibitor.•All the molecules were tested for the inhibitory activities against PTP1B and CDC25B.•Compound 14i containing pyrrole and thiazolidine-4-one showed IC50 = 6.09 μM for PTP1B.•Compound 14b displayed IC50 of 8.66 μM and 1.66 μM for PTP1B and CDC25B respectively.•Dock modes between 14b/14h/14i and PTP1B, ADMET properties and SAR were analyzed.A new series of 2-substituted imino-3-substituted-5- heteroarylidene-1,3-thiazolidine-4-ones as the potent bidentate PTP1B inhibitors were designed and synthesized in this paper. All of the new compounds were characterized and identified by spectra analysis. The biological screening test against PTP1B showed that some of these compounds have the positive inhibitory activity against PTP1B. The activity of the compounds with 5-substituted pyrrole on 5-postion of 1,3-thiazolidine-4-one are more potent than that of those compounds with 5-substituted pyridine group. Compound 14b, 14h and 14i showed IC50 values of 8.66 μM, 6.83 μM and 6.09 μM against PTP1B, respectively. Docking analysis of these active compounds with PTP1B showed the possible interaction modes of these biheterocyclic compounds with the active sites of PTP1B. The inhibition tests against oncogenetic CDC25B were also conducted on this set of compounds to evaluate the selectivity and possible anti-neoplastic activity. Compound 14b also showed the lowest IC50 of 1.66 μM against CDC25B among all the possible inhibitors, including 14g, 14h, 14i and 15c. Some pharmacological parameters including VolSurf, steric and electric descriptors of all the compounds were calculated to give some hints about the relative relationship with the biological activity. The result of this study might give some light on designing the possible anti-cancer drugs targeting at phosphatases. The most active compound 14i might be used as the lead compound for further structure modification of the new low molecular weight PTP1B inhibitors with the N-containing heterocyclic skeleton.Figure optionsDownload full-size imageDownload as PowerPoint slide
Co-reporter:Jing Chen, Li-Xin Gao, Jing-Xu Gong, Cheng-Shi Jiang, Li-Gong Yao, Jing-Ya Li, Jia Li, Wei Xiao, Yue-Wei Guo
Bioorganic & Medicinal Chemistry Letters 2015 Volume 25(Issue 10) pp:2211-2216
Publication Date(Web):15 May 2015
DOI:10.1016/j.bmcl.2015.03.060
A series of novel 1,2-dithiolan-4-yl benzoate compounds were synthesized and evaluated for in vitro PTP1B inhibitory activity. Some derivatives exhibited improved PTP1B inhibitory activity and selectivity compared to hit 6a, a compound from in-house library screening inspired by marine cyclic disulfide. The preliminary SAR analysis with assistance of molecular modeling approach revealed 6j (IC50 = 0.59 μM) as the most potent PTP1B inhibitor among all derivatives.
Co-reporter:Wei Yang, Lixuan Li, Yulan Wang, Xiaowei Wu, Tingting Li, Nan Yang, Mingbo Su, Li Sheng, Mingyue Zheng, Yi Zang, Jia Li, Hong Liu
Bioorganic & Medicinal Chemistry 2015 23(17) pp: 5881-5890
Publication Date(Web):
DOI:10.1016/j.bmc.2015.06.071
Co-reporter:Xiao-Peng He, Qiong Deng, Liang Cai, Chang-Zheng Wang, Yi Zang, Jia Li, Guo-Rong Chen, and He Tian
ACS Applied Materials & Interfaces 2014 Volume 6(Issue 8) pp:5379
Publication Date(Web):April 4, 2014
DOI:10.1021/am5010909
Developing an effective means for the real-time probing of amyloid β (Aβ) that is closely implicated in Alzheimer’s disease (AD) could help better understand and monitor the disease. Here we describe an economic approach based on the simple composition of a natural product, resveratrol (Res), with graphene oxide (GO) for the rapid, fluorogenic recognition of Aβ. The Res@GO composite has proved specific for Aβ over a range of proteins and ions, and could sensitively capture both Aβ monomers and fibers in a physiological buffer solution within only 3 min. The composite can also fluorescently image amyloid deposits in a mouse brain section within 30 min. This new protocol is much cheaper and more timesaving than the conventional immunofluorescence staining technique employed clinically, providing an economic tool for the concise detection of AD.Keywords: Alzheimer’s disease; amyloid β; detection; fluorogenic; graphene; resveratrol;
Co-reporter:Wei Ma, Hui-Ting Liu, Xiao-Peng He, Yi Zang, Jia Li, Guo-Rong Chen, He Tian, and Yi-Tao Long
Analytical Chemistry 2014 Volume 86(Issue 11) pp:5502
Publication Date(Web):May 7, 2014
DOI:10.1021/ac501463u
Here, we describe a novel “switch-on” biosensor based on quinonyl glycosides functionalized quantum dots (QDs) for the specific targeting and imaging of transmembrane glycoprotein receptors on the surface of cancer cells. The design of the quinonyl glycosides lies in that the quinone moiety serves as a quencher of QDs and the glycoside moiety as a biospecific ligand for targeting a receptor. We observed that the quenched photoluminescence of the quinone glycosides functionalized QDs could be significantly recovered by a specific lectin that selectively binds to the glycosides clustering the QDs but was not affected by a panel of nonspecific lectins. Moreover, we determined that quinonyl galactoside functionalized QDs could optically image the asialoglycoprotein receptors of a hepatoma cell line in a target-specific manner. This system might provide new insights into the fabrication of photoluminogenic biosensors for the analysis of the universal ligand–receptor recognitions in nature.
Co-reporter:Xun Ji, Chunmei Xia, Jiang Wang, Mingbo Su, Lei Zhang, Tiancheng Dong, Zeng Li, Xia Wan, Jingya Li, Jia Li, Linxiang Zhao, Zhaobing Gao, Hualiang Jiang, Hong Liu
European Journal of Medicinal Chemistry 2014 Volume 86() pp:242-256
Publication Date(Web):30 October 2014
DOI:10.1016/j.ejmech.2014.08.059
•The DPP4 inhibitory activities and selectivity of compounds 8 and 9 were tested.•Compounds 8l and 9l showed good efficacy in OGTTs in ICR mice.•Compound 9l reduced the blood glucose level in diabetic BKS db/db mice with multiple doses for 5 weeks.•Compounds 8l and 9l did not block hERG channel and displayed no inhibition of liver metabolic enzymes.Based on the previous work in our group and the principle of computer-aided drug design, a series of novel β-amino pyrrole-2-carbonitrile derivatives was designed and synthesized. Compounds 8l and 9l were efficacious and selective DPP4 inhibitors resulting in decreased blood glucose in vivo. Compound 8l had moderate DPP4 inhibitory activity (IC50 = 0.05 μM) and good oral bioavailability (F = 53.2%). Compound 9l showed excellent DPP4 inhibitory activity (IC50 = 0.01 μM), good selectivity (selective ratio: DPP8/DPP4 = 898.00; DPP9/DPP4 = 566.00) against related peptidases, and good efficacy in an oral glucose tolerance tests in ICR mice and moderate PK profiles (F = 22.8%, t1/2 = 2.74 h). Moreover, compound 9l did not block hERG channel and exhibited no inhibition of liver metabolic enzymes such as CYP2C9.By rational design and modification, compound 9l showed excellent DPP4 inhibitory activity, good selectivity and efficacy in OGTTs in ICR mice, did not block the hERG channel and no inhibition of live metabolic enzyme like CYP2C9.
Co-reporter:Ben-Ren Liao, Hai-Bing He, Ling-Ling Yang, Li-Xin Gao, Liang Chang, Jie Tang, Jing-Ya Li, Jia Li, Fan Yang
European Journal of Medicinal Chemistry 2014 Volume 83() pp:15-25
Publication Date(Web):18 August 2014
DOI:10.1016/j.ejmech.2014.06.011
•Non-phosphorus-based fructose-1,6-bisphosphatase (FBPase) inhibitors, 2,5-diphenyl-1,3,4-oxadiazoles, were explored.•Compounds bearing a linear alkyl with terminal hydrogen-bonding acceptor showed potent inhibition at molecular level.•Compound 22 and 27b inhibit the glucose production in primary rat hepatocytes with an IC50 value of 167.3 μM and 112.5 μM.•Both inhibition and binding mode to the enzyme were investigated by enzymatic kinetics and in silico experiments.With the aim of discovering a novel class of non-phosphorus-based fructose-1,6-bisphosphatase (FBPase) inhibitors, a series of 2,5-diphenyl-1,3,4-oxadiazoles were synthesized based on the hit compound (1) resulting from a high-throughput screening (HTS). Structure–activity relationship (SAR) studies led to the identification of several compounds with comparable inhibitory activities to AMP, the natural allosteric inhibitor of FBPase. Notably, compound 22 and 27b, bearing a terminal carboxyl or 1H-tetrazole, demonstrated remarkable inhibition to gluconeogenesis (GNG). In addition, both inhibition and binding mode to the enzyme were investigated by enzymatic kinetics and in silico experiments for representative compounds 16 and 22.Novel non-phosphorus-based FBPase inhibitors were explored, and some compounds showed comparable inhibitory activities to AMP and remarkable inhibition to gluconeogenesis. Both inhibition and binding mode to the enzyme were investigated.
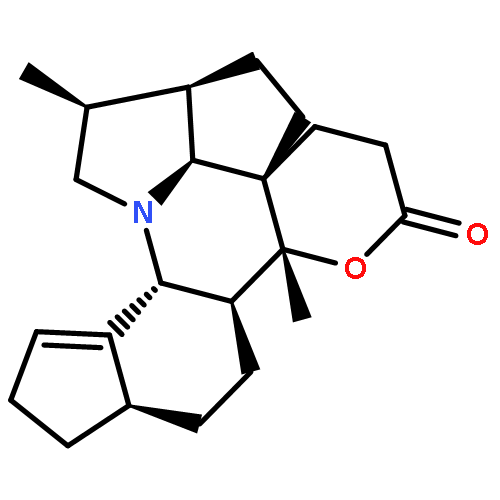
Co-reporter:Jian-Peng Yin, Chun-Lan Tang, Li-Xin Gao, Wei-Ping Ma, Jing-Ya Li, Ying Li, Jia Li and Fa-Jun Nan
Organic & Biomolecular Chemistry 2014 vol. 12(Issue 21) pp:3441-3445
Publication Date(Web):25 Mar 2014
DOI:10.1039/C4OB00214H
A series of structurally related analogues of the natural product paracaseolide A were synthesized and identified as potent PTP1B inhibitors. Among these analogues, compound 10 in particular showed improved PTP1B enzyme inhibitory activity, high selectivity for PTP1B over TC-PTP, and improved cellular effects.
Co-reporter:Fei Chen, Hui Chai, Ming-Bo Su, Yang-Ming Zhang, Jia Li, Xin Xie, and Fa-Jun Nan
ACS Medicinal Chemistry Letters 2014 Volume 5(Issue 6) pp:628
Publication Date(Web):April 4, 2014
DOI:10.1021/ml400470s
Inspired by the thiazole–thiazoline cap group in natural product largazole, a series of structurally simplified bisthiazole-based histone deacetylase inhibitors were prepared and evaluated. Compound 8f was evaluated in vivo in an experimental autoimmune encephalomyelitis (EAE) model and found to be orally efficacious in ameliorating clinical symptoms of EAE mice.Keywords: bisthiazole; Histone deacetylase inhibitors; largazole
Co-reporter:Wei Yang, Lixuan Li, Xun Ji, Xiaowei Wu, Mingbo Su, Li Sheng, Yi Zang, Jia Li, Hong Liu
Bioorganic & Medicinal Chemistry 2014 Volume 22(Issue 21) pp:6146-6155
Publication Date(Web):1 November 2014
DOI:10.1016/j.bmc.2014.08.030
A series of 4-anilinothieno[2,3-d]pyrimidine-based hydroxamic acid derivatives as novel HDACs inhibitors were designed, synthesized and evaluated. Most of these compounds displayed good to excellent inhibitory activities against HDAC1, 3, 6. The IC50 values of compound 10r against HDAC1, HDAC3, HDAC6 was 1.14 ± 0.03 nM, 3.56 ± 0.08 nM, 11.43 ± 0.12 nM. Compound 10r noticeably up-regulated the level of histone H3 acetylation compared to the SAHA. Most of the compounds showed the strong anti-proliferative activity against human cancer cell lines including RMPI8226 and HCT-116. The IC50 values of Compounds 10r and 10t against RPMI8226 was 2.39 ± 0.20 μM, 1.41 ± 0.44 μM, respectively, and the HCT-116 was sensitive to the compounds 10h, 10m, 10r, 10w with the IC50 values <1.9 μM.
Co-reporter:Wen-Long Wang, Chao Huang, Li-Xin Gao, Chun-Lan Tang, Jun-Qing Wang, Min-Chen Wu, Li Sheng, Hai-Jun Chen, Fa-Jun Nan, Jing-Ya Li, Jia Li, Bainian Feng
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 8) pp:1889-1894
Publication Date(Web):15 April 2014
DOI:10.1016/j.bmcl.2014.03.015
Co-reporter:Ying Zhi, Li-Xin Gao, Yi Jin, Chun-Lan Tang, Jing-Ya Li, Jia Li, Ya-Qiu Long
Bioorganic & Medicinal Chemistry 2014 22(14) pp: 3670-3683
Publication Date(Web):
DOI:10.1016/j.bmc.2014.05.028
Co-reporter:Ying Zhi, Li-Xin Gao, Yi Jin, Chun-Lan Tang, Jing-Ya Li, Jia Li, Ya-Qiu Long
Bioorganic & Medicinal Chemistry 2014 22(15) pp: 4347
Publication Date(Web):
DOI:10.1016/j.bmc.2014.06.026
Co-reporter:Yiquan Zou, Lei Xu, Wuyan Chen, Yiping Zhu, Tiantian Chen, Yan Fu, Li Li, Lanping Ma, Bing Xiong, Xin Wang, Jian Li, Jianhua He, Haiyan Zhang, Yechun Xu, Jia Li, Jingkang Shen
European Journal of Medicinal Chemistry 2013 Volume 68() pp:270-283
Publication Date(Web):October 2013
DOI:10.1016/j.ejmech.2013.06.027
•We explored the SAR by combining click chemistry and in situ screening assay.•Pro70 and Thr72 were the critical components for binding with these inhibitors.•Based on crystal structures, pyrazole was discovered as a novel C-terminus.•Pyrazole derivatives showed good activities and better selectivity for BACE1.We recently discovered and reported dual inhibitor 5 of AChE and BACE1 with N-benzylpiperidine ethyl as C-terminus. Compound 5 showed potent inhibitory activities for BACE1, and could reduce endogenous Aβ1–40 production in APP transgenic mice. In present work, we rapidly identified substituted triazole as the C-terminus of compound 5 by replacing the benzylpiperidine ethyl group with click chemistry and tested these synthesized compounds by in situ screening assay. As revealed by the crystal structures of BACE1 in complex with our triazole compound 12, we found that Pro70 and Thr72 located in the flap region were the critical components for binding with these inhibitors. With the aid of the crystal structure, a new series of five-membered heterocyclic compounds was prepared in order to explore the structure–activity relationship (SAR) of this class of molecules. From these efforts, pyrazole was discovered as a novel C-terminus of BACE1 inhibitors. After further modification of pyrazole with variable substituents, compound 37 exhibited good potency in enzyme inhibition assay (IC50 = 0.025 μM) and compound 33 showed moderate inhibition effects on Aβ production of APP transfected HEK293 cells. Moreover, these pyrazole derivatives demonstrated good selectivity versus cathepsin D. Our results indicated that the vicinity of Pro70 and Thr72 might be utilized as a subsite, and the discovered pyrazole derivatives might provide useful hints for developing novel BACE1 inhibitors as anti-AD drugs.We combined the click chemistry and in situ screening technologies to fast explore the SAR of C terminus of our previous compound. Further modification identified the pyrazole group as a good substituent for BACE1 inhibitors in the enzymatic, cellular inhibition assays.
Co-reporter:Qing Ye, Yanhong Shen, Yubo Zhou, Dan Lv, Jianrong Gao, Jia Li, Yongzhou Hu
European Journal of Medicinal Chemistry 2013 Volume 68() pp:361-371
Publication Date(Web):October 2013
DOI:10.1016/j.ejmech.2013.07.046
•A series of 7-azaindazolyl-indolyl-maleimides were synthesized.•Most compounds showed potent activity towards GSK-3β.•Selected compounds 17a, 17b, 17g, 17i, 29a and 30 significantly reduced Aβ-induced Tau hyperphosphorylation.•The structure–activity relationships of compounds have been discussed.A series of 7-azaindazolyl-indolyl-maleimides were designed, synthesized and evaluated for their GSK-3β inhibitory activity. Most compounds exhibited potent activity against GSK-3β. Among them, compounds 17a, 17b, 17g, 17i, 29a and 30 significantly reduced Aβ-induced Tau hyperphosphorylation, showin;g the inhibition of GSK-3β at the cell level. Preliminary structure–activity relationships were discussed based on the experimental data obtained.
Co-reporter:Kai-Bin Li, Xiao-Li Wei, Yi Zang, Xiao-Peng He, Guo-Rong Chen, Jia Li and Kaixian Chen
Analyst 2013 vol. 138(Issue 23) pp:7087-7089
Publication Date(Web):12 Sep 2013
DOI:10.1039/C3AN01379K
This study reveals that a dipropargyl rhodamine B derivative previously described as a reaction-based irreversible palladium probe responds, however, more sensitively to mercury with a reversible “turn-on” fluorescence. The probe also shows a much better imaging ability for mercury than for palladium in live cells.
Co-reporter:Lei Zhang;Mingbo Su;Jingya Li;Xun Ji;Jiang Wang;Zeng Li;Jia Li;Hong Liu
Chemical Biology & Drug Design 2013 Volume 81( Issue 2) pp:198-207
Publication Date(Web):
DOI:10.1111/cbdd.12058
Dipeptidyl peptidase-4 inhibitors hold great potential for the treatment of type 2 diabetes. A series of 1-(γ-1,2,3-triazol substituted prolyl)-(S)-3,3-difluoropyrrolidines were designed, synthesized, and evaluated as novel dipeptidyl peptidase-4 inhibitors. Most of the compounds exhibited good in vitro potency against dipeptidyl peptidase-4. Among these, compounds 7j, 7q, and 7s displayed good dipeptidyl peptidase-4 activity and excellent selectivity versus other proteases including dipeptidyl peptidase-8, dipeptidyl peptidase-9, and FAP. The possible binding modes of compounds 7j, 7q, and 7s with dipeptidyl peptidase-4 were also explored by molecular docking simulation.
Co-reporter:Li-Fang Yu, Yuan-Yuan Li, Ming-Bo Su, Mei Zhang, Wei Zhang, Li-Na Zhang, Tao Pang, Run-Tao Zhang, Bing Liu, Jing-Ya Li, Jia Li, and Fa-Jun Nan
ACS Medicinal Chemistry Letters 2013 Volume 4(Issue 5) pp:475-480
Publication Date(Web):March 25, 2013
DOI:10.1021/ml400028q
Adenosine 5′-monophosphate-activated protein kinase (AMPK) is emerging as a promising drug target for its regulatory function in both glucose and lipid metabolism. Compound PT1 (5) was originally identified from high throughput screening as a small molecule activator of AMPK through the antagonization of the autoinhibition in α subunits. In order to enhance its potency at AMPK and bioavailability, structure–activity relationship studies have been performed and resulted in a novel series of AMPK activators based on an alkene oxindole scaffold. Following their evaluation in pharmacological AMPK activation assays, lead compound 24 was identified to possess improved potency as well as favorable pharmacokinetic profile. In the diet-induced obesity (DIO) mouse model, compound 24 was found to improve glucose tolerance and alleviate insulin resistance. The in vitro and in vivo data for these alkene oxindoles warrant further studies for their potential therapeutic medications in metabolic associated diseases.Keywords: alkene oxindole; AMPK activator; diabetes; DIO mouse model; insulin sensitivity;
Co-reporter:Lei Cui;Xiao-Peng He;Li-Xin Gao;Jia Li;Guo-Rong Chen
Journal of Heterocyclic Chemistry 2013 Volume 50( Issue 3) pp:684-688
Publication Date(Web):
DOI:10.1002/jhet.1517
The efficient construction of triazolyl peptidomimetics via the powerful click chemistry for the discovery of small molecule-based chemotherapeutic agents represents a promising strategy in drug development today. Herein, the synthesis of novel mono-triazolyl or bis-triazolyl amino acid derivatives was rapidly achieved via microwave-assisted Cu(I)-catalyzed azide-alkyne 1,3-dipolar cycloaddition (CuAAC). Subsequent in vitro enzymatic assay on several homologous protein tyrosine phosphatases (PTPs) identified the triazolyl dimers as new specific inhibitors of Cell Cycle Division 25B (CDC25B) phosphatase and Protein Tyrosine Phosphatase 1B (PTP1B).
Co-reporter:Linrong Zhu;Dr. Yuanyuan Li;Dr. Ling Qiu;Dr. Mingbo Su; Xin Wang;Chunmei Xia;Dr. Yi Qu; Jingya Li; Jia Li; Bing Xiong; Jingkang Shen
ChemMedChem 2013 Volume 8( Issue 7) pp:1104-1116
Publication Date(Web):
DOI:10.1002/cmdc.201300104
Abstract
