Co-reporter:Justin M. Hoffman, Allen G. Oliver, and Seth N. Brown
Journal of the American Chemical Society March 29, 2017 Volume 139(Issue 12) pp:4521-4521
Publication Date(Web):March 3, 2017
DOI:10.1021/jacs.7b00985
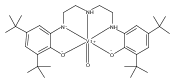
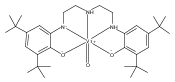
The rhenium(V) oxo complex oxo(triphenylphosphine) (bis(3,5-di-tert-butyl-2-phenoxo)amido)rhenium(V), (ONOCat)ReO(PPh3), reacts with molecular oxygen to give triphenylphosphine oxide and the dimeric rhenium(VII) complex fac,anti-(ONOCat)Re(O)(μ-O)2Re(O)(ONOCat). The ONO ligand adopts an unusual fac geometry, presumably to maximize π donation to rhenium; strong π donation is substantiated by the intraligand bond distances (metrical oxidation state = −2.24(9)). Addition of the N-heterocyclic carbene ligand IMes to fac,anti-(ONOCat)Re(O)(μ-O)2Re(O)(ONOCat) cleaves the dimer into monomeric C1-symmetric fac-(ONOCat)ReO2(IMes). The monorhenium(VII) complex is deoxygenated by PMe2Ph to give the rhenium(V) compound (ONOCat)ReO(IMes), which can be independently prepared by ligand substitution of (ONOCat)ReO(PPh3). The degree of stereochemical rigidity exhibited by the dioxo compound, as established by dynamic NMR spectroscopy, excludes the intermediacy of mer-(ONOQ)ReVO2(IMes) in this oxygen atom transfer reaction. Thus, oxygen atom transfer takes place preferentially by direct reduction of the oxorhenium(VII) moiety (classical oxygen atom transfer) rather than through initial internal electron transfer and ligand-centered reduction of an oxorhenium(V)-iminoquinone.
Co-reporter:Daniel D. Swanson;Kyle M. Conner;Seth N. Brown
Dalton Transactions 2017 vol. 46(Issue 28) pp:9049-9057
Publication Date(Web):2017/07/18
DOI:10.1039/C7DT01945A
1,1′-Bis(4-(2-hydroxy-3,5-di-tert-butylphenyl)aminophenyl)ferrocene (pFlipH4) is prepared in three steps from commercially available 1,1′-ferrocenediboronic acid or its pinacol ester. The suitability of the ligand to bind as a tetradentate ligand in a cis, planar fashion has been confirmed by formation of a square planar palladium bis-iminosemiquinone (pFlip)Pd. The linker unit appears to be structurally similar to 1,1′-ferrocenediyl, but the electronic interaction of the ferrocene with the aminophenols is minimal.
Co-reporter:Travis Marshall-Roth and Seth N. Brown
Dalton Transactions 2015 vol. 44(Issue 2) pp:677-685
Publication Date(Web):04 Nov 2014
DOI:10.1039/C4DT02936D
A tris(aminophenol), tris(2-(3′,5′-di-tert-butyl-2′-hydroxyphenyl)amino-4-methylphenyl)amine, MeClampH6, is prepared in three steps from tri-p-tolylamine. The ligand reacts with dioxomolybdenum(VI) bis(acetylacetonate) to form an oxo-free heptadentate complex, (MeClamp)Mo, with a capped octahedral geometry. The molybdenum is formally in the +6 oxidation state, with significant π donation of the amidophenolates, as judged by intraligand bond distances. Two ligand-based oxidations and one metal-centered reduction are observed by cyclic voltammetry. Analysis of the optical spectrum of the compound gives an estimate of the energetic stabilization of the ligand π orbitals by bonding to the molybdenum of approximately 0.9 eV, corresponding to about 40 kcal mol−1 per π bond.
Co-reporter:Jacqueline Cipressi and Seth N. Brown
Chemical Communications 2014 vol. 50(Issue 59) pp:7956-7959
Publication Date(Web):05 Jun 2014
DOI:10.1039/C4CC03404J
Ru(ONO)2 and Os(ONO)2 distort from octahedral towards trigonal prismatic geometry in order to relieve π antibonding due to donation from the second-highest ligand orbital to the metal. Increasing oxidation of the ONO ligand suppresses distortion by increasing σ* interactions in the trigonal prism.
