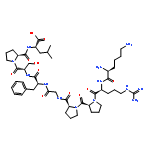
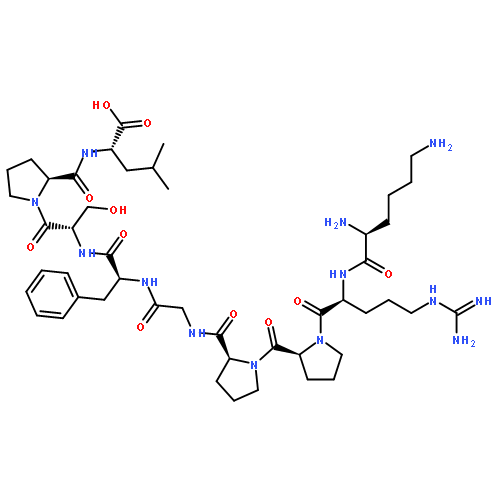
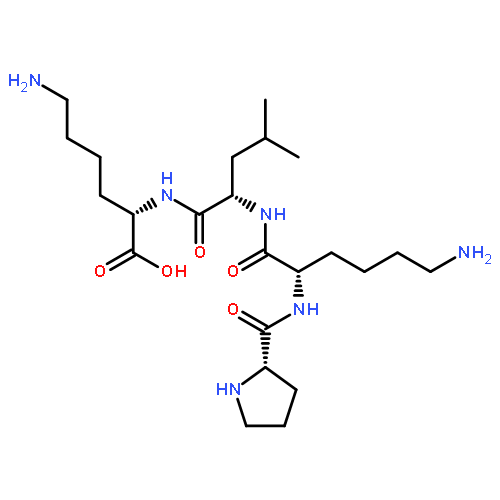
Co-reporter:Michael Remesic, Victor J. Hruby, Frank Porreca, and Yeon Sun Lee
ACS Chemical Neuroscience June 21, 2017 Volume 8(Issue 6) pp:1147-1147
Publication Date(Web):April 3, 2017
DOI:10.1021/acschemneuro.7b00090
Opioids, and more specifically μ-opioid receptor (MOR) agonists such as morphine, have long been clinically used as therapeutics for severe pain states but often come with serious side effects such as addiction and tolerance. Many studies have focused on bringing about analgesia from the MOR with attenuated side effects, but its underlying mechanism is not fully understood. Recently, focus has been geared toward the design and elucidation of the orthosteric site with ligands of various biological profiles and mixed subtype opioid activities and selectivities, but targeting the allosteric site is an area of increasing interest. It has been shown that allosteric modulators play key roles in influencing receptor function such as its tolerance to a ligand and affect downstream pathways. There has been a high variance of chemical structures that provide allosteric modulation at a given receptor, but recent studies and reviews tend to focus on the altered cellular mechanisms instead of providing a more rigorous description of the allosteric ligand’s structure–function relationship. In this review, we aim to explore recent developments in the structural motifs that potentiate orthosteric binding and their influences on cellular pathways in an effort to present novel approaches to opioid therapeutic design.Keywords: Allosteric modulation; Cannabinoid receptors; Heterodimers; Opioid receptors; Opioid side effects; Pain therapeutics; Sigma receptors;
Co-reporter:Cyf Ramos-Colon;Sara M. Hall;Frank Porreca;Lindsay LeBaron;Josephine Lai;Chaoling Qu;Jennifer Yanhua Xie;Victor J. Hruby
ACS Chemical Neuroscience December 21, 2016 Volume 7(Issue 12) pp:1746-1752
Publication Date(Web):September 12, 2016
DOI:10.1021/acschemneuro.6b00258
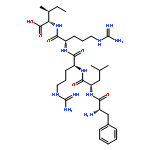
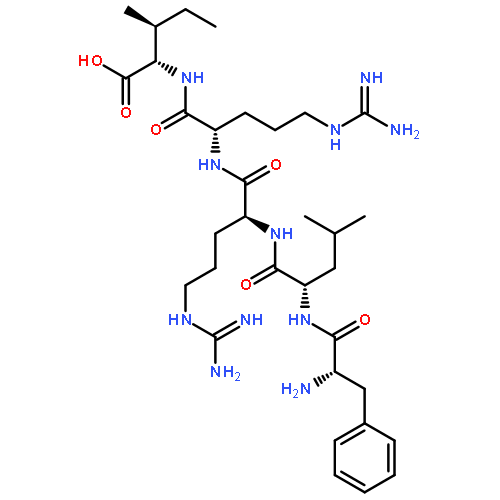
Dynorphin A (Dyn A) is a unique endogenous ligand that possesses well-known neuroinhibitory effects via opioid receptors with a preference for the kappa receptor but also neuroexcitatory effects, which cause hyperalgesia. We have shown that the neuroexcitatory effects are mediated through bradykinin (BK) receptors and that intrathecal (i.th.) administration of our lead ligand 1, [des-Arg7]-Dyn A-(4–11), which shows good binding affinity (IC50 = 150 nM) at the BK receptors, blocks Dyn A-induced hyperalgesia in naïve animals and reverses thermal and tactile hypersensitivities in a dose-dependent manner in nerve-injured animals. However, 1 has a serious drawback as a potential drug candidate for the treatment of neuropathic pain because of its susceptibility to enzymatic degradation. In an effort to increase its stability, we modified ligand 1 using non-natural amino acids and found that analogues substituted at or near the N-terminus with a d-isomer retain binding at the receptor and provide a large increase in stability. In