Co-reporter:Pokpong Rungthanaphatsophon;Adrien Bathelier;Dr. Ludovic Castro;Dr. Andrew C. Behrle;Dr. Charles L. Barnes;Dr. Laurent Maron;Dr. Justin R. Walensky
Angewandte Chemie International Edition 2017 Volume 56(Issue 42) pp:12925-12929
Publication Date(Web):2017/10/09
DOI:10.1002/anie.201706496
AbstractThe reaction of (C5Me5)2Th(CH3)2 with the phosphonium salts [CH3PPh3]X (X=Cl, Br, I) was investigated. When X=Br and I, two equivalents of methane are liberated to afford (C5Me5)2Th[CHPPh3]X, rare terminal phosphorano-stabilized carbenes with thorium. These complexes feature the shortest thorium–carbon bonds (≈2.30 Å) reported to date, and electronic structure calculations show some degree of multiple bonding. However, when X=Cl, only one equivalent of methane is lost with concomitant formation of benzene from an unstable phosphorus(V) intermediate, yielding (C5Me5)2Th[κ2-(C,C′)-(CH2)(CH2)PPh2]Cl. Density functional theory (DFT) investigations of the reaction energy profiles for [CH3PPh3]X, X=Cl and I showed that in the case of iodide, thermodynamics prevents the production of benzene and favors formation of the carbene.
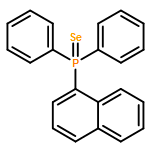
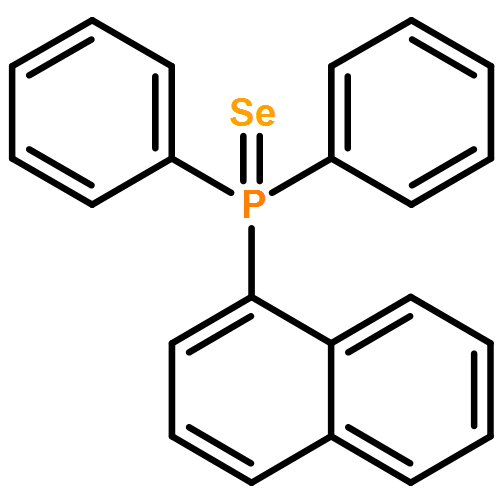
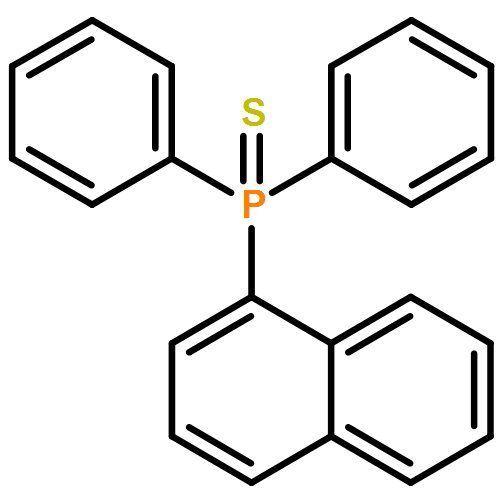
Co-reporter:Kira Behm;Jeremy B. Essner;Charles L. Barnes;Gary A. Baker
Dalton Transactions 2017 vol. 46(Issue 33) pp:10867-10875
Publication Date(Web):2017/08/22
DOI:10.1039/C7DT01953J
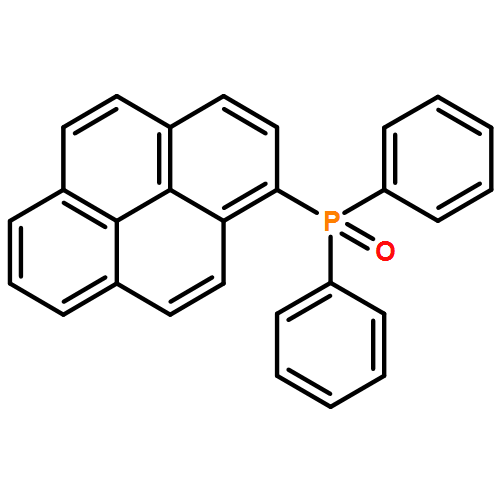
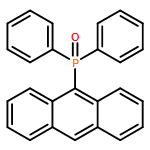
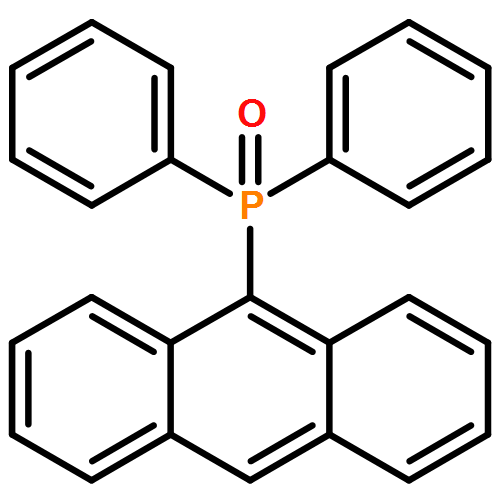