Co-reporter:Leila G. Ranis, Kalpani Werellapatha, Nicholas J. Pietrini, Bruce A. Bunker, and Seth N. Brown
Inorganic Chemistry 2014 Volume 53(Issue 19) pp:10203-10216
Publication Date(Web):September 23, 2014
DOI:10.1021/ic501222n
Group 6 complexes M(ONO)2 (M = Cr, Mo, W; ONO = bis(2-oxy-3,5-di-tert-butylphenyl)amide) are prepared by the reaction of divalent metal halide precursors with Pb(ONOQ)2. Analogous complexes containing the 2,4,6,8-tetra-tert-butyl-1,9-dioxophenoxazinate ligand (DOPO) are prepared by protonolysis of chromocene with H(DOPOQ) or by reaction of Pb(DOPOQ)2 with M2Br4(CO)8 (M = Mo, W). The molybdenum and tungsten complexes are symmetrical, octahedral compounds for which spectroscopic data are consistent with M(VI) complexes with fully reduced [LCat]3– ligands. Quantitative analysis of the intraligand bond lengths, by comparison with literature standards, allows calculation of metrical oxidation states (MOS) for the ONO ligands. The MOS values of the tungsten and molybdenum complexes indicate that π donation from the ligand is weak and that differences between the ONO and DOPO ligands are small. In both the solid state and in solution, Cr(DOPO)2 is paramagnetic with localized quinone and semiquinone ligands bound to Cr(III). The geometry and electronic structure of Cr(ONO)2 differ in the solid state and in solution, as determined by crystallography, magnetic measurements, and Cr K-edge X-ray absorption spectroscopy. In solution, the structure resembles that of the DOPO analogue. In contrast, solid Cr(ONO)2 is a singlet, and X-ray absorption near-edge spectroscopy indicates that the chromium is significantly more oxidized in the solid state than in solution. An electronic description compounds to that of the tungsten and molybdenum analogues, but with considerably more charge transfer from the ligand to chromium via π donation, is in agreement with the experimental observations.
Co-reporter:Sukesh Shekar and Seth N. Brown
Dalton Transactions 2014 vol. 43(Issue 9) pp:3601-3611
Publication Date(Web):07 Jan 2014
DOI:10.1039/C3DT53496K
The dioxomolybdenum(VI) complex (tBuClipH2)MoO2 (tBuClipH4 = 4,4′-di-tert-butyl-N,N′-bis(3,5-di-tert-butyl-2-hydroxyphenyl)-2,2′-diaminobiphenyl) reacts with 3,5-di-tert-butylcatechol to form oxo-free (tBuClip)Mo(3,5-tBu2Cat). The bis(amidophenoxide)-monocatecholate complex is monomeric and exhibits a cis-β geometry in the solid state. Variable-temperature NMR data are consistent with two fluxional processes, one that interconverts several geometric isomers at low temperature, and a second that interchanges the ends of the tBuClip ligand at ambient temperatures. The high-temperature fluxional process can be explained by a single Bailar trigonal twist coupled with atropisomerization of the chiral diaminobiaryl backbone. Addition of excess catechol to (tBuClipH2)MoO2 results in formation of a dimolybdenum mono-oxo complex (tBuClip)Mo(μ-3,5-tBu2Cat)2Mo(O)(3,5-tBu2Cat). This complex, which contains a seven-coordinate bis(amidophenoxide)molybdenum center and a six-coordinate oxomolybdenum center, represents a structural hybrid between dimeric oxomolybdenumbis(catecholate) and molybdenum tris(catecholate) complexes. Both mono- and dimolybdenum complexes are best formulated as containing Mo(VI), but there is structural evidence for significant π donation from the amidophenolates. (tBuClip)Mo(3,5-tBu2Cat) binds pyridine to form a mixture of isomeric seven-coordinate adducts. The Lewis acidity of the mixed amidophenoxide-catecholate appears to be lower than its tris(catecholate) or oxobis(amidophenoxide) analogues, which manifests itself principally in relatively slow binding of pyridine to the six-coordinate complex (k = 8 × 104 L mol−1 s−1 at 0 °C) rather than in the rate of dissociation of pyridine from the seven-coordinate adduct.
Co-reporter:Sukesh Shekar and Seth N. Brown
The Journal of Organic Chemistry 2014 Volume 79(Issue 24) pp:12047-12055
Publication Date(Web):October 7, 2014
DOI:10.1021/jo501888r
Chlorosilanes R(X)(Y)SiCl (R = Me, Ph; X, Y = Me, Ph, Cl) have been reported to react with Pb(ONOQ)2 (ONOQ = 3,5-di-tert-butyl-1,2-quinone-(3,5-di-tert-butyl-2-oxy-1-phenyl)imine) to give five-coordinate (X)(Y)Si(ON[R]O), in which the R group has migrated from silicon to nitrogen. This migration is intramolecular, as confirmed by the lack of crossover between (CH3)3SiCl and (CD3)3SiCl in their reaction with Pb(ONOQ)2. Reaction of PhSiMeCl2 takes place with high kinetic stereoselectivity to produce isomer Ph(Cl)Si(ON[Me]O) in which the phenyl is axial in the trigonal bipyramid, which subsequently isomerizes to the thermodynamic isomer with axial chlorine. This indicates that migration takes place preferentially from the stereoisomer of the octahedral intermediate, κ3-Ph(CH3)(Cl)Si(ONOQ), in which the phenyl and methyl groups are mutually trans, indicating that the observed complete selectivity for methyl over phenyl migration is due to intrinsic differences in migratory aptitude. DFT calculations suggest that migration takes place from this isomer not because it undergoes migration faster than other possible stereoisomers, but because it is formed most rapidly, and migration occurs faster than isomerization.