The worldwide prevalence of diabetes has spurred numerous studies on the development of new antidiabetic medicines. As a result, dipeptidyl peptidase IV (DPP4) has been recognized as a validated target. In our efforts to discover new DPP4 inhibitors, we analyzed the complexed structures of DPP4 available in Protein Data Bank and designed a series of triazole compounds. After enzyme activity assays and crystallographic verification of the binding interaction patterns, we found that the triazole compounds can inhibit DPP4 with micromolar IC50 values. Liver microsome stability and cytochrome P450 metabolic tests were performed on this series, revealing undesirable pharmacokinetic profiles for the triazole compounds. To overcome this liability, we substituted the triazole ring with an amide or urea group to produce a new series of DPP4 inhibitors. Based on its enzyme activity, metabolic stability, and selectivity over DPP8 and DPP9, we selected compound 21 r for further study of its in vivo effects in mice using an oral glucose tolerance test (OGTT). The results show that 21 r has efficacy similar to that of sitagliptin at a dose of 3 mg kg−1. The crystal structure of 21 r bound to DPP4 also reveals that the trifluoromethyl group is directed toward a subpocket different from the subsite bound by sitagliptin, providing clues for the design of new DPP4 inhibitors.
Co-reporter:Qiuhua Zhu, Lixin Gao, Zhipeng Chen, Sichao Zheng, Huafei Shu, Jia Li, Huanfeng Jiang, Shuwen Liu
European Journal of Medicinal Chemistry 2012 Volume 54() pp:232-238
Publication Date(Web):August 2012
DOI:10.1016/j.ejmech.2012.05.001
A series of tetra- and pentasubstituted polyfunctional dihydropyrroles 5 and 6 were synthesized via practical multicomponent reactions (MCRs) for research on their structure–activity relationship as caspase-3 inhibitors. Among 39 compounds evaluated, 14 of them exhibited inhibition against caspase-3 with IC50 ranging from 5 to 20 μM. The inhibitory activities of 5 and 6 depend on the nature of substituents on different positions. 5 and 6 possess a different scaffold from those previously reported and are the first caspase-3 inhibitors prepared via MCRs. The most active compounds 5k (IC50 = 5.27 μM) could therefore be used as a lead for the development of highly potent caspase-3 inhibitors as drug candidates for therapeutic agents by taking advantage of MCRs.Graphical abstractTetra- and pentasubstituted dihydropyrroles 5 and 6 were synthesized via practical one-pot multicomponent reactions (MCRs). 14 of them exhibited inhibition against caspase-3 with IC50 ranging from 5 to 20 μM.Highlights► The MCR scope for the synthesis of pentasubstituted polyfunctional dihydropyrroles 6 were expanded. ► Tetra- and pentasubstituted dihydropyrroles 5 and 6 are a novel series of caspase-3 inhibitors. ► The inhibitory activity of 5 and 6 depend on the nature of substituents on different positions.
Co-reporter:Liang-Peng Sun;Li-Xin Gao;Wei-Ping Ma;Jia Li;Hu-Ri Piao
Chemical Biology & Drug Design 2012 Volume 80( Issue 4) pp:584-590
Publication Date(Web):
DOI:10.1111/j.1747-0285.2012.01431.x
A series of 2,4,6-trihydroxychalcone derivatives were synthesized and identified as reversible and competitive protein tyrosine phosphatase (PTP) 1B inhibitors with IC50 values in the micromolar range. Compound 4a had the greatest in vitro inhibition activity against PTP1B (IC50 = 0.27 ± 0.01 μm) and the best selectivity (6.9-fold) for PTP1B relative to T-cell protein tyrosine phosphatases. The compounds identified herein provide a foundation on which to design specific inhibitors of PTP1B and other PTPs.
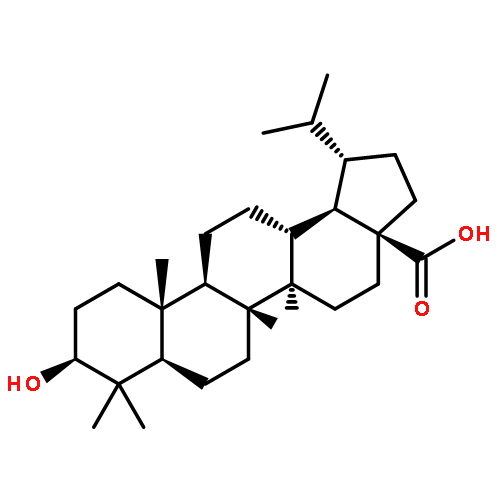
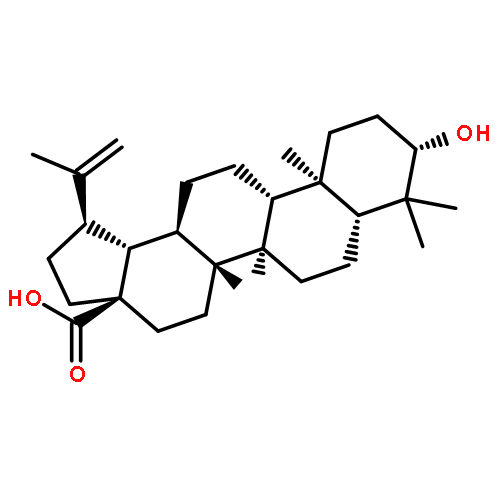
Co-reporter:Hai-Bing He, Li-Xin Gao, Qi-Feng Deng, Wei-Ping Ma, Chun-Lan Tang, Wen-Wei Qiu, Jie Tang, Jing-Ya Li, Jia Li, Fan Yang
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 23) pp:7237-7242
Publication Date(Web):1 December 2012
DOI:10.1016/j.bmcl.2012.09.040
Protein tyrosine phosphatase 1B (PTP1B) is a major negative regulator of both insulin and leptin signals. For years, inhibiting of PTP1B has been considered to be a potential therapeutics for treating Type 2 diabetes and obesity. Recently, we recognized lithocholic acid (LCA) as a natural inhibitor against PTP1B (IC50 = 12.74 μM) by a vertical screen for the first time. Further SAR research was carried out by synthesizing and evaluating a series of compounds bearing two methyls at C-4 position and a fused heterocycle to ring A. Among them, compound 14b achieved a PTP1B inhibitory activity about eightfold than LCA and a 14-fold selectivity over the homogenous enzyme TCPTP.The synthesis and biological evaluation of a series of heterocyclic ring-substituted 4,4-dimethyl lithocholic acid derivatives are reported for the first time. The active compound 14b displayed potent inhibitory activity on protein tyrosine phosphatase 1B with IC50 value of approximately 1.62 μM.
Co-reporter:Hou-ling Dai, Li-xin Gao, Ying Yang, Jing-ya Li, Jia-gao Cheng, Jia Li, Ren Wen, Yan-qing Peng, Jian-bin Zheng
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 24) pp:7440-7443
Publication Date(Web):15 December 2012
DOI:10.1016/j.bmcl.2012.10.054
A series of di-indolinone derivatives was designed and synthesized to optimize our lead compounds basing on molecular docking study as PTP1B inhibitors. Successive enzymatic assay identified the synthetic di-indolinone as novel PTP1B inhibitors with low micromole-ranged inhibitory activity and at least several-fold selectivity over other tested homologous PTPs.
Co-reporter:Guanghua Fang, Mengzhu Xue, Mingbo Su, Dingyu Hu, Yanlian Li, Bing Xiong, Lanping Ma, Tao Meng, Yuelei Chen, Jingya Li, Jia Li, Jingkang Shen
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 14) pp:4540-4545
Publication Date(Web):15 July 2012
DOI:10.1016/j.bmcl.2012.05.123
The introduction of the multi-objective optimization has dramatically changed the virtual combinatorial library design, which can consider many objectives simultaneously, such as synthesis cost and drug-likeness, thus may increase positive rates of biological active compounds. Here we described a software called CCLab (Combinatorial Chemistry Laboratory) for combinatorial library design based on the multi-objective genetic algorithm. Tests of the convergence ability and the ratio to re-take the building blocks in the reference library were conducted to assess the software in silico, and then it was applied to a real case of designing a 5 × 6 HDAC inhibitor library. Sixteen compounds in the resulted library were synthesized, and the histone deactetylase (HDAC) enzymatic assays proved that 14 compounds showed inhibitory ratios more than 50% against tested 3 HDAC enzymes at concentration of 20 μg/mL, with IC50 values of 3 compounds comparable to SAHA. These results demonstrated that the CCLab software could enhance the hit rates of the designed library and would be beneficial for medicinal chemists to design focused library in drug development (the software can be downloaded at: http://202.127.30.184:8080/drugdesign.html).
Co-reporter:Liang-Peng Sun, Qiang Shen, Hong-Hua Piao, Wei-Ping Ma, Li-Xin Gao, Wei Zhang, Fa-Jun Nan, Jia Li, Hu-Ri Piao
European Journal of Medicinal Chemistry 2011 Volume 46(Issue 9) pp:3630-3638
Publication Date(Web):September 2011
DOI:10.1016/j.ejmech.2011.05.027
A series of novel nonphosphonate-based pTyr mimetics comprised (±)-3-(2-(2-fluorobenzyloxy)naphthalen-6-yl)-2-aminopropanoic acid derivatives were identified as reversible and competitive PTP1B inhibitors via a structure-based design approach. Among the compounds studied, 12h was found to have the best in vitro inhibition activity against PTP1B (IC50 = 1.25 ± 0.24 μM) and the best selectivity (3-fold) between PTP1B and TCPTP. These results should provide suitable druglike lead compounds for the design of inhibitors of PTP1B as well as other PTPs.Structure-based design led to the discovery of novel nonphosphonate-based pTyr mimetics comprised (±)-3-(2-(2-fluorobenzyloxy)naphthalen-6-yl)-2-aminopropanoic acid derivatives (12a–r) as inhibitors of protein tyrosine phosphatase 1B (PTP1B).Highlights► Structure-based design led to the discovery of novel nonphosphonate-based pTyr mimetics. ► These derivatives are potent, competitive, and reversible inhibitors of PTP1B. ► These results should provide suitable druglike lead compounds.
Co-reporter:Cui Li, Xiao-Peng He, Yin-Jie Zhang, Zhen Li, Li-Xin Gao, Xiao-Xin Shi, Juan Xie, Jia Li, Guo-Rong Chen, Yun Tang
European Journal of Medicinal Chemistry 2011 Volume 46(Issue 9) pp:4212-4218
Publication Date(Web):September 2011
DOI:10.1016/j.ejmech.2011.06.025
With an aim of developing novel protein tyrosine phosphatase (PTP) 1B inhibitors based on sugar scaffolds, a focused library of benzyl 6-triazolo(hydroxy)benzoic glucosides was efficiently constructed via the modular and selective Cu(I)-catalyzed azide-alkyne 1,3-dipolar cycloaddtion (click chemistry). These glycoconjugates bearing alkyl chain length-varied bridges between the sugar and (hydroxy)-benzoic moieties were identified as new PTP1B inhibitors with selectivity over T-Cell PTP (TCPTP), SH2-Containing PTP-1 (SHP-1), SHP-2 and Leukocyte Antigen-Related Tyrosine Phosphatase (LAR). Molecular docking study sequentially elaborated the plausible binding modes of the structurally diverse sugar-based inhibitors with PTP1B.Novel PTP1B inhibitors based on a sugar scaffold were efficiently constructed via click chemistry. Molecular docking study sequentially elaborated their plausible binding modes with PTP1B.Highlights► Novel PTP1B inhibitors based on a glucosyl scaffold were developed. ► Click chemistry was employed to modularly construct the focused library. ► The inhibitors were identified with high selectivity on PTP1B over others. ► Plausible PTP1B inhibitor interactions were suggested by molecular docking.
Co-reporter:Gui-Hua Tang, Dong-Mei Chen, Bei-Ying Qiu, Li Sheng, Yue-Hu Wang, Guang-Wan Hu, Fu-Wei Zhao, Li-Juan Ma, Huan Wang, Qiao-Qin Huang, Jin-Jin Xu, Chun-Lin Long, and Jia Li
Journal of Natural Products 2011 Volume 74(Issue 1) pp:45-49
Publication Date(Web):December 15, 2010
DOI:10.1021/np100606u
Eight new amide alkaloids (1−8) and 19 known ones were isolated from the whole plant of Piper boehmeriaefolium. Their structures were determined through spectroscopic data analyses. Cytotoxic activity of these amides against human cervical carcinoma HeLa cells was evaluated, and 1-[(9E)-10-(3,4-methylenedioxyphenyl)-9-decenoyl]pyrrolidine (9) exhibited significant inhibitory activity with an IC50 value of 2.7 μg/mL.
Co-reporter:Xiao-Peng He, Qiong Deng, Li-Xin Gao, Cui Li, Wei Zhang, Yu-Bo Zhou, Yun Tang, Xiao-Xin Shi, Juan Xie, Jia Li, Guo-Rong Chen, Kaixian Chen
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 13) pp:3892-3900
Publication Date(Web):1 July 2011
DOI:10.1016/j.bmc.2011.05.049
Protein tyrosine phosphatases (PTPs) are well-validated therapeutic targets for many human major diseases. The development of their potent inhibitors has therefore become a main focus of both academia and the pharmaceutical industry. We report herein a facile strategy toward the fabrication of new and competent PTP inhibitor entities by simply ’clicking’ alkynyl amino acids onto diverse azido sugar templates. Triazolyl glucosyl, galactosyl, and mannosyl serine and threonine derivatives were efficiently synthesized via click reaction, which were then identified as potent CDC25B and PTP1B inhibitors selective over a panel of homologous PTPs tested. Their inhibitory activity and selectivity were found to largely lie on the structurally and configurationally diversified monosaccharide moieties whereon serinyl and threoninyl residues were introduced. In addition, MTT assay revealed the triazole-connected sugar-amino acid hybrids may also inhibit the growth of several human cancer cell lines including A549, Hela, and especially HCT-116. On the basis of such compelling evidence, we consider that this compound series could furnish promising chemical entities serving as new CDC25B and PTP1B inhibitors with potential cellular activity. Furthermore, the ‘click’ strategy starting from easily accessible and biocompatible amino acids and sugar templates would allow the modular fabrication of a rich library of new PTP inhibitors efficaciously and productively.
Co-reporter:Zhuo Song, Xiao-Peng He, Cui Li, Li-Xin Gao, Zhao-Xia Wang, Yun Tang, Juan Xie, Jia Li, Guo-Rong Chen
Carbohydrate Research 2011 Volume 346(Issue 1) pp:140-145
Publication Date(Web):3 January 2011
DOI:10.1016/j.carres.2010.10.023
The synthesis of triazole-linked glycosyl acetophenone, benzoic acid, and α-ketocarboxylic acid derivatives was readily achieved via Cu(I)-catalyzed azide–alkyne cycloaddition (‘click’ reaction) in excellent yields of 93–97%. Among the synthesized glycoconjugates, the triazolyl α-ketocarboxylic acids were identified as the most potent protein tyrosine phosphatase 1B (PTP1B) inhibitors with micromole-ranged IC50 values and moderate-to-good selectivity over a panel of homologous PTPs including TCPTP (4.6-fold), LAR (>30-fold), SHP-1 (>30-fold) and SHP-2 (>30-fold). Moreover, a docking simulation was conducted to propose a plausible binding mode of the glucosyl α-ketocarboxylic acid triazole with the enzymatic target.Triazole-linked glycosyl acetophenone, benzoic acid and α-ketocarboxylic acid derivatives have been efficiently synthesized by click chemistry. Glycosyl α-ketocarboxylic acids exhibited micromolar inhibition towards PTP1B. Docking simulation was conducted to propose a plausible binding mode with the enzyme.
Co-reporter:Yi Wei, Yue-Ting Chen, Lei Shi, Li-Xin Gao, Shen Liu, Yong-Mei Cui, Wei Zhang, Qiang Shen, Jia Li and Fa-Jun Nan
MedChemComm 2011 vol. 2(Issue 11) pp:1104-1109
Publication Date(Web):23 Sep 2011
DOI:10.1039/C1MD00153A
A series of compounds synthesized from a key intermediate in the total synthesis of Scleritodermin A containing a novel conjugated thiazole moiety, 2-(1-amino-2-p-hydroxyphenylethane)-4-(4-carboxy-2,4-dimethyl-2Z,4E-propadiene)-thiazole (ACT), were discovered to be potent inhibitors of protein tyrosine phosphatase 1B, with IC50 values in the low micromolar range. Structure–activity relationships around the scaffold were investigated and some compounds exhibited more potent PTP1B inhibitory activity and improved specificities compared with the original hit.
Co-reporter:Jin-Wei Yang, Xiao-Peng He, Cui Li, Li-Xin Gao, Li Sheng, Juan Xie, Xiao-Xin Shi, Yun Tang, Jia Li, Guo-Rong Chen
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 4) pp:1092-1096
Publication Date(Web):15 February 2011
DOI:10.1016/j.bmcl.2010.12.126
There has been considerable interest in the development of protein tyrosine phosphatase (PTP) inhibitors since many of the PTP members are tightly associated with major human diseases including autoimmune disorders, diabetes and cancer. We report here a unique and rapid approach toward the development of novel PTP inhibitor entities based on triazolyl pseudo-glycopeptides. By employing microwave-accelerated Cu(I)-catalyzed azide-alkyne 1,3-dipolar cycloaddition (CuAAC or ‘click reaction’), a series of triazole-linked serinyl, threoninyl, phenylalaninyl and tyrosinyl 1-O-gluco- or galactosides have been efficiently synthesized in high yields within only ∼30 min. Successive biological assay identified these glycopeptidotriazoles as favorable PTP1B and CDC25B inhibitors with selectivity over TCPTP, LAR, SHP-1 and SHP-2. Both the structural diversity of the amino acid (Ser, Thr, Phe and Tyr) introduced and the epimeric identity (Glc or Gal) on monosaccharide scaffold were determined to impact the corresponding inhibitory activity and selectivity. In addition, the benzylated sugar scaffold was demonstrated to act as a crucial role for enhancing the binding affinity of the inhibitors with the targeted PTP. Docking simulation was eventually conducted to propose plausible binding modes of this compound series with PTP1B and CDC25B. Our approach readily realized from naturally abundant raw materials (sugar and amino acid) and via facile, regioselective and expeditious synthetic method (microwave-assisted click reaction) might provide new insights toward the ‘click’ fabrication of structurally diverse PTP inhibitors.We report the ‘click’ fabrication of triazolyl glycopeptidomimetics as novel PTP1B and CDC25B inhibitors and their plausible binding modes with the targeted PTP via docking simulation.
Co-reporter:Zhuo Song, Xiao-Peng He, Xiao-Ping Jin, Li-Xin Gao, Li Sheng, Yu-Bo Zhou, Jia Li, Guo-Rong Chen
Tetrahedron Letters 2011 Volume 52(Issue 8) pp:894-898
Publication Date(Web):23 February 2011
DOI:10.1016/j.tetlet.2010.12.055
Bidentate 1-O-methyl-α-d-pyranoglucosides bearing two triazolyl α-ketoester groups on the 2,6- or 3,4-positions of sugar scaffold were efficiently synthesized via Cu(I)-catalyzed azide-alkyne 1,3-dipolar cycloaddition (click reaction) in good yields. These newly featured sugar derivatives displayed favorable inhibitory activity on protein tyrosine phosphatase 1B (PTP1B) and unexpected selective fluorescence quenching in the presence of Ni2+.
Co-reporter:Jia Wang;Xiaopeng He;Lixin Gao;Li Sheng;Xiaoxin Shi;Jia Li;Guorong Chen
Chinese Journal of Chemistry 2011 Volume 29( Issue 6) pp:1227-1232
Publication Date(Web):
DOI:10.1002/cjoc.201190228
Abstract
Triazolyl phenylalanine and tyrosine-aryl C-glycoside hybrids were readily synthesized via microwave-assisted Cu(I)-catalyzed azide-alkyne 1,3-dipolar cycloaddition in high yields. Successive enzymatic assay identified the synthesized glycoconjugates as novel PTP1B inhibitors with low micromole-ranged inhibitory activity and at least several-fold selectivity over other homologous PTPs tested. In addition, the benzyl groups on glucosyl moiety were found crucial toward PTP1B inhibition.
Co-reporter:Yu Luo, Hao-Min Liu, Ming-Bo Su, Li Sheng, Yu-Bo Zhou, Jia Li, Wei Lu
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 16) pp:4844-4846
Publication Date(Web):15 August 2011
DOI:10.1016/j.bmcl.2011.06.046
Two natural piperamides (piperlonguminine and refrofractamide A) and their derivatives were synthesized and evaluated for inhibitory activity against histone deacetylases, as well as the HCT-116 human colon cancer cell line. The preliminary structure activity relationship was discussed. Compounds featuring a hydroxamic acid moiety exhibited moderate HDAC activity and in vitro cytotoxicity.
Co-reporter:Xiao-Peng He;Cui Li;Zhi-Zhou Wang;Li-Xin Gao;Xiao-Xin Shi
Glycoconjugate Journal 2011 Volume 28( Issue 7) pp:
Publication Date(Web):2011/10/01
DOI:10.1007/s10719-011-9347-0
There has been increasing interest in the development of drug candidates based on sugar templates that possess rich structural and, especially, configurational diversities. We disclose herein that the epimeric identity between methyl 3,4-bis-phenylalanyl/tyrosinyl triazolyl-alpha-D-galactopyranoside and glucopyranoside may lead to their distinct inhibitory effects on specific protein tyrosine phosphatases (PTPs). Subsequently performed molecular docking study elucidated the plausible binding behaviors of the more potent galactosyl inhibitors with their primary PTP target, i.e. Cell Division Cycle 25B (CDC25B) phosphatase.