particular when Leu5 was modified, with either the d-isomer or N-methylation, there was a large increase in stability (t1/2 = 0.7–160 h in rat plasma) observed. From these studies, we have developed a very stable Dyn A analogue 16, [d-Leu5,des-Arg7]-Dyn A-(4–11), that binds to BK receptors (IC50 = 130 nM) in the same range as ligand 1 and shows good antihyperalgesic effects in both naïve rats and L5/L6 spinal nerve ligation rats.Keywords: Antihyperalgesic effect; Bradykinin receptors; Neuropathic pain; Non-opioid dynorphin A; Peptide stability; Structure−activity relationship;
Co-reporter:Cyf N. Ramos-Colon, Yeon Sun Lee, Michael Remesic, Sara M. Hall, Justin LaVigne, Peg Davis, Alexander J. Sandweiss, Mary I. McIntosh, Jessica Hanson, Tally M. Largent-Milnes, Todd W. Vanderah, John Streicher, Frank Porreca, and Victor J. Hruby
Journal of Medicinal Chemistry 2016 Volume 59(Issue 22) pp:10291-10298
Publication Date(Web):October 31, 2016
DOI:10.1021/acs.jmedchem.6b01411
Dynorphin A (Dyn A) is an endogenous ligand for the opioid receptors with preference for the κ opioid receptor (KOR), and its structure–activity relationship (SAR) has been extensively studied at the KOR to develop selective potent agonists and antagonists. Numerous SAR studies have revealed that the Arg7 residue is essential for KOR activity. In contrast, our systematic SAR studies on [des-Arg7]Dyn A analogues found that Arg7 is not a key residue and even deletion of the residue does not affect biological activities at the KOR. In addition, it was also found that [des-Arg7]Dyn A(1–9)-NH2 is a minimum pharmacophore and its modification at the N-terminus leads to selective KOR antagonists. A lead ligand, 14, with high affinity and antagonist activity showed improved metabolic stability and could block antinociceptive effects of a KOR selective agonist, FE200665, in vivo, indicating high potential to treat KOR mediated disorders such as stress-induced relapse.
Co-reporter:Yeon Sun Lee, Michael Remesic, Cyf Ramos-Colon, Sara M. Hall, Alexander Kuzmin, David Rankin, Frank Porreca, Josephine Lai, Victor J. Hruby
Bioorganic & Medicinal Chemistry Letters 2016 Volume 26(Issue 22) pp:5513-5516
Publication Date(Web):15 November 2016
DOI:10.1016/j.bmcl.2016.10.010
Nerve injury and inflammation cause up-regulation of an endogenous opioid ligand, dynorphin A (Dyn A), in the spinal cord resulting in hyperalgesia via the interaction with bradykinin receptors (BRs). This is a non-opioid neuroexcitatory effect that cannot be blocked by opioid antagonists. Our systematic structure–activity relationships study on Dyn A identified lead ligands 1 and 4, along with the key structural feature (i.e. amphipathicity) for the BRs. However, the ligands showed very low metabolic stability in plasma (t1/2 <1 h) and therefore, in order to improve their metabolic stabilities with retained biological activities, various modifications were performed. Cyclization of ligand 4 afforded a cyclic Dyn A analogue 5 that retained the same range of binding affinity as the linear ligand with improved metabolic stability (t1/2 >5 h) and therefore possesses the potential as a pharmacophoric scaffold to be utilized for drug development.