Co-reporter:Daniel D. Wright and Seth N. Brown
Inorganic Chemistry 2013 Volume 52(Issue 14) pp:7831-7833
Publication Date(Web):July 1, 2013
DOI:10.1021/ic4010592
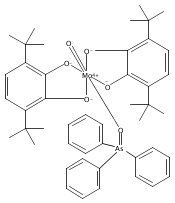
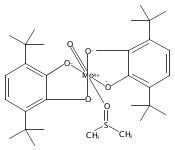
Oxo(triphenylphosphine)[bis(3,5-di-tert-butyl-2-phenoxo)amido]rhenium(V) [(ONOCat)ReO(PPh3)] is prepared by the reaction of iododioxobis(triphenylphosphine)rhenium(V) [ReO2(PPh3)2I] with lead bis(3,5-di-tert-butyl-1,2-quinone-1-(2-oxy-3,5-di-tert-butylphenyl)imine) [Pb(ONOQ)2]. In this reaction, the ONO ligand undergoes a two-electron reduction, with concomitant oxidation of PPh3 to OPPh3 and transformation of the dioxorhenium(V) fragment into a monooxorhenium(V) fragment, constituting a net nonclassical oxygen atom transfer. (ONOCat)ReO(PPh3) adopts a square pyramidal geometry with an apical oxo group [dReO = 1.6873(14) Å] and a highly folded ONO ligand [O–Re–O = 129.55(6)°]. The fully reduced, trianionic oxidation state of the ONO ligand is confirmed by spectroscopic and metrical data.
Co-reporter:Amanda H. Randolph ; Nicholas J. Seewald ; Karl Rickert
Inorganic Chemistry 2013 Volume 52(Issue 21) pp:12587-12598
Publication Date(Web):October 22, 2013
DOI:10.1021/ic401736f
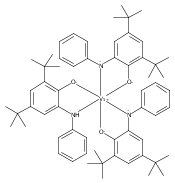
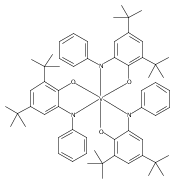
In the solid state, tris(3,5-di-tert-butylcatecholato)molybdenum(VI) forms a dimer with seven-coordinate molybdenum and bridging catecholates. NMR spectroscopy indicates that the dimeric structure is retained in solution. The molybdenum center has a high affinity for Lewis bases such as pyridine or pyridine-N-oxide, forming seven-coordinate monomers with a capped octahedral geometry, as illustrated by the solid-state structure of (3,5-tBu2Cat)3Mo(py). Structural data indicate that the complexes are best considered as Mo(VI) with substantial π donation from the nonbridging catecholates to molybdenum. Both the dimeric and the monomeric tris(catecholates) react rapidly with water to form free catechol and oxomolybdenum bis(catecholate) complexes. Monooxomolybdenum complexes are also obtained, more slowly, on reaction with dioxygen, with organic products consisting mostly of 3,5-di-tert-butyl-1,2-benzoquinone with minor amounts of the extradiol oxidation product 4,6-di-tert-butyl-1-oxacyclohepta-4,6-diene-2,3-dione. The pyridine-N-oxide complex reacts on heating (with excess pyO) to form initially (3,5-tBu2Cat)2MoO(Opy) and ultimately MoO3(Opy), with quinone and free pyridine as the only organic products. The decay of (3,5-tBu2Cat)3Mo(Opy) shows an accelerated, autocatalytic profile because the oxidation of its product, (3,5-tBu2Cat)2MoO(Opy), produces an oxo-rich, catecholate-poor intermediate which rapidly conproportionates with (3,5-tBu2Cat)3Mo(Opy), providing an additional pathway for its conversion to the mono-oxo product. The tris(catecholate) fragment Mo(3,5-tBu2Cat)3 deoxygenates Opy in this nonclassical oxygen atom transfer reaction slightly less rapidly than does its oxidized product, MoO(3,5-tBu2Cat)2.
Co-reporter:Sukesh Shekar and Seth N. Brown
Organometallics 2013 Volume 32(Issue 2) pp:556-564
Publication Date(Web):January 14, 2013
DOI:10.1021/om301028c
Silylation of the oxidized ligand in lead(II) bis(3,5-tert-butyl-1,2-quinone-(3,5-di-tert-butyl-2-hydroxy-1-phenyl)imine), Pb(ONOQ)2, with chlorosilanes RSiX2Cl (R = Me, Ph; X = Me, Ph, Cl) results in the tetracyclic, pentacoordinate silicon compounds (ON[R]O)SiX2 with a reduced, dianionic amine-bisphenolate ligand in which the methyl or phenyl group has migrated to nitrogen. Methyl migration is observed exclusively over phenyl migration, though phenyl migration is observed if no alkyl groups are present on silicon. Chlorotriphenylstannane reacts similarly, with migration of the phenyl group from tin to nitrogen. The five-coordinate products adopt a trigonal-bipyramidal geometry with long bonds to the apical amine donors (d(Si–N) = 2.3–2.9 Å). Chlorine occupies the other axial position in strong preference to methyl or phenyl, while methyl has a modest preference for the axial position in comparison to phenyl (8.7:1).
Co-reporter:Travis Marshall-Roth, Sean C. Liebscher, Karl Rickert, Nicholas J. Seewald, Allen G. Oliver and Seth N. Brown
Chemical Communications 2012 vol. 48(Issue 63) pp:7826-7828
Publication Date(Web):19 Jun 2012
DOI:10.1039/C2CC33523A
Mechanistic studies indicate that the oxomolybdenum(VI) bis(3,5-di-tert-butylcatecholate) fragment deoxygenates pyridine-N-oxides in a reaction where the oxygen is delivered to molybdenum but the electrons for substrate reduction are drawn from the bound catecholate ligands, forming 3,5-di-tert-butyl-1,2-benzoquinone.