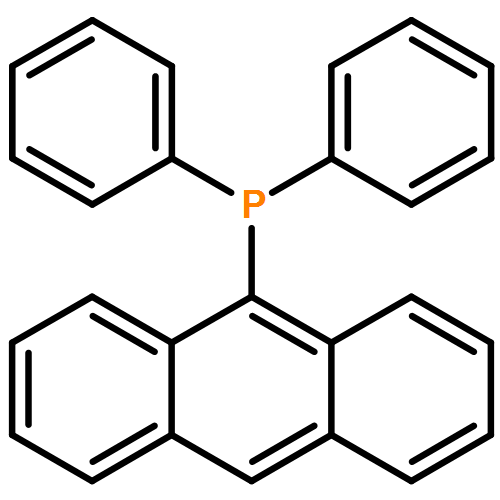
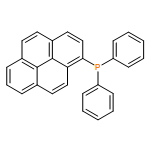
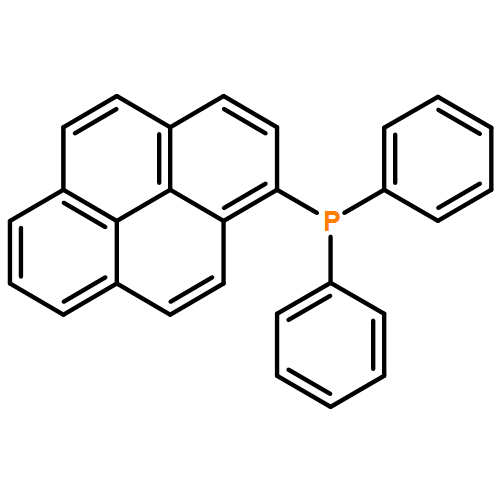
The synthesis, characterization, and fluorescence spectroscopy of E(C16H9)3 complexes (E = P, As, Sb, and Bi), as well as OP(C16H9)3, OAs(C16H9)3, and SP(C16H9)3, is reported. These compounds exhibit fluorescence quantum yields that span two orders of magnitude, from well below 1% to ca. 14%.
Co-reporter:Pokpong Rungthanaphatsophon;Adrien Bathelier;Dr. Ludovic Castro;Dr. Andrew C. Behrle;Dr. Charles L. Barnes;Dr. Laurent Maron;Dr. Justin R. Walensky
Angewandte Chemie 2017 Volume 129(Issue 42) pp:13105-13109
Publication Date(Web):2017/10/09
DOI:10.1002/ange.201706496
AbstractThe reaction of (C5Me5)2Th(CH3)2 with the phosphonium salts [CH3PPh3]X (X=Cl, Br, I) was investigated. When X=Br and I, two equivalents of methane are liberated to afford (C5Me5)2Th[CHPPh3]X, rare terminal phosphorano-stabilized carbenes with thorium. These complexes feature the shortest thorium–carbon bonds (≈2.30 Å) reported to date, and electronic structure calculations show some degree of multiple bonding. However, when X=Cl, only one equivalent of methane is lost with concomitant formation of benzene from an unstable phosphorus(V) intermediate, yielding (C5Me5)2Th[κ2-(C,C′)-(CH2)(CH2)PPh2]Cl. Density functional theory (DFT) investigations of the reaction energy profiles for [CH3PPh3]X, X=Cl and I showed that in the case of iodide, thermodynamics prevents the production of benzene and favors formation of the carbene.