Co-reporter:Wan-Yi Tai;Run-Tao Zhang;Yi-Ming Ma;Min Gu;Gang Liu;Dr. Jia Li;Dr. Fa-Jun Nan
ChemMedChem 2011 Volume 6( Issue 9) pp:1555-1558
Publication Date(Web):
DOI:10.1002/cmdc.201100164
Co-reporter:Hai-Lin Zhang;Xiao-Peng He;Li Sheng;Yuan Yao;Wei Zhang
Molecular Diversity 2011 Volume 15( Issue 4) pp:889-900
Publication Date(Web):2011 November
DOI:10.1007/s11030-011-9318-1
Series of novel 6-triazole-linked galacto- or glucolipids were efficiently synthesized from O-benzylated sugar azides and various lipid alkynes via Cu(I)-catalyzed azide-alkyne 1,3-dipolar cycloaddition (click chemistry) followed by hydrogenolysis with PdCl2/H2. Subsequent MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide) assay toward a panel of human cancer cell lines revealed that these triazologlycolipids possess low-to-modest toxicity on A549 (lung), MCF-7 (breast), HeLa (cervix), and HepG2 (liver). Furthermore, both the carbon chain length at the lipid end and the epimeric identity of the glycosyl moiety were determined to impact their corresponding bioactivity.
Co-reporter:Juan Zhang, Qiang Shen, Jin-Cai Lu, Jing-Ya Li, Wen-Yong Liu, Jian-Jun Yang, Jia Li, Kai Xiao
Food Chemistry 2010 Volume 119(Issue 4) pp:1491-1496
Publication Date(Web):15 April 2010
DOI:10.1016/j.foodchem.2009.09.031
Phenolic compounds were separated from the leaves of Cyclocarya paliurus (Batal.) Ijinskaja and their bioactivities were evaluated through an in vitro PTP1B inhibitory assay. Bioassay-guided fractionation of the ethanol extract has resulted in the isolation of a naphthoquinone derivative, (1R, 2R, 4R)-1,2,4-trihydroxy-1,2,3,4-tetrahydro-naphthalene-1-O-β-d-glucopyranoside, named cyclonoside A, and a lactone, (4R, 5S, 6R)-8,9,10-trihydroxy-4-[3′,4′-dihydroxyphenyl]-1,6-dioxaspiro[4,5]decan-2-one, named cyclospirolide, along with 10 known phenolic compounds: quercetin-3-O-α-d-glucuronide, quercetin-3-O-β-d-glucuronide, myricetin-3-O-β-d-glucuronide, 1-caffeoylquinic acid, 3-caffeoylquinic acid, 4-caffeoylquinic acid, 5-caffeoylquinic acid, caffeic acid, 5-hydroxynaphthalene-1, 4-di-O-β-d-glucopyranoside and piceid. The structures of these compounds were established by means of spectroscopic methods including extensive 2D NMR techniques and chemical evidence. Among all the compounds, 1, 2, 4, 5, 10 and 11 showed strong inhibition against PTP1B, with IC50 values ranging from 1.922 ± 0.480 to 10.50 ± 2.67 μg/mL. The results suggested that the extract from this plant could be used as a potential source for functional food ingredient with anti-obesity.
Co-reporter:Zhe Cheng, An-Feng Chen, Fang Wu, Li Sheng, Han-Kun Zhang, Min Gu, Yuan-Yuan Li, Li-Na Zhang, Li-Hong Hu, Jing-Ya Li, Jia Li
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 16) pp:5915-5924
Publication Date(Web):15 August 2010
DOI:10.1016/j.bmc.2010.06.085
The clinical use of the natural alkaloid berberine (BBR) as an antidiabetic reagent is limited by its poor bioavailability. Our previous work demonstrated that dihydroberberine (dhBBR) has enhanced bioavailability and in vivo efficacy compared with berberine. Here we synthesized the 8,8-dimethyldihydroberberine (Di-Me) with improved stability, and bioavailability over dhBBR. Similar to BBR and dhBBR, Di-Me inhibited mitochondria respiration, increased AMP:ATP ratio, activated AMPK and stimulated glucose uptake in L6 myotubes. In diet-induced obese (DIO) mice, Di-Me counteracted the increased adiposity, tissue triglyceride accumulation and insulin resistance, and improved glucose tolerance at a dosage of 15 mg/kg. Administered to db/db mice with a dosage of 50 mg/kg, Di-Me effectively reduced random fed and fasting blood glucose, improved glucose tolerance, alleviated insulin resistance and reduced plasma triglycerides, with better efficacy than dhBBR at the same dosage. Our work highlights the importance of dihydroberberine analogs as potential therapeutic reagents for type 2 diabetes treatment.8,8-Disubstituted dihydroberberine hydrochlorides were synthesized and evaluated. Di-Me shows enhanced chemical stability, bioavailability and oral efficacy on obese and diabetic mouse models.
Co-reporter:Ji-Qing Xu, Qiang Shen, Jia Li, Li-Hong Hu
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 11) pp:3934-3939
Publication Date(Web):1 June 2010
DOI:10.1016/j.bmc.2010.04.073
Protein tyrosine phosphatase 1B (PTP1B) is a key factor in the negative regulation of insulin pathway and a promising target for treatment of diabetes and obesity. Herein, the sapogenin 2b, prepared from the natural triterpene saponin 1b, was modified at 3-position to establish the dammarane derivatives library via esterification, oxidation and reductive amination reaction and evaluated as PTP1B inhibitors. 3-O-para-Carboxylphenyl substituted derivative 5b was found with the best in vitro inhibition activity to protein tyrosine phosphatase 1B (IC50 = 0.27 μM), where 3-O-meta-carboxylphenyl substituted 5a exhibited the best selectivity (nearly fivefolds) between PTP1B and T-cell protein tyrosine phosphatase.We reported the dammaranes inhibitors of PTP1B and concluded the SAR. Compound 5b was found with the best activity (IC50 = 0.27 μM) and 5a exhibited the best selectivity (nearly fivefolds) between PTP1B and TCPTP.
Co-reporter:Hai-Jun Chen, Yong Liu, Li-Na Wang, Qiang Shen, Jia Li, Fa-Jun Nan
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 9) pp:2876-2879
Publication Date(Web):1 May 2010
DOI:10.1016/j.bmcl.2010.03.040
Structural optimization and preliminary structure–activity relationship studies of a series of N-substituted maleimide fused-pyrazole analogues with Cdc25B inhibitory activity, starting from a high-throughput screening hit, are illustrated. A simplified 3,5-diacyl pyrazole analogue was obtained as the most potent compound (118, IC50 = 0.12 μM) with a 270-fold increase in potency.
Co-reporter:Qing Ye, Guiqing Xu, Dan Lv, Zhe Cheng, Jia Li, Yongzhou Hu
Bioorganic & Medicinal Chemistry 2009 Volume 17(Issue 13) pp:4302-4312
Publication Date(Web):1 July 2009
DOI:10.1016/j.bmc.2009.05.031
A series of novel 4-azaindolyl-indolyl-maleimides were synthesized and evaluated for their GSK-3β inhibitory activity. Most compounds exhibited high potency to GSK-3β. Among them, compound 7c was the most promising GSK-3β inhibitor. Preliminary structure–activity relationships were discussed based on the experimental data obtained and showed that different substituents on the indole ring and side chains at 1-position of indole had varying degrees of influence on the GSK-3β inhibitory potency. In a cell-based functional assay, compounds 7c and 15a significantly reduced Aβ-induced Tau hyperphosphorylation by inhibiting GSK-3β.A novel series of 4-azaindolyl-indolyl-maleimides were synthesized and evaluated for their GSK-3β inhibitory activity. Most compounds showed potent activity towards GSK-3β. Among them, compound 7c was the most promising GSK-3β inhibitor.
Co-reporter:Yiping Zhu, Kun Xiao, Lanping Ma, Bin Xiong, Yan Fu, Haiping Yu, Wei Wang, Xin Wang, Dingyu Hu, Hongli Peng, Jingya Li, Qi Gong, Qian Chai, Xican Tang, Haiyan Zhang, Jia Li, Jingkang Shen
Bioorganic & Medicinal Chemistry 2009 Volume 17(Issue 4) pp:1600-1613
Publication Date(Web):15 February 2009
DOI:10.1016/j.bmc.2008.12.067
To explore novel effective drugs for the treatment of Alzheimer’s disease (AD), a series of dual inhibitors of acetylcholineterase (AChE) and β-secretase (BACE-1) were designed based on the multi-target-directed ligands strategy. Among them, inhibitor 28 exhibited good dual potency in enzyme inhibitory potency assay (BACE-1: IC50 = 0.567 μM; AChE: IC50 = 1.83 μM), and also showed excellent inhibitory effects on Aβ production of APP transfected HEK293 cells (IC50 = 98.7 nM) and mild protective effect against hydrogen peroxide (H2O2)-induced PC12 cell injury. Encouragingly, intracerebroventricular injection of 28 into amyloid precursor protein (APP) transgenic mice caused a 29% reduction of Aβ1–40 production. Therefore, 28 was demonstrated as a good lead compound for the further study and more importantly, the strategy of AChE and BACE-1 dual inhibitors might be a promising direction for developing novel drugs for AD patients.Novel dual inhibitors of acetylcholinesterase and β-secretase were design, synthesis and biological evaluation. Among them, compound 28 exhibited good dual potency in enzyme inhibitory potency assay (BACE-1: IC50 = 0.567 μM; AChE: IC50 = 1.83 μM), and also showed excellent inhibitory effects on Aβ production of APP transfected HEK293 cells (IC50 = 98.7 nM) and mild protective effect against hydrogen peroxide (H2O2)-induced PC12 cell injury. Encouragingly, intracerebroventricular injection of 28 into amyloid precursor protein (APP) transgenic mice caused a 29% reduction of Aβ1–40 production.
Co-reporter:Wen-Wei Qiu, Qiang Shen, Fan Yang, Bo Wang, Hui Zou, Jing-Ya Li, Jia Li, Jie Tang
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 23) pp:6618-6622
Publication Date(Web):1 December 2009
DOI:10.1016/j.bmcl.2009.10.017
A series of maslinic acid derivatives have been synthesized by introducing various fused heterocyclic rings at C-2 and C-3 positions. Their inhibitory effects on PTP1B, TCPTP and related PTPs are evaluated. Most of the compounds exhibited a dramatic increase in inhibitory potency and selectivity, the two most potent PTP1B inhibitors 20 (IC50 = 0.61 μM) and 29 (IC50 = 0.64 μM) showed about 10-fold more potent than lead compound maslinic acid. More importantly, 29 possesses the best selectivity of 6.9-fold for PTP1B over TCPTP.A series of heterocyclic ring-substituted maslinic acid derivatives were prepared and subsequently evaluated on PTP1B, TCPTP and related PTPs in order to increase PTP1B inhibitory activity and especially selectivity for PTP1B over TCPTP.
Co-reporter:Wenwei QIU;Jie XU;Xin LI;Li ZHONG;Jingya LI;Jia LI;Fajun NAN
Chinese Journal of Chemistry 2009 Volume 27( Issue 4) pp:825-833
Publication Date(Web):
DOI:10.1002/cjoc.200990138
Abstract
To explore the molecular mechanism of the matrix metalloproteinases (MMPs) in tumor processes, two photoaffinity trimodular probes were designed and synthesized based on the structure activity relationship and the following photoaffinity labelling experiments afforded positive results.
Co-reporter:Qing Ling, Yue Huang, Yueyang Zhou, Zhengliang Cai, Bing Xiong, Yahui Zhang, Lanping Ma, Xin Wang, Xin Li, Jia Li, Jingkang Shen
Bioorganic & Medicinal Chemistry 2008 Volume 16(Issue 15) pp:7399-7409
Publication Date(Web):1 August 2008
DOI:10.1016/j.bmc.2008.06.014
A novel synthesis of the human leukocyte common antigen-related (LAR) phosphatase inhibitor, illudalic acid, has been achieved by a route more amenable to structure modifications. A series of simpler analogues of illudalic acid was synthesized and evaluated for potency in inhibiting LAR. The structure–activity relationship (SAR) study has shown that the 5-formyl group and the hemi-acetal lactone are crucial for effective inhibition of LAR activity, and are the key pharmacophores of illudalic acid. The fused dimethylcyclopentene ring moiety evidently helps to enhance the potency of illudalic acid against LAR. A preliminary study of the mechanism of action of illudalic acid against LAR was conducted using electrospray ionization mass spectrometry (ESI-MS) and molecular docking techniques. The results are in full agreement with the described mechanism.A novel synthesis of the human leukocyte common antigen-related phosphatase (LAR) inhibitor, illudalic acid, has been achieved by a route more amenable to structure modifications. A preliminary study of the structure–activity relationship (SAR) and of the mechanism of action of illudalic acid was conducted.
Co-reporter:Yi-Nan Zhang, Wei Zhang, Di Hong, Lei Shi, Qiang Shen, Jing-Ya Li, Jia Li, Li-Hong Hu
Bioorganic & Medicinal Chemistry 2008 Volume 16(Issue 18) pp:8697-8705
Publication Date(Web):15 September 2008
DOI:10.1016/j.bmc.2008.07.080
Protein tyrosine phosphatase 1B is a key factor in the negative regulation of insulin pathway and a promising target for treatment of diabetes and obesity. Herein, a series of competitive inhibitors were optimized from oleanolic acid, a natural triterpenoid identified against PTP1B by screening libraries of traditional Chinese medicinal herbs. Modifying at 3 and 28 positions, we obtained compound 13 with a Ki of 130 nM, which exhibited good selectivity between other phosphatases involved in insulin pathway except T-cell protein tyrosine phosphatase. Further evaluation in cell models illustrated that the derivatives enhanced insulin receptor phosphorylation in CHO/hIR cells and also stimulated glucose uptake in L6 myotubes with or addition of without insulin.
Co-reporter:Yingdong Zhu, Ping Zhang, Haiping Yu, Jia Li, Ming-Wei Wang and Weimin Zhao
Journal of Natural Products 2007 Volume 70(Issue 10) pp:1570-1577
Publication Date(Web):September 21, 2007
DOI:10.1021/np070260v
Chemical studies on the constituents of Dracaena cochinchinensis led to the discovery of eight new flavonoid derivatives (1–8) along with 14 known compounds (9–22). The identification and structural elucidation of these isolates were based on spectral analyses. All isolates were tested for antibacterial activities against Helicobacter pylori (ATCC43504) and thrombin inhibitory effects. As a result, new flavonoid derivatives 6 and 7 and (2S)-4′,7-dihydroxy-8-methylflavan (11) were found to be most efficacious against H. pylori (ATCC43504) with MIC values of 29.5, 29.5, and 31.3 µM, respectively, and the seven new flavonoid derivatives (1–7) and one known biflavonoid (9) were observed to exhibit moderate thrombin inhibitory activity.
Co-reporter:Li Zhang, Beiying Qiu, Bing Xiong, Xin Li, Jingya Li, Xin Wang, Jia Li, Jingkang Shen
Bioorganic & Medicinal Chemistry Letters 2007 Volume 17(Issue 8) pp:2118-2122
Publication Date(Web):15 April 2007
DOI:10.1016/j.bmcl.2007.01.094
A series of quinoxalinylurea-based inhibitors are synthesized and shown to be the novel and potent inhibitors against Jnk Stimulatory Phosphatase-1 (JSP-1), which is a special member of dual-specificity protein phosphatase (DSP) family. Biological assay and computational modeling studies showed the compounds were reversible and noncompetitive inhibitors of JSP-1. JSP-1 inhibitors may be useful for the treatment of inflammatory, vascular, neurodegenerative, metabolic, and oncological diseases in humans associated with dysfunctional Jnk signaling.A series of quinoxalinylurea-based inhibitors are synthesized and shown to be the novel and potent inhibitors against JSP-1. Compound A17 showed the most potent activity to inhibit JSP-1.
Co-reporter:Wen-Wei Qiu Dr.;Jie Xu;Jing-Ya Li Dr.;Jia Li Dr. Dr.
ChemBioChem 2007 Volume 8(Issue 12) pp:
Publication Date(Web):10 JUL 2007
DOI:10.1002/cbic.200700148
Three in one. A trimodular probe (1) with three functional groups (for target recognition, proximal target crosslinking, and distal reporter tag attachment via click chemistry of a photostable azido-acetyl group) has been designed. We demonstrate its specificity, sensitivity, and potential for general application in activity-based protein profiling for type I methionine aminopeptidases.
Co-reporter:Weigang Huang, Jingya Li, Wei Zhang, Yueyang Zhou, Chuanming Xie, Yu Luo, Yunfei Li, Jingli Wang, Jia Li, Wei Lu
Bioorganic & Medicinal Chemistry Letters 2006 Volume 16(Issue 7) pp:1905-1908
Publication Date(Web):1 April 2006
DOI:10.1016/j.bmcl.2005.12.080
Miltirone analogues were synthesized and evaluated for inhibitory activity against Cdc25 and PTP1B. Most of the compounds demonstrated potent Cdc25 inhibitory activity, and several exhibited higher selectivity for Cdc25 than for PTP1B. In a cytotoxic assay, most of the compounds displayed cytotoxicity against the tumor cell lines A549 and HCT-116, producing IC50 values in the micromolar range.
Co-reporter:Yong-Mei Cui, Qing-Qing Huang, Jie Xu, Ling-Ling Chen, Jing-Ya Li, Qi-Zhuang Ye, Jia Li, Fa-Jun Nan
Bioorganic & Medicinal Chemistry Letters 2005 Volume 15(Issue 16) pp:3732-3736
Publication Date(Web):15 August 2005
DOI:10.1016/j.bmcl.2005.05.055
A series of thiazole-4-carboxylic acid thiazol-2-ylamide (TCAT, 4) derivatives were designed and synthesized according to simple bioisosteric replacement from previously reported pyridine-2-carboxylic acid thiazol-2-ylamide (PCAT) MetAP inhibitors. The preliminary SAR studies demonstrated that these TCAT series of compounds showed different activity and selectivity compared with those of the corresponding PCAT compounds. These findings provide useful information for the design and discovery of more potent inhibitors of type I MetAPs.A new series of potent type I MetAP inhibitors were obtained by simple bioisosteric replacement of previously reported pyridine-2-carboxylic acid thiazol-2-ylamide (PCAT) MetAP inhibitors.
Co-reporter:Yong-Mei Cui, Qing-Qing Huang, Jie Xu, Ling-Ling Chen, Jing-Ya Li, Qi-Zhuang Ye, Jia Li, Fa-Jun Nan
Bioorganic & Medicinal Chemistry Letters 2005 Volume 15(Issue 18) pp:4130-4135
Publication Date(Web):15 September 2005
DOI:10.1016/j.bmcl.2005.06.005
Systematic SAR studies on the thiazole ring 5-substituent of TCAT derivatives revealed that the introduction of a β-alkoxy or an amino group enhanced the inhibitory activity significantly. The present compounds are representative of specific Co(II)-MetAP1 inhibitors. Before the physiologically relevant metal ions for MetAPs are established, these small molecular compounds could be used as tools for detailed biological studies.Systematic SAR studies on the thiazole ring 5-substituent of TCAT inhibitors of EcMetAP1 and ScMetAP1 revealed that the introduction of a β-alkoxy or an amino group enhanced the inhibitory activity significantly.
Co-reporter:Jean-Michel Garcia, Anhui Gao, Pei-Lan He, Joyce Choi, Wei Tang, Roberto Bruzzone, Olivier Schwartz, Hugo Naya, Fa-Jun Nan, Jia Li, Ralf Altmeyer, Jian-Ping Zuo
Antiviral Research (March 2009) Volume 81(Issue 3) pp:239-247
Publication Date(Web):March 2009
DOI:10.1016/j.antiviral.2008.12.004
Co-reporter:Suzhen Dong, Yubing Lei, Shikun Jia, Lixin Gao, Jia Li, Tong Zhu, Shunying Liu, Wenhao Hu
Bioorganic & Medicinal Chemistry Letters (15 February 2017) Volume 27(Issue 4) pp:1105-1108
Publication Date(Web):15 February 2017
DOI:10.1016/j.bmcl.2016.11.055
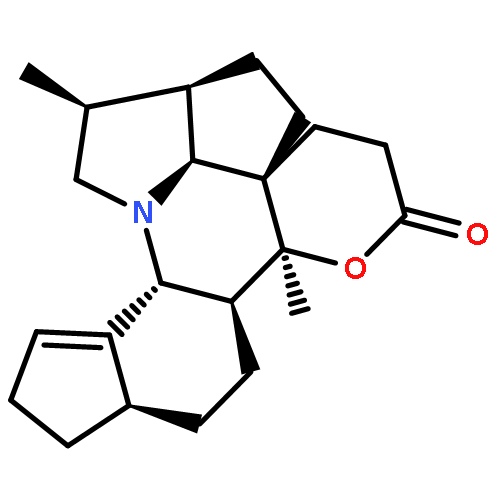
Co-reporter:Jian-Peng Yin, Chun-Lan Tang, Li-Xin Gao, Wei-Ping Ma, Jing-Ya Li, Ying Li, Jia Li and Fa-Jun Nan
Organic & Biomolecular Chemistry 2014 - vol. 12(Issue 21) pp:NaN3445-3445
Publication Date(Web):2014/03/25
DOI:10.1039/C4OB00214H
A series of structurally related analogues of the natural product paracaseolide A were synthesized and identified as potent PTP1B inhibitors. Among these analogues, compound 10 in particular showed improved PTP1B enzyme inhibitory activity, high selectivity for PTP1B over TC-PTP, and improved cellular effects.