Co-reporter:Seth N. Brown
Inorganic Chemistry 2012 Volume 51(Issue 3) pp:1251-1260
Publication Date(Web):January 19, 2012
DOI:10.1021/ic202764j
Catecholates and 2-amidophenoxides are prototypical “noninnocent” ligands which can form metal complexes where the ligands are best described as being in the monoanionic (imino)semiquinone or neutral (imino)quinone oxidation state instead of their closed-shell dianionic form. Through a comprehensive analysis of structural data available for compounds with these ligands in unambiguous oxidation states (109 amidophenolates, 259 catecholates), the well-known structural changes in the ligands with oxidation state can be quantified. Using these correlations, an empirical “metrical oxidation state” (MOS) which gives a continuous measure of the apparent oxidation state of the ligand can be determined based on least-squares fitting of its C–C, C–O, and C–N bond lengths to this single parameter (a simple procedure for doing so is provided via a spreadsheet in the Supporting Information). High-valent d0 metal complexes, particularly those of vanadium(V) and molybdenum(VI), have ligands with unexpectedly positive, and generally nonintegral, MOS values. The structural effects in these complexes are attributed not to electron transfer, but rather to amidophenoxide- or catecholate-to-metal π bonding, an interpretation supported by the systematic variation of the MOS values as a function of the degree of competition with the other π-donating groups in the structures.
Co-reporter:Jason A. Kopec, Sukesh Shekar, and Seth N. Brown
Inorganic Chemistry 2012 Volume 51(Issue 3) pp:1239-1250
Publication Date(Web):January 19, 2012
DOI:10.1021/ic201736h
The 2,2′-biphenyl-bridged bis(2-aminophenol) ligand 4,4′-di-tert-butyl-N,N′-bis(3,5-di-tert-butyl-2-hydroxyphenyl)-2,2′-diaminobiphenyl (tBuClipH4) reacts with MoO2(acac)2 to form (tBuClipH2)MoO2, where the diarylamines remain protonated and bind trans to the terminal oxo groups. This complex readily loses water on treatment with pyridine or 3,5-lutidine to form mono-oxo complexes (tBuClip)MoO(L), which exhibit predominantly a cis-β geometry with an aryloxide trans to the oxo group. Exchange of the pyridine ligands is rapid and takes place by a dissociative mechanism, which occurs with retention of stereochemistry at molybdenum. Oxo-free alkoxide complexes (tBuClip)Mo(OR)2 are formed from (tBuClipH2)MoO2 and ROH. Treatment of NMo(OtBu)3 with tBuClipH4 results in complete deprotonation of the bis(aminophenol) and formation of a dimolybdenum complex (tBuClip)Mo(μ-N)(μ-NH2)Mo(tBuClip) containing both a bridging nitride (Mo–N = 1.848 Å, Mo–N–Mo = 109.49°) and a bridging amide group. The strong π bonding of this bis(amidophenoxide) ligand allows the molybdenum center to interconvert readily among species forming three, two, one, or zero π bonds from multiply bonded ligands.
Co-reporter:Seth N. Brown
The Journal of Physical Chemistry Letters 2012 Volume 3(Issue 2) pp:278-279
Publication Date(Web):January 19, 2012
DOI:10.1021/jz201672n
Co-reporter:Natcharee Kongprakaiwoot, Mauricio Quiroz-Guzman, Allen G. Oliver and Seth N. Brown
Chemical Science 2011 vol. 2(Issue 2) pp:331-336
Publication Date(Web):27 Oct 2010
DOI:10.1039/C0SC00468E
3,3′,5,5′-Tetrasubstituted-2,2′-biphenolate complexes of titanium(IV) with bis(diketonate) (Bob), bis(hydroxamate) (Hox) and mixed diketonate–hydroxamate (Hob) ligands have been prepared from the corresponding diisopropoxide complexes. Four of the twelve compounds have been characterized crystallographically, and in the solid state all show the (Δ,R)/(Λ,S) relative stereochemistry at titanium and the biaryloxide, respectively, as previously observed in (acac)2Ti(1,1′-bi-2-naphtholate) complexes. In solution the compounds epimerize by atropisomerization of the biphenolate moiety with ΔG‡ ≈ 14 kcal mol−1. The bis(diketonate) complexes show high diastereoselectivity except for the most electron-poor tetranitrobiphenolate. In contrast, the bis(hydroxamate) shows low to moderate selectivity which correlates with the steric but not electronic properties of the biphenolates (Br < CH3 < NO2 < tBu). The mixed diketonate–hydroxamate complexes show intermediate behaviour. These observations are rationalized on the basis of MO arguments regarding ligand-metal π bonding. Symmetrical chelates such as diketonates foster mixing of two dπ orbitals and create a dissymmetric electronic environment. This mixing does not take place with unsymmetrical ligands such as hydroxamates, which therefore do not create an environment where electronic effects contribute significantly to binding stereoselectivity.