Co-reporter:Andrew C. Behrle and Justin R. Walensky
Dalton Transactions 2016 vol. 45(Issue 24) pp:10042-10049
Publication Date(Web):28 Apr 2016
DOI:10.1039/C6DT00776G
The reactivity of thorium–phosphido and thorium–arsenido bonds was probed using tert-butyl isocyanide, tBuNC. Reaction of (C5Me5)2Th[E(H)R]2, E = P, As; R = 2,4,6-iPr3C6H2, 2,4,6-Me3C6H2 with tBuNC affords the first phosphaazaallene and arsaazaallene moieties with an f-element.
Co-reporter:Andrew C. Behrle; Ludovic Castro; Laurent Maron
Journal of the American Chemical Society 2015 Volume 137(Issue 47) pp:14846-14849
Publication Date(Web):November 17, 2015
DOI:10.1021/jacs.5b09826
The synthesis and structural determination of the first thorium phosphinidene complex are reported. The reaction of 2 equiv of (C5Me5)2Th(CH3)2 with H2P(2,4,6-iPr3C6H2) at 95 °C produces [(C5Me5)2Th]2(μ2-P[(2,6-CH2CHCH3)2-4-iPrC6H2] as well as 4 equiv of methane, 2 equiv from deprotonation of the phosphine and 2 equiv from C–H bond activation of one methyl group of each of the isopropyl groups at the 2- and 6-positions. Transition state calculations indicate that the steps in the mechanism are P–H, C–H, C–H, and then P–H bond activation to form the phosphinidene.
Co-reporter:Andrew C. Lane; Charles L. Barnes; William E. Antholine; Denan Wang; Adam T. Fiedler
Inorganic Chemistry 2015 Volume 54(Issue 17) pp:8509-8517
Publication Date(Web):August 7, 2015
DOI:10.1021/acs.inorgchem.5b01161
Molecular examples of mixed-valence copper complexes through chemical oxidation are rare but invoked in the mechanism of substrate activation, especially oxygen, in copper-containing enzymes. To examine the cooperative chemistry between two metals in close proximity to each other we began studying the reactivity of a dinuclear Cu(I) amidinate complex. The reaction of [(2,6-Me2C6H3N)2C(H)]2Cu2, 1, with I2 in tetrahydrofuran (THF), CH3CN, and toluene affords three new mixed-valence copper complexes [(2,6-Me2C6H3N)2C(H)]2Cu2(μ2-I3)(THF)2, 2, [(2,6-Me2C6H3N)2C(H)]2Cu2(μ2-I) (NCMe)2, 3, and [(2,6-Me2C6H3N)2C(H)]3Cu3(μ3-I)2, 4, respectively. The first two compounds were characterized by UV–vis and electron paramagnetic resonance spectroscopies, and their molecular structure was determined by X-ray crystallography. Both di- and trinuclear mixed-valence intermediates were characterized for the reaction of compound 1 to compound 4, and the molecular structure of 4 was determined by X-ray crystallography. The electronic structure of each of these complexes was also investigated using density functional theory.