![2H,11H-Naphtho[2,1-b]pyrano[3,4-e]pyran-11-one,3,6-bis(acetyloxy)-4-[(acetyloxy)methyl]-1,3,4,4a,5,6,6a,12,12a,12b-decahydro-12-hydroxy-4,6a,12b-trimethyl-9-(3-pyridinyl)-,(3S,4R,4aR,6S,6aS,12R,12aS,12bS)-](http://img.cochemist.com/ccimg/147500/147444-03-9.png)
![2H,11H-Naphtho[2,1-b]pyrano[3,4-e]pyran-11-one,3,6-bis(acetyloxy)-4-[(acetyloxy)methyl]-1,3,4,4a,5,6,6a,12,12a,12b-decahydro-12-hydroxy-4,6a,12b-trimethyl-9-(3-pyridinyl)-,(3S,4R,4aR,6S,6aS,12R,12aS,12bS)-](http://img.cochemist.com/ccimg/147500/147444-03-9_b.png)
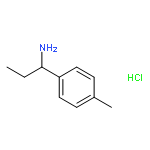
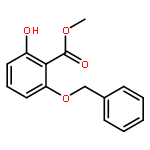
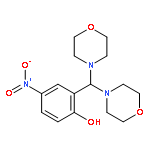
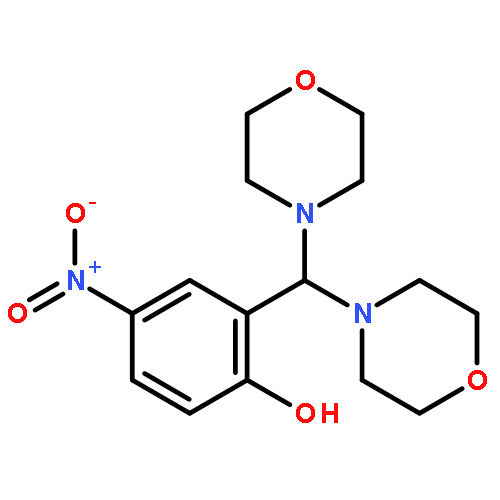
![Phenol, 2-[(2-methyl-1H-indol-3-yl)-4-morpholinylmethyl]-4-nitro-](http://img.cochemist.com/ccimg/511300/511295-38-8.png)
![Phenol, 2-[(2-methyl-1H-indol-3-yl)-4-morpholinylmethyl]-4-nitro-](http://img.cochemist.com/ccimg/511300/511295-38-8_b.png)
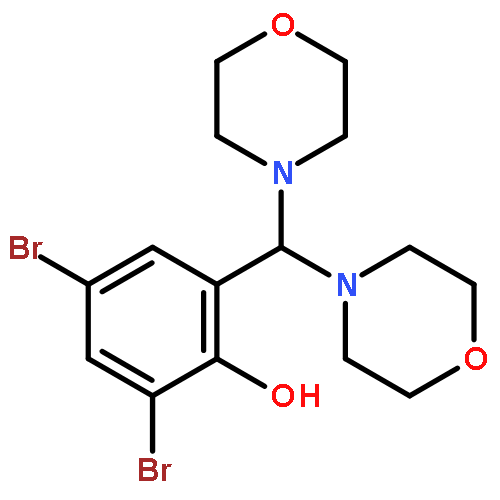
![Phenol, 2,4-dibromo-6-[(2-methyl-1H-indol-3-yl)-4-morpholinylmethyl]-](http://img.cochemist.com/ccimg/372600/372508-77-5.png)
![Phenol, 2,4-dibromo-6-[(2-methyl-1H-indol-3-yl)-4-morpholinylmethyl]-](http://img.cochemist.com/ccimg/372600/372508-77-5_b.png)
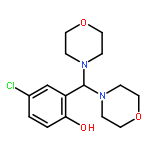
![Phenol, 4-chloro-2-[(2-methyl-1H-indol-3-yl)-4-morpholinylmethyl]-](http://img.cochemist.com/ccimg/298700/298685-88-8.png)
![Phenol, 4-chloro-2-[(2-methyl-1H-indol-3-yl)-4-morpholinylmethyl]-](http://img.cochemist.com/ccimg/298700/298685-88-8_b.png)
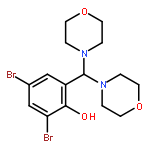