Co-reporter:Mauricio Quiroz-Guzman, Allen G. Oliver, Andrew J. Loza and Seth N. Brown
Dalton Transactions 2011 vol. 40(Issue 43) pp:11458-11468
Publication Date(Web):21 Sep 2011
DOI:10.1039/C1DT11228G
A diarylamino-substituted N-methyl tetrahydrosalen (salan) ligand, An2NLH2, is prepared in four steps and overall 53% yield from 5-bromosalicylaldehyde, with the key step a palladium-catalysed Hartwig–Buchwald amination of the tert-butyldimethylsilyl-protected 5-bromo-N-methylsalan ligand. Reaction of An2NLH2 or its bromo analogue with Ti(OiPr)4 or TiF4 results in metalation of the ligand. The isopropoxide groups are readily exchanged with α- or β-hydroxyacids to form chelated complexes. X-ray crystallography and NMR spectroscopy indicate that the salan ligands are quite flexible, with An2NLTiF2, for example, showing four stereoisomers in its 19F NMR spectrum. The major stereoisomer of (salan)Ti(X)(Y) depends principally on the trans influence of the X and Y groups. Complexes of An2NL show two reversible, closely spaced redox couples at approximately + 0.1 V vs.ferrocene/ferrocenium, and a second set of two closely spaced redox couples at ∼ + 0.8 V vs. Fc/Fc+.
Co-reporter:Davide Lionetti ; Andrew J. Medvecz ; Vesela Ugrinova ; Mauricio Quiroz-Guzman ; Bruce C. Noll ;Seth N. Brown
Inorganic Chemistry 2010 Volume 49(Issue 10) pp:4687-4697
Publication Date(Web):April 16, 2010
DOI:10.1021/ic100347h
New sterically encumbered tripodal aminetris(aryloxide) ligands N(CH2C6H2-3-tBu-5-X-2-OH)3 (tBu,XLH3) with relatively electron-rich phenols are prepared by Mannich condensation (X = OCH3) or by a reductive amination/Hartwig−Buchwald amination sequence on the benzyl-protected bromosalicylaldehyde (X = N[C6H4-p-OCH3]2), followed by debenzylation using Pearlman’s catalyst (Pd(OH)2/C). The analogous dianisylamino-substituted compound lacking the tert-butyl group ortho to the phenol (H,An2NLH3) is also readily prepared. The ligands are metalated by titanium(IV) tert-butoxide to form the five-coordinate alkoxides LTi(OtBu). Treatment of the tert-butoxides with aqueous HCl yields the five-coordinate chlorides LTiCl, and with acetylacetone gives the six-coordinate diketonates LTi(acac). The diketonate complexes tBu,XLTi(acac) show reversible ligand-based oxidations with first oxidation potentials of +0.57, +0.33, and −0.09 V (vs ferrocene/ferrocenium) for X = tBu, MeO, and An2N, respectively. Both dianisylamine-substituted complexes R,An2NLTi(acac) (R = tBu, H) show similar electrochemistry, with three one-electron oxidations closely spaced at ∼0 V and three oxidations due to removal of a second electron from each diarylaminoaryloxide arm at ∼ + 0.75 V. The new electron-rich tripodal ligands therefore have the capacity to release multiple electrons at unusually low potentials for aryloxide groups.
Co-reporter:Natcharee Kongprakaiwoot, Jack B. Armstrong, Bruce C. Noll and Seth N. Brown
Dalton Transactions 2010 vol. 39(Issue 42) pp:10105-10115
Publication Date(Web):07 Sep 2010
DOI:10.1039/C0DT00828A
The 2,2′-bis(methylene)biphenyl bridged bis(diketonate) (Tol2Bob)TiIV fragment is resolved by reaction of the isopropoxide complex with (R)-1,1′-bi-2-naphthol, which selectively forms (S,Δ)-(Tol2Bob)Ti(R-BINOL). The binaphtholate ligand can be cleaved from titanium by careful treatment with trifluoromethanesulfonic acid to give (S,Δ)-(Tol2Bob)Ti(OTf)2, which has a half-life toward racemization of at least 34 h at 51 °C. Racemization of the (Tol2Bob)TiIV fragment is strongly accelerated under protic conditions, probably due to protonolysis of one of the diketonate ligands. Analogous optically active titanium bis(diketonates) can also be prepared by using an optically active 2,2′-bis(methylene)-1,1′-binaphthyl bridged bis(diketonate) ligand, Tol2Bobbinap, prepared from (R)-BINOLH2. Complexation of (R)-Tol2BobbinapH2 with Ti(OiPr)4 gives only a single diastereomer with the Λ configuration at titanium. The bis(diketonate)titanium(IV) fragment gives rise to characteristic signals in circular dichroism spectra which can be used to identify the configuration at the metal centre.
Co-reporter:Erin R. Hurley ; Xuyang He ; Seth N. Brown ;Kenneth W. Henderson
Journal of the American Chemical Society 2009 Volume 131(Issue 17) pp:6056-6057
Publication Date(Web):April 8, 2009
DOI:10.1021/ja809716j
Thermodynamic stereocontrol of the (hexamethyldisilazide)magnesium enolates of propiophenone in THF is reported. The overall stereoselectivity proves to be very sensitive to concentration, since dimeric species with bridging enolates show no stereoselectivity while monomeric enolates show a very strong thermodynamic preference for the Z enolate. Kinetically, interconversion among aggregates is remarkably slow, whereas stereoisomerization of the monomer, even in the absence of a proton source such as ketone or amine, is remarkably fast. Furthermore, stereoisomerization takes place in the absence of a proton source or excess ketone. These observations contrast with accepted views of these fundamentally important processes and have implications for understanding the identity and reactivity of metal enolates.