Co-reporter:Mehr-un-Nisa, Munawar A. Munawar, Yeon Sun Lee, David Rankin, Jawaria Munir, Josephine Lai, Misbahul A. Khan, Victor J. Hruby
Bioorganic & Medicinal Chemistry 2015 Volume 23(Issue 6) pp:1251-1259
Publication Date(Web):15 March 2015
DOI:10.1016/j.bmc.2015.01.047
A series of opioid and serotonin re-uptake inhibitors (SSRIs) bifunctional ligands have been designed, synthesized, and tested for their activities and efficacies at μ-, δ- and κ opioid receptors and SSRIs receptors. Most of the compounds showed high affinities for μ- and δ-opioid receptors and lower affinities for SSRIs and κ opioid receptors. A docking study on the μ-opioid receptor binding pocket has been carried out for ligands 3–11. The ligands 7 and 11 have displayed the highest binding profiles for the μ-opioid receptor binding site with ΔGbind (−12.14 kcal/mol) and Ki value (1.0 nM), and ΔGbind (−12.41 kcal/mol) and Ki value (0.4 nM), respectively. Ligand 3 was shown to have the potential of dual acting serotonin/norepinephrine re-uptake inhibitor (SNRI) antidepressant activity in addition to opioid activities, and thus could be used for the design of multifunctional ligands in the area of a novel approach for the treatment of pain and depression.
Co-reporter:Yeon Sun Lee, Sara M. Hall, Cyf Ramos-Colon, Michael Remesic, Lindsay LeBaron, Ann Nguyen, David Rankin, Frank Porreca, Josephine Lai, Victor J. Hruby
Bioorganic & Medicinal Chemistry Letters 2015 Volume 25(Issue 1) pp:30-33
Publication Date(Web):1 January 2015
DOI:10.1016/j.bmcl.2014.11.026
It has been shown that under chronic pain or nerve injury conditions, up-regulated dynorphin A (Dyn A) interacts with bradykinin receptors (BRs) to cause hyperalgesia in the spinal cord. Thus BRs antagonist can modulate hyperalgesia by blocking Dyn A’s interaction with the BRs in the central nervous system. In our earlier structure–activity relationship (SAR) study, [des-Arg7]-Dyn A-(4–11) 13 was discovered as a minimum pharmacophore for rat brain BRs with its antagonist activity (anti-hyperalgesic effect) in in vivo tests using naïve or injured animals. We have pursued further modification on the [des-Arg7]-Dyn A analogues and identified a key insight into the pharmacophore of the rat brain BRs: amphipathicity.
Co-reporter:Steve M. Fernandes, Yeon Sun Lee, Robert J. Gillies, Victor J. Hruby
Bioorganic & Medicinal Chemistry 2014 Volume 22(Issue 22) pp:6360-6365
Publication Date(Web):15 November 2014
DOI:10.1016/j.bmc.2014.09.055
Membrane proteins, especially G-protein coupled receptors (GPCRs), are interesting and important theragnostic targets since many of them serve in intracellular signaling critical for all aspects of health and disease. The potential utility of designed bivalent ligands as targeting agents for cancer diagnosis and/or therapy can be evaluated by determining their binding to the corresponding receptors. As proof of concept, GPCR cell surface proteins are shown to be targeted specifically using multivalent ligands. We designed, synthesized, and tested a series of bivalent ligands targeting the over-expressed human melanocortin 4 receptor (hMC4R) in human embryonic kidney (HEK) 293 cells. Based on our data suggesting an optimal linker length of 25 ± 10 Å inferred from the bivalent melanocyte stimulating hormone (MSH) agonist, the truncated heptapeptide, referred to as MSH(7): Ac-Ser-Nle-Glu-His-D-Phe-Arg-Trp-NH2 was used to construct a set of bivalent ligands incorporating a hMC4R antagonist, SHU9119: Ac-Nle-c[Asp-His-2′-D-Nal-Arg-Trp-Lys]-NH2 and another set of bivalent ligands containing the SHU9119 antagonist pharmacophore on both side of the optimized linkers. These two binding motifs within the bivalent constructs were conjoined by semi-rigid (Pro-Gly)3 units with or without the flexible poly(ethylene glycol) (PEGO) moieties. Lanthanide-based competitive binding assays showed bivalent ligands binds to the hMC4R with up to 240-fold higher affinity than the corresponding linked monovalent ligands.
Co-reporter:Yeon Sun Lee, David Rankin, Sara M. Hall, Cyf Ramos-Colon, Jose Juan Ortiz, Robert Kupp, Frank Porreca, Josephine Lai, Victor J. Hruby
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 21) pp:4976-4979
Publication Date(Web):1 November 2014
DOI:10.1016/j.bmcl.2014.09.033
In our earlier studies, bradykinin receptors (BRs) were identified as a potential target for the neuroexcitatory effects of dynorphin A (Dyn A) in the central nervous system (CNS), and [des-Arg7]-Dyn A-(4–11) (6) was discovered as a lead ligand to modulate Dyn A-(2–13) induced neuroexcitatory effects in the CNS as an antagonist. In an effort to gain insights into key structural features of the Dyn A for the BRs, we pursued further structure–activity relationships (SAR) study on the [des-Arg7]-Dyn A analogs and confirmed that all of the [des-Arg7]-Dyn A analogues showed good binding affinities at the BRs.