![Urea, N-(4-chlorophenyl)-N'-[(4-methylphenyl)methyl]-](http://img.cochemist.com/ccimg/92300/92277-80-0.png)
![Urea, N-(4-chlorophenyl)-N'-[(4-methylphenyl)methyl]-](http://img.cochemist.com/ccimg/92300/92277-80-0_b.png)



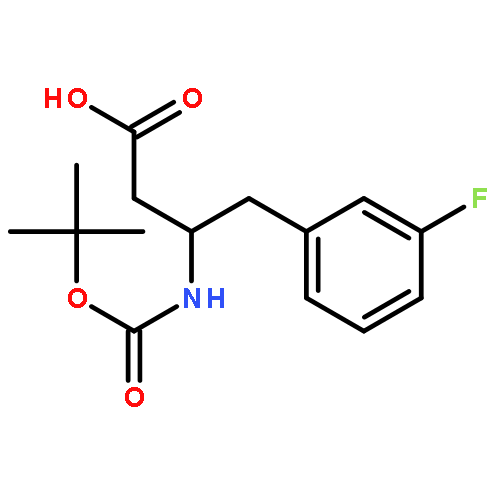
![Carbamic acid, [3-(3-formylphenoxy)propyl]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/897500/897404-12-5.png)
![Carbamic acid, [3-(3-formylphenoxy)propyl]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/897500/897404-12-5_b.png)
![Benzonitrile, 4-[3-(1-piperazinyl)propoxy]-](http://img.cochemist.com/ccimg/360600/360553-67-9.png)
![Benzonitrile, 4-[3-(1-piperazinyl)propoxy]-](http://img.cochemist.com/ccimg/360600/360553-67-9_b.png)