Co-reporter:Lea T. Dulatas, Seth N. Brown, Edema Ojomo, Bruce C. Noll, Matthew J. Cavo, Paul B. Holt and Matthew M. Wopperer
Inorganic Chemistry 2009 Volume 48(Issue 22) pp:10789-10799
Publication Date(Web):October 21, 2009
DOI:10.1021/ic901683c
A tetradentate bis(ferrocenyldiketonate) ligand, Fc2BobH2, is prepared via Claisen condensation of acetylferrocene and 2,2′-biphenyldiacetyl chloride, and is metalated with titanium(IV) isopropoxide to give (Fc2Bob)Ti(OiPr)2 in good yield. The isopropoxide groups are replaced with di(4-nitrophenyl)phosphate groups on treatment with the corresponding acid, and with chlorides on treatment with trimethylsilyl chloride. Metathesis with catechol leads to the bis(o-hydroxyphenoxide) complex rather than the chelating catecholate complex. Hydrolysis selectively gives the μ-oxo trimer (Δ,Δ,Δ)/(Λ,Λ,Λ)-{(Fc2Bob)Ti(μ-O)}3. The solid-state structures of the μ-oxo trimer and the bis(o-hydroxyphenoxide) complex show that the ferrocene substituents are oriented proximal to the biphenyl backbone rather than pointed out toward the exogenous groups. The complexes show dramatic changes in color depending on the bound anions, ranging from the red isopropoxide (λmax = 489 nm) to the green bis(di(4-nitrophenyl)phosphate) (λmax = 653 nm). The oxidation potentials of the ferrocenes show modest shifts based on the titanium environment, but the redox potentials of the two ferrocenes are never separated by more than 60 mV. These results and those of density-functional theory (DFT) calculations indicate that the titanium interacts principally with the lowest unoccupied molecular orbital (LUMO) of the ferrocenyldiketonate and very little with its highest occupied molecular orbital (HOMO).
Co-reporter:Natcharee Kongprakaiwoot, Bruce C. Noll and Seth N. Brown
Inorganic Chemistry 2008 Volume 47(Issue 24) pp:11902-11909
Publication Date(Web):November 12, 2008
DOI:10.1021/ic8016479
A 2,2′-bis(methylene)biphenyl-bridged bis(hydroxamic acid) (HoxH2) is prepared by reaction of 2,2′-biphenyldiacetyl chloride with 2 equiv of N-methylhydroxylamine. Use of 1 equiv of CH3NHOH gives the cyclic diacylhydroxylamine, which is selectively ring-opened to give a mixed monohydroxamate-monodiketonate ligand HobH2. Both ligands are metalated by Ti(OiPr)4 to give the corresponding LTi(OiPr)2 complexes as exclusively the cis-α, (R)-Λ/(S)-Δ isomers, similar to the previously prepared bis(diketonate) analogues (Bob)TiX2. The carbonyl oxygens of the hydroxamates in the Hox ligand are constrained to be cis to each other, and the crystal structure of (Hob)Ti(OiPr)2 suggests that the carbonyl oxygen is a slightly weaker donor than the diketonate oxygen, based on a modest difference in their trans influences. A differential trans effect is also manifest in the observation of only a single geometric isomer of (Hob)Ti(OiPr)(O3SCF3) and in a 15.6:1 preference for the isomer of (Hob)Ti(OCH2CMe2CO2) in which the alkoxide is trans to the hydroxamate ligand.
Co-reporter:Vesela Ugrinova, Gregory A. Ellis and Seth N. Brown
Chemical Communications 2004 (Issue 4) pp:468-469
Publication Date(Web):22 Jan 2004
DOI:10.1039/B315092E
The monomeric titanium(IV) hydroxide complex, LTi(OH)
(LH3
= tris(2-hydroxy-3,5-di-tert-butylbenzyl)amine), which is sterically inhibited from condensation to a μ-oxo dimer, cannot be prepared by hydrolysis of the alkoxide, with Keq
= 0.012 for hydrolysis of the titanium methoxide in THF.
Co-reporter:Natcharee Kongprakaiwoot, Mauricio Quiroz-Guzman, Allen G. Oliver and Seth N. Brown
Chemical Science (2010-Present) 2011 - vol. 2(Issue 2) pp:NaN336-336
Publication Date(Web):2010/10/27
DOI:10.1039/C0SC00468E
3,3′,5,5′-Tetrasubstituted-2,2′-biphenolate complexes of titanium(IV) with bis(diketonate) (Bob), bis(hydroxamate) (Hox) and mixed diketonate–hydroxamate (Hob) ligands have been prepared from the corresponding diisopropoxide complexes. Four of the twelve compounds have been characterized crystallographically, and in the solid state all show the (Δ,R)/(Λ,S) relative stereochemistry at titanium and the biaryloxide, respectively, as previously observed in (acac)2Ti(1,1′-bi-2-naphtholate) complexes. In solution the compounds epimerize by atropisomerization of the biphenolate moiety with ΔG‡ ≈ 14 kcal mol−1. The bis(diketonate) complexes show high diastereoselectivity except for the most electron-poor tetranitrobiphenolate. In contrast, the bis(hydroxamate) shows low to moderate selectivity which correlates with the steric but not electronic properties of the biphenolates (Br < CH3 < NO2 < tBu). The mixed diketonate–hydroxamate complexes show intermediate behaviour. These observations are rationalized on the basis of MO arguments regarding ligand-metal π bonding. Symmetrical chelates such as diketonates foster mixing of two dπ orbitals and create a dissymmetric electronic environment. This mixing does not take place with unsymmetrical ligands such as hydroxamates, which therefore do not create an environment where electronic effects contribute significantly to binding stereoselectivity.