![1H-Cyclopenta[b]benzofuran,3a-(bromomethyl)-2,3,3a,8b-tetrahydro-3,6,8b-trimethyl-, (3S,3aS,8bS)-](http://img.cochemist.com/ccimg/68800/68773-08-0.png)
![1H-Cyclopenta[b]benzofuran,3a-(bromomethyl)-2,3,3a,8b-tetrahydro-3,6,8b-trimethyl-, (3S,3aS,8bS)-](http://img.cochemist.com/ccimg/68800/68773-08-0_b.png)
![Phenol,4-bromo-2-[(1R,3S)-1,3-dimethyl-2-methylenecyclopentyl]-5-methyl-](http://img.cochemist.com/ccimg/29500/29424-17-7.png)
![Phenol,4-bromo-2-[(1R,3S)-1,3-dimethyl-2-methylenecyclopentyl]-5-methyl-](http://img.cochemist.com/ccimg/29500/29424-17-7_b.png)

![Phenol, 2-[(1S,2R,5R)-1,2-dimethylbicyclo[3.1.0]hex-2-yl]-5-methyl-](http://img.cochemist.com/ccimg/10600/10539-88-5.png)
![Phenol, 2-[(1S,2R,5R)-1,2-dimethylbicyclo[3.1.0]hex-2-yl]-5-methyl-](http://img.cochemist.com/ccimg/10600/10539-88-5_b.png)
![Phenol,4-bromo-2-[(1S,2R,5R)-1,2-dimethylbicyclo[3.1.0]hex-2-yl]-5-methyl-](http://img.cochemist.com/ccimg/10600/10539-87-4.png)
![Phenol,4-bromo-2-[(1S,2R,5R)-1,2-dimethylbicyclo[3.1.0]hex-2-yl]-5-methyl-](http://img.cochemist.com/ccimg/10600/10539-87-4_b.png)
![[(3S,3aS,8bS)-7-bromo-3,6,8b-trimethyl-1,2,3,8b-tetrahydro-3aH-benzo[b]cyclopenta[d]furan-3a-yl]methanol](http://img.cochemist.com/ccimg/6800/6790-64-3.png)
![[(3S,3aS,8bS)-7-bromo-3,6,8b-trimethyl-1,2,3,8b-tetrahydro-3aH-benzo[b]cyclopenta[d]furan-3a-yl]methanol](http://img.cochemist.com/ccimg/6800/6790-64-3_b.png)
![7-bromo-3,3a,6,8b-tetramethyl-2,3,3a,8b-tetrahydro-1H-benzo[b]cyclopenta[d]furan](http://img.cochemist.com/ccimg/6800/6790-63-2.png)
![7-bromo-3,3a,6,8b-tetramethyl-2,3,3a,8b-tetrahydro-1H-benzo[b]cyclopenta[d]furan](http://img.cochemist.com/ccimg/6800/6790-63-2_b.png)