Co-reporter:Sukesh Shekar and Seth N. Brown
Dalton Transactions 2014 - vol. 43(Issue 9) pp:NaN3611-3611
Publication Date(Web):2014/01/07
DOI:10.1039/C3DT53496K
The dioxomolybdenum(VI) complex (tBuClipH2)MoO2 (tBuClipH4 = 4,4′-di-tert-butyl-N,N′-bis(3,5-di-tert-butyl-2-hydroxyphenyl)-2,2′-diaminobiphenyl) reacts with 3,5-di-tert-butylcatechol to form oxo-free (tBuClip)Mo(3,5-tBu2Cat). The bis(amidophenoxide)-monocatecholate complex is monomeric and exhibits a cis-β geometry in the solid state. Variable-temperature NMR data are consistent with two fluxional processes, one that interconverts several geometric isomers at low temperature, and a second that interchanges the ends of the tBuClip ligand at ambient temperatures. The high-temperature fluxional process can be explained by a single Bailar trigonal twist coupled with atropisomerization of the chiral diaminobiaryl backbone. Addition of excess catechol to (tBuClipH2)MoO2 results in formation of a dimolybdenum mono-oxo complex (tBuClip)Mo(μ-3,5-tBu2Cat)2Mo(O)(3,5-tBu2Cat). This complex, which contains a seven-coordinate bis(amidophenoxide)molybdenum center and a six-coordinate oxomolybdenum center, represents a structural hybrid between dimeric oxomolybdenumbis(catecholate) and molybdenum tris(catecholate) complexes. Both mono- and dimolybdenum complexes are best formulated as containing Mo(VI), but there is structural evidence for significant π donation from the amidophenolates. (tBuClip)Mo(3,5-tBu2Cat) binds pyridine to form a mixture of isomeric seven-coordinate adducts. The Lewis acidity of the mixed amidophenoxide-catecholate appears to be lower than its tris(catecholate) or oxobis(amidophenoxide) analogues, which manifests itself principally in relatively slow binding of pyridine to the six-coordinate complex (k = 8 × 104 L mol−1 s−1 at 0 °C) rather than in the rate of dissociation of pyridine from the seven-coordinate adduct.
Co-reporter:Mauricio Quiroz-Guzman, Allen G. Oliver, Andrew J. Loza and Seth N. Brown
Dalton Transactions 2011 - vol. 40(Issue 43) pp:NaN11468-11468
Publication Date(Web):2011/09/21
DOI:10.1039/C1DT11228G
A diarylamino-substituted N-methyl tetrahydrosalen (salan) ligand, An2NLH2, is prepared in four steps and overall 53% yield from 5-bromosalicylaldehyde, with the key step a palladium-catalysed Hartwig–Buchwald amination of the tert-butyldimethylsilyl-protected 5-bromo-N-methylsalan ligand. Reaction of An2NLH2 or its bromo analogue with Ti(OiPr)4 or TiF4 results in metalation of the ligand. The isopropoxide groups are readily exchanged with α- or β-hydroxyacids to form chelated complexes. X-ray crystallography and NMR spectroscopy indicate that the salan ligands are quite flexible, with An2NLTiF2, for example, showing four stereoisomers in its 19F NMR spectrum. The major stereoisomer of (salan)Ti(X)(Y) depends principally on the trans influence of the X and Y groups. Complexes of An2NL show two reversible, closely spaced redox couples at approximately + 0.1 V vs.ferrocene/ferrocenium, and a second set of two closely spaced redox couples at ∼ + 0.8 V vs. Fc/Fc+.
Co-reporter:Natcharee Kongprakaiwoot, Jack B. Armstrong, Bruce C. Noll and Seth N. Brown
Dalton Transactions 2010 - vol. 39(Issue 42) pp:NaN10115-10115
Publication Date(Web):2010/09/07
DOI:10.1039/C0DT00828A
The 2,2′-bis(methylene)biphenyl bridged bis(diketonate) (Tol2Bob)TiIV fragment is resolved by reaction of the isopropoxide complex with (R)-1,1′-bi-2-naphthol, which selectively forms (S,Δ)-(Tol2Bob)Ti(R-BINOL). The binaphtholate ligand can be cleaved from titanium by careful treatment with trifluoromethanesulfonic acid to give (S,Δ)-(Tol2Bob)Ti(OTf)2, which has a half-life toward racemization of at least 34 h at 51 °C. Racemization of the (Tol2Bob)TiIV fragment is strongly accelerated under protic conditions, probably due to protonolysis of one of the diketonate ligands. Analogous optically active titanium bis(diketonates) can also be prepared by using an optically active 2,2′-bis(methylene)-1,1′-binaphthyl bridged bis(diketonate) ligand, Tol2Bobbinap, prepared from (R)-BINOLH2. Complexation of (R)-Tol2BobbinapH2 with Ti(OiPr)4 gives only a single diastereomer with the Λ configuration at titanium. The bis(diketonate)titanium(IV) fragment gives rise to characteristic signals in circular dichroism spectra which can be used to identify the configuration at the metal centre.
Co-reporter:Travis Marshall-Roth and Seth N. Brown
Dalton Transactions 2015 - vol. 44(Issue 2) pp:NaN685-685
Publication Date(Web):2014/11/04
DOI:10.1039/C4DT02936D
A tris(aminophenol), tris(2-(3′,5′-di-tert-butyl-2′-hydroxyphenyl)amino-4-methylphenyl)amine, MeClampH6, is prepared in three steps from tri-p-tolylamine. The ligand reacts with dioxomolybdenum(VI) bis(acetylacetonate) to form an oxo-free heptadentate complex, (MeClamp)Mo, with a capped octahedral geometry. The molybdenum is formally in the +6 oxidation state, with significant π donation of the amidophenolates, as judged by intraligand bond distances. Two ligand-based oxidations and one metal-centered reduction are observed by cyclic voltammetry. Analysis of the optical spectrum of the compound gives an estimate of the energetic stabilization of the ligand π orbitals by bonding to the molybdenum of approximately 0.9 eV, corresponding to about 40 kcal mol−1 per π bond.
Co-reporter:Travis Marshall-Roth, Sean C. Liebscher, Karl Rickert, Nicholas J. Seewald, Allen G. Oliver and Seth N. Brown
Chemical Communications 2012 - vol. 48(Issue 63) pp:NaN7828-7828
Publication Date(Web):2012/06/19
DOI:10.1039/C2CC33523A
Mechanistic studies indicate that the oxomolybdenum(VI) bis(3,5-di-tert-butylcatecholate) fragment deoxygenates pyridine-N-oxides in a reaction where the oxygen is delivered to molybdenum but the electrons for substrate reduction are drawn from the bound catecholate ligands, forming 3,5-di-tert-butyl-1,2-benzoquinone.
Co-reporter:Jacqueline Cipressi and Seth N. Brown
Chemical Communications 2014 - vol. 50(Issue 59) pp:NaN7959-7959
Publication Date(Web):2014/06/05
DOI:10.1039/C4CC03404J
Ru(ONO)2 and Os(ONO)2 distort from octahedral towards trigonal prismatic geometry in order to relieve π antibonding due to donation from the second-highest ligand orbital to the metal. Increasing oxidation of the ONO ligand suppresses distortion by increasing σ* interactions in the trigonal prism.
Co-reporter:Daniel D. Swanson, Kyle M. Conner and Seth N. Brown
Dalton Transactions 2017 - vol. 46(Issue 28) pp:NaN9057-9057
Publication Date(Web):2017/06/20
DOI:10.1039/C7DT01945A
1,1′-Bis(4-(2-hydroxy-3,5-di-tert-butylphenyl)aminophenyl)ferrocene (pFlipH4) is prepared in three steps from commercially available 1,1′-ferrocenediboronic acid or its pinacol ester. The suitability of the ligand to bind as a tetradentate ligand in a cis, planar fashion has been confirmed by formation of a square planar palladium bis-iminosemiquinone (pFlip)Pd. The linker unit appears to be structurally similar to 1,1′-ferrocenediyl, but the electronic interaction of the ferrocene with the aminophenols is minimal.
Co-reporter:Tobias Schroeder, Vesela Ugrinova, Bruce C. Noll and Seth N. Brown
Dalton Transactions 2006(Issue 8) pp:NaN1040-1040
Publication Date(Web):2005/11/17
DOI:10.1039/B511915D
Dibenzoylmethane derivatives with one (L1H2) or both (L2H3, L3H3) benzenes linked at their ortho positions to 4,6-di-tert-butylphenol moieties by two-carbon linkers have been synthesized. The mono-β-diketone-monophenol ligand L1H2 is metalated by titanium alkoxides to form the homoleptic complex (L1)2Ti and heteroleptic complexes (L1)Ti([OCH2CH2]2NR) (R = H, CH3), and reacts with Cp3Sc to form CpSc(L1). These are the first examples of complexes of a β-diketonate ligand which is further chelating to a single metal center. Crystallographic analysis of (L1)2Ti indicates that the 10-membered ring allows chelation of the phenoxide with little strain, and both fac and mer geometries are accessible in solution. Protonolysis of the second cyclopentadienyl ring of Cp3Sc appears to take place by an indirect, Cp3Sc-catalyzed pathway.
.jpg)






















![(+)-4,5-Bis[hydroxy(diphenyl)methyl]-2,2-dimethyl-1,3-dioxolane](http://img.cochemist.com/ccimg/93400/93379-49-8.png)
![(+)-4,5-Bis[hydroxy(diphenyl)methyl]-2,2-dimethyl-1,3-dioxolane](http://img.cochemist.com/ccimg/93400/93379-49-8_b.png)


![[1,1'-Biphenyl]-2,2'-diol,3,3',5,5'-tetrabromo-](http://img.cochemist.com/ccimg/22000/21987-62-2.png)
![[1,1'-Biphenyl]-2,2'-diol,3,3',5,5'-tetrabromo-](http://img.cochemist.com/ccimg/22000/21987-62-2_b.png)