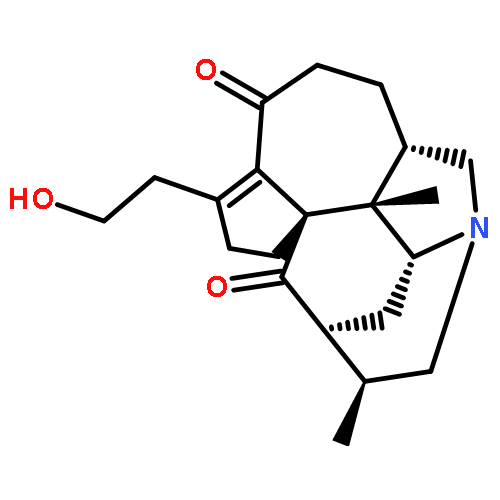
![Methyl (1R,2S,3R,5R,6S,10S,16R,17R)-2,6-dimethyl-20-oxo-8-azahexacyclo[11.5.1.11,5.02,10.03,8.016,19]icos-13(19)-ene-17-carboxylate](http://img.cochemist.com/ccimg/881400/881388-87-0.png)
![Methyl (1R,2S,3R,5R,6S,10S,16R,17R)-2,6-dimethyl-20-oxo-8-azahexacyclo[11.5.1.11,5.02,10.03,8.016,19]icos-13(19)-ene-17-carboxylate](http://img.cochemist.com/ccimg/881400/881388-87-0_b.png)
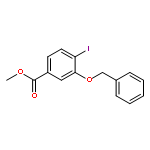
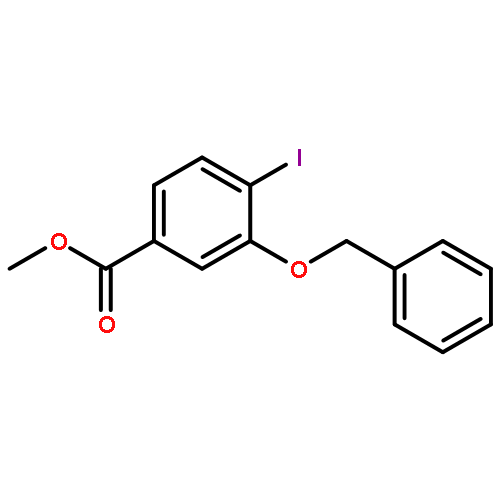
![Methyl (1S,2S,3R,5R,6S,10S)-2,6-dimethyl-21-oxo-14-oxa-8-azahexacyclo[11.6.1.11,5.02,10.03,8.017,20]henicosa-13(20),17-diene-18-carboxylate](http://img.cochemist.com/ccimg/874300/874201-05-5.png)
![Methyl (1S,2S,3R,5R,6S,10S)-2,6-dimethyl-21-oxo-14-oxa-8-azahexacyclo[11.6.1.11,5.02,10.03,8.017,20]henicosa-13(20),17-diene-18-carboxylate](http://img.cochemist.com/ccimg/874300/874201-05-5_b.png)
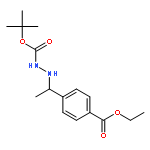
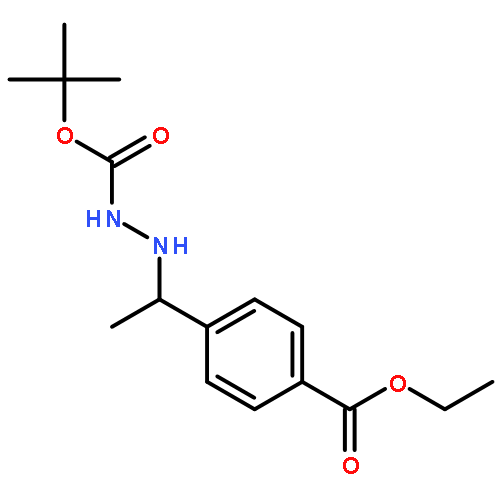
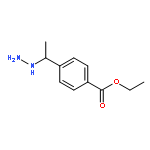
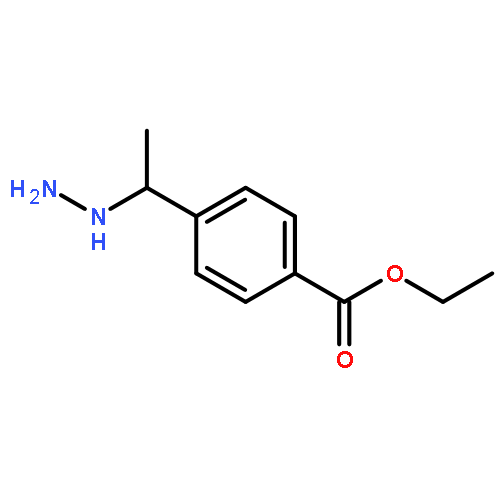
![Hydrazinecarboxylic acid, [1-[4-(ethoxycarbonyl)phenyl]ethylidene]-,
1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/870900/870822-87-0.png)
![Hydrazinecarboxylic acid, [1-[4-(ethoxycarbonyl)phenyl]ethylidene]-,
1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/870900/870822-87-0_b.png)
![Methyl (1R,2S,3R,5R,6S,10S)-2,6-dimethyl-20-oxo-8-azahexacyclo[11.5.1.11,5.02,10.03,8.016,19]icosa-13(19),16-diene-17-carboxylate](http://img.cochemist.com/ccimg/857700/857672-34-5.png)
![Methyl (1R,2S,3R,5R,6S,10S)-2,6-dimethyl-20-oxo-8-azahexacyclo[11.5.1.11,5.02,10.03,8.016,19]icosa-13(19),16-diene-17-carboxylate](http://img.cochemist.com/ccimg/857700/857672-34-5_b.png)




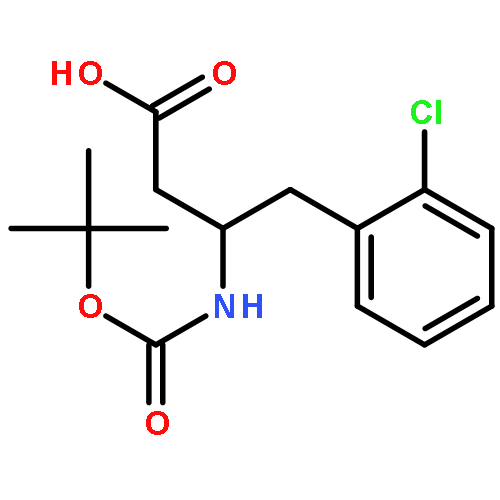


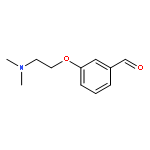
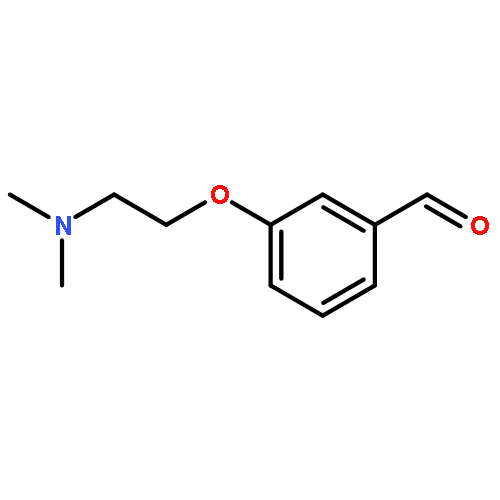
![Benzaldehyde, 3-[(1-methyl-4-piperidinyl)oxy]-](http://img.cochemist.com/ccimg/186200/186191-30-0.png)
![Benzaldehyde, 3-[(1-methyl-4-piperidinyl)oxy]-](http://img.cochemist.com/ccimg/186200/186191-30-0_b.png)


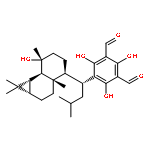
![1,3-Benzenedicarboxaldehyde,5-[(1S)-1-[(1R)-2-[(1R,3R)-2,2-dimethyl-3-(3-oxobutyl)cyclopropyl]-1-methyl-3-oxocyclopentyl]-3-methylbutyl]-2,4,6-trihydroxy-](http://img.cochemist.com/ccimg/172700/172617-99-1.png)
![1,3-Benzenedicarboxaldehyde,5-[(1S)-1-[(1R)-2-[(1R,3R)-2,2-dimethyl-3-(3-oxobutyl)cyclopropyl]-1-methyl-3-oxocyclopentyl]-3-methylbutyl]-2,4,6-trihydroxy-](http://img.cochemist.com/ccimg/172700/172617-99-1_b.png)














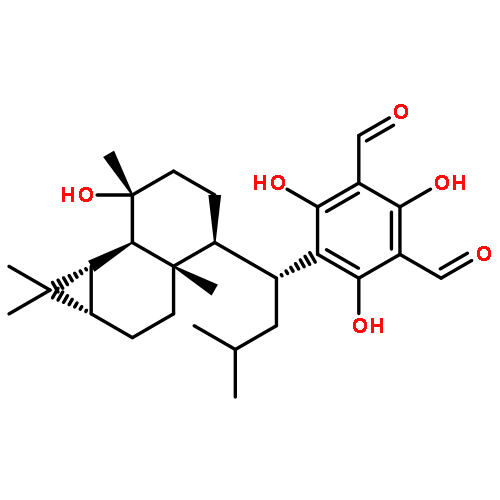
![1,3-Benzenedicarboxaldehyde,2,4,6-trihydroxy-5-[(1R)-3-methyl-1-[(1R,3aR,4R,7R)-1,2,3,3a,4,5,6,7-octahydro-7-(1-hydroxy-1-methylethyl)-1,4-dimethyl-1-azulenyl]butyl]-](http://img.cochemist.com/ccimg/142700/142647-71-0.png)
![1,3-Benzenedicarboxaldehyde,2,4,6-trihydroxy-5-[(1R)-3-methyl-1-[(1R,3aR,4R,7R)-1,2,3,3a,4,5,6,7-octahydro-7-(1-hydroxy-1-methylethyl)-1,4-dimethyl-1-azulenyl]butyl]-](http://img.cochemist.com/ccimg/142700/142647-71-0_b.png)
![2,4,6-trihydroxy-5-{1-[6-(2-hydroxypropan-2-yl)-4,8a-dimethyl-1,2,3,4,6,7,8,8a-octahydronaphthalen-1-yl]-3-methylbutyl}benzene-1,3-dicarbaldehyde](http://img.cochemist.com/ccimg/142700/142628-54-4.png)
![2,4,6-trihydroxy-5-{1-[6-(2-hydroxypropan-2-yl)-4,8a-dimethyl-1,2,3,4,6,7,8,8a-octahydronaphthalen-1-yl]-3-methylbutyl}benzene-1,3-dicarbaldehyde](http://img.cochemist.com/ccimg/142700/142628-54-4_b.png)
![1,3-Benzenedicarboxaldehyde,5-[(1R)-1-[(1aR,4aR,7S,7aR,7bR)-decahydro-1,1,7-trimethyl-4-methylene-1H-cycloprop[e]azulen-7-yl]-3-methylbutyl]-2,4,6-trihydroxy-](http://img.cochemist.com/ccimg/142700/142628-53-3.png)
![1,3-Benzenedicarboxaldehyde,5-[(1R)-1-[(1aR,4aR,7S,7aR,7bR)-decahydro-1,1,7-trimethyl-4-methylene-1H-cycloprop[e]azulen-7-yl]-3-methylbutyl]-2,4,6-trihydroxy-](http://img.cochemist.com/ccimg/142700/142628-53-3_b.png)










![Cyclopenta[b]pyrrole-1,2(2H)-dicarboxylic acid, hexahydro-, 1-(1,1-dimethylethyl) ester, (2S,3aS,6aS)-](/data/chemimg/1071600/124002-32-0.png)
![Cyclopenta[b]pyrrole-1,2(2H)-dicarboxylic acid, hexahydro-, 1-(1,1-dimethylethyl) ester, (2S,3aS,6aS)-](/data/chemimg/1071600/124002-32-0_b.png)






![Benzaldehyde, 4-[(6-hydroxyhexyl)oxy]-](/data/chemimg/996000/96735-91-0.png)
![Benzaldehyde, 4-[(6-hydroxyhexyl)oxy]-](/data/chemimg/996000/96735-91-0_b.png)
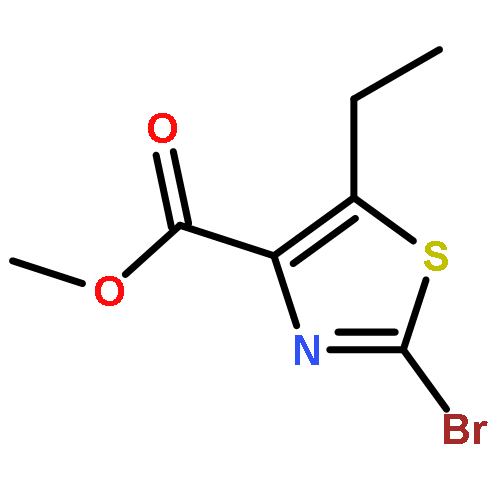
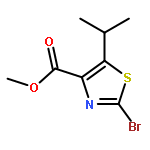
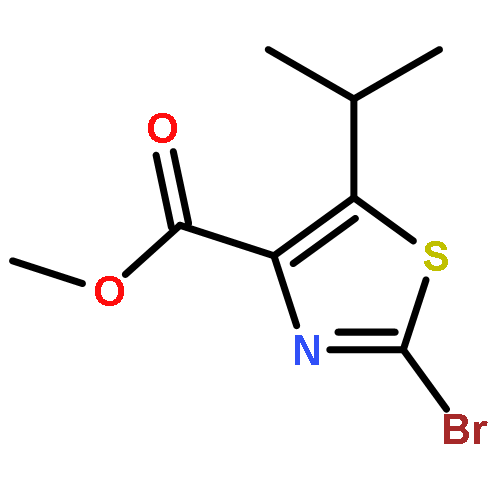
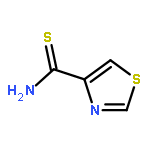
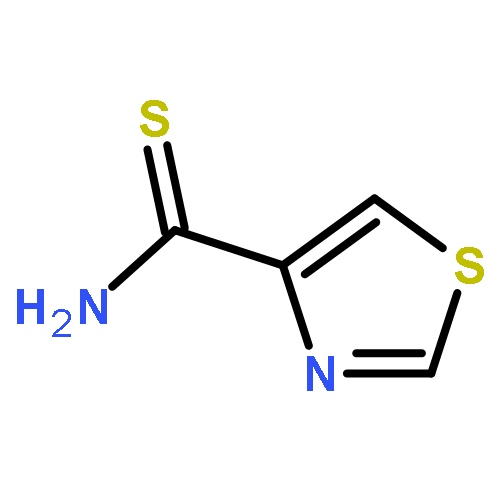
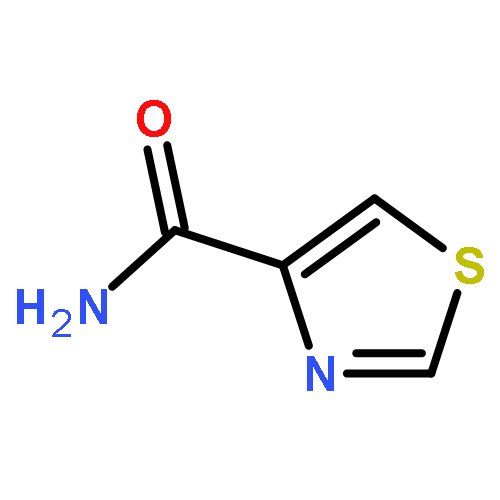
![[2,4'-Bithiazole]-4-carboxylic acid, 2'-methyl-](http://img.cochemist.com/ccimg/91600/91520-83-1.png)
![[2,4'-Bithiazole]-4-carboxylic acid, 2'-methyl-](http://img.cochemist.com/ccimg/91600/91520-83-1_b.png)














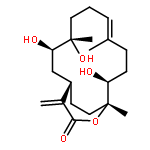
![(1R,4S,6R,13S,14R)-13-hydroxy-4,9,13-trimethyl-17-methylidene-5,15-dioxatricyclo[12.3.1.0~4,6~]octadec-9-en-16-one](http://img.cochemist.com/ccimg/65700/65669-72-9.png)
![(1R,4S,6R,13S,14R)-13-hydroxy-4,9,13-trimethyl-17-methylidene-5,15-dioxatricyclo[12.3.1.0~4,6~]octadec-9-en-16-one](http://img.cochemist.com/ccimg/65700/65669-72-9_b.png)
![(2E)-1-{3-[(2E)-3,7-dimethylocta-2,6-dien-1-yl]-2,4-dihydroxyphenyl}-3-(4-hydroxyphenyl)prop-2-en-1-one](http://img.cochemist.com/ccimg/63000/62949-76-2.png)
![(2E)-1-{3-[(2E)-3,7-dimethylocta-2,6-dien-1-yl]-2,4-dihydroxyphenyl}-3-(4-hydroxyphenyl)prop-2-en-1-one](http://img.cochemist.com/ccimg/63000/62949-76-2_b.png)
![6-NITRO-3H-THIENO[2,3-D]PYRIMIDIN-4-ONE](http://img.cochemist.com/ccimg/56900/56844-41-8.png)
![6-NITRO-3H-THIENO[2,3-D]PYRIMIDIN-4-ONE](http://img.cochemist.com/ccimg/56900/56844-41-8_b.png)
![Thieno[2,3-d]pyrimidine, 4-chloro-6-nitro-](http://img.cochemist.com/ccimg/56900/56844-13-4.png)
![Thieno[2,3-d]pyrimidine, 4-chloro-6-nitro-](http://img.cochemist.com/ccimg/56900/56844-13-4_b.png)
![(E)-1-[2-HYDROXY-4-METHOXY-3-(3-METHYLBUT-2-ENYL)PHENYL]-3-(4-HYDROXYPHENYL)PROP-2-EN-1-ONE](http://img.cochemist.com/ccimg/56000/55912-03-3.png)
![(E)-1-[2-HYDROXY-4-METHOXY-3-(3-METHYLBUT-2-ENYL)PHENYL]-3-(4-HYDROXYPHENYL)PROP-2-EN-1-ONE](http://img.cochemist.com/ccimg/56000/55912-03-3_b.png)




![3H-Cyclotridec[d]oxireno[f]isoindole-8,11,12(13H)-trione,4,7,14,14a,15,15a,16a,16b-octahydro-7-hydroxy-14-(1H-indol-3-ylmethyl)-4,6,15,15a-tetramethyl-,(1E,4S,5E,7R,9E,11aR,14S,14aR,15S,15aR,16aS,16bR)-](http://img.cochemist.com/ccimg/50400/50335-03-0.png)
![3H-Cyclotridec[d]oxireno[f]isoindole-8,11,12(13H)-trione,4,7,14,14a,15,15a,16a,16b-octahydro-7-hydroxy-14-(1H-indol-3-ylmethyl)-4,6,15,15a-tetramethyl-,(1E,4S,5E,7R,9E,11aR,14S,14aR,15S,15aR,16aS,16bR)-](http://img.cochemist.com/ccimg/50400/50335-03-0_b.png)




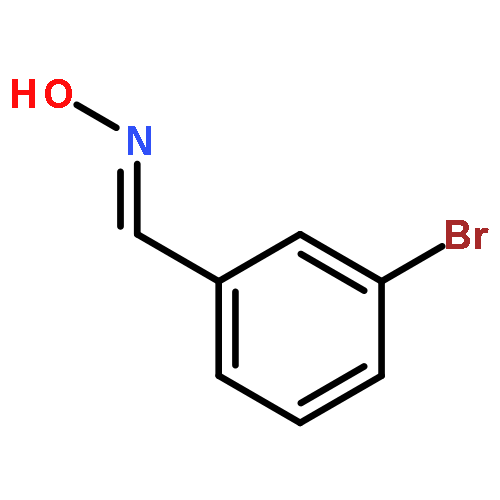
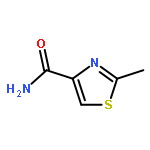
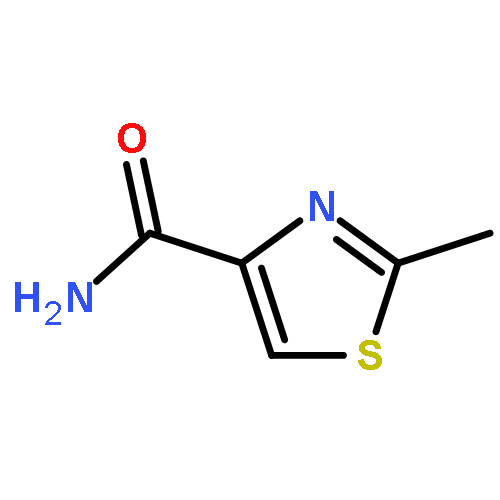
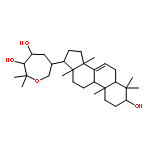


![(9R)-8,8-dimethyl-2-oxo-9,10-dihydro-2H,8H-pyrano[2,3-f]chromen-9-yl (2Z)-2-methylbut-2-enoate](http://img.cochemist.com/ccimg/19500/19427-82-8.png)
![(9R)-8,8-dimethyl-2-oxo-9,10-dihydro-2H,8H-pyrano[2,3-f]chromen-9-yl (2Z)-2-methylbut-2-enoate](http://img.cochemist.com/ccimg/19500/19427-82-8_b.png)

![[1,1':2',1''-Terphenyl]-4'-amine](http://img.cochemist.com/ccimg/10600/10569-67-2.png)
![[1,1':2',1''-Terphenyl]-4'-amine](http://img.cochemist.com/ccimg/10600/10569-67-2_b.png)


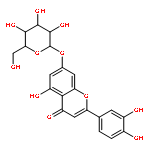

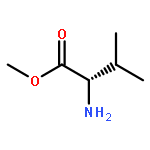
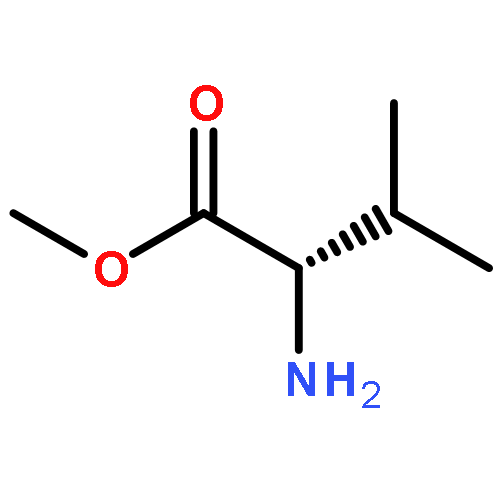


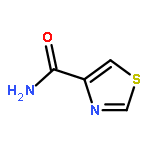



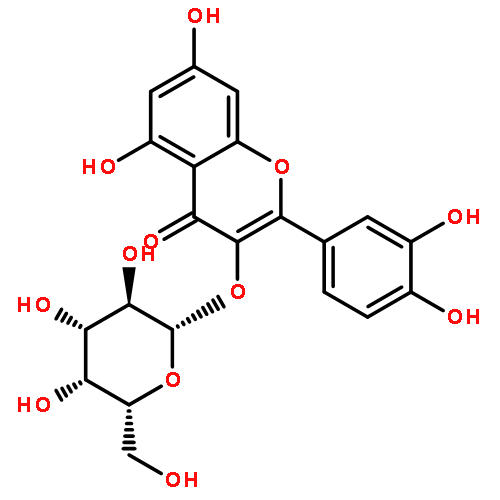
![2-(3,4-dihydroxyphenyl)-5,7-dihydroxy-3-[(2R,3S,4R,5S,6S)-3,4,5-trihydroxy-6-(hydroxymethyl)tetrahydropyran-2-yl]oxy-chromen-4-one](http://img.cochemist.com/ccimg/500/482-35-9.png)
![2-(3,4-dihydroxyphenyl)-5,7-dihydroxy-3-[(2R,3S,4R,5S,6S)-3,4,5-trihydroxy-6-(hydroxymethyl)tetrahydropyran-2-yl]oxy-chromen-4-one](http://img.cochemist.com/ccimg/500/482-35-9_b.png)