Co-reporter:Andrew C. Behrle, Jessica R. Levin, Jee Eon Kim, Jonathan M. Drewett, Charles L. Barnes, Eric J. Schelter and Justin R. Walensky
Dalton Transactions 2015 vol. 44(Issue 6) pp:2693-2702
Publication Date(Web):11 Sep 2014
DOI:10.1039/C4DT01798F
We report M(IV) M = Ti, Zr, Hf, Ce, and Th, complexes of a selenium bis(phenolate) ligand, 2,2′-selenobis(4,6-di-tert-butylphenol), (H2ArOSeO), 1. Reaction of Ti(NEt2)4 with two equivalents of 1 affords Ti(ArOSeO)2, 2. Salt metathesis of ZrCl4 and HfCl4 with two equivalents of Na2ArOSeO produces Zr(ArOSeO)2(THF), 3, and Hf(ArOSeO)2(THF), 4, respectively. Protonolysis of ThCl[N(SiMe3)2]3 with two equivalents of 1 yields Th(ArOSeO)2(THF)2, 5. Salt metathesis of Ce(OTf)3 and two equivalents of Na2ArOSeO produces [Na(THF)3][Ce(ArOSeO)2], which was oxidized in situ using 0.5 equivalents of I2 to yield the diamagnetic Ce(IV) product, Ce(ArOSeO)2(THF)2, 6. Addition of 2,2′-bipyridyl to 6 forms Ce(ArOSeO)2(bipy), 6a. Each diamagnetic complex was characterized using 1H, 13C, and 77Se NMR and IR spectroscopy and the structures of 2–6a were established with X-ray crystallography. Electrochemical measurements using cyclic voltammetry on complexes 2, 5, and 6 are also reported.
Co-reporter:Ashley N. Dame;Mohan S. Bharara;Charles L. Barnes
European Journal of Inorganic Chemistry 2015 Volume 2015( Issue 18) pp:2996-3005
Publication Date(Web):
DOI:10.1002/ejic.201500378
Abstract
To explore the coordination chemistry of salicylaldiminate ligands as a non-cyclopentadienyl ligand framework, we report the synthesis of their ThIV and UIV complexes. Deprotonation of (NC5H4)N=C(H)(3,5-tBu2C6H2)OH (1), (NC9H6)N=C(H)(3,5-tBu2C6H2)OH (2), and (CH3SC6H4)N=C(H)(3,5-tBu2C6H2)OH (3) with sodium hydride and the subsequent addition of 0.5 equivalents of [UI4(1,4-dioxane)2] yielded [{(NC5H4)N=C(H)(3,5-tBu2C6H2)O-κ3(O,N,N′)}2UI2] (4), [{(NC9H6)N=C(H)(3,5-tBu2C6H2)O-κ3(O,N,N′)}2UI2] (5), and [{(CH3SC6H4)N=C(H)(3,5-tBu2C6H2)O-κ3(O,N,S)}2UI2] (6), respectively. The reaction of 3 equivalents of the sodium salt of 1 afforded [{(NC5H4)N=C(H)(3,5-tBu2C6H2)O-κ3(O,N,N′)}3U]I3 (7). By using [ThCl4(dme)2], [{(NC5H4)N=C(H)(3,5-tBu2C6H2)O-κ3(O,N,N′)}2ThCl2] (8) and [{(NC9H6)N=C(H)(3,5-tBu2C6H2)O-κ3(O,N,N′)}2ThCl2] (9) were isolated. The metathesis of both chloride ligands in 9 with NaN3 led to a rare thorium azide product, [{(NC9H6)N=C(H)(3,5-tBu2C6H2)O-κ3(O,N,N′)}2Th(N3)2] (10). The reaction of 5 with AgOTf (OTf = O3SCF3) afforded [{(NC9H6)N=C(H)(3,5-tBu2C6H2)O-κ3(O,N,N′)}2U(OTf)2] (11), and the subsequent reaction of 11 with 2 equivalents of NaN3 provided [{(NC9H6)N=C(H)(3,5-tBu2C6H2)O-κ3(O,N,N′)}2U(N3)2] (12). The reaction of 5 with 2 equivalents of AgNO3 produced [{(NC9H6)N=C(H)(3,5-tBu2C6H2)O-κ3(O,N,N′)}2U{NO3-κ2(O,O)}2] (13). Each complex was characterized by 1H NMR and IR spectroscopy and, with the exception of 11, their structures were determined by X-ray crystallographic analysis.
Co-reporter:Matthew V. Vollmer, Charles W. Machan, Melissa L. Clark, William E. Antholine, Jay Agarwal, Henry F. Schaefer III, Clifford P. Kubiak, and Justin R. Walensky
Organometallics 2015 Volume 34(Issue 1) pp:3-12
Publication Date(Web):December 9, 2014
DOI:10.1021/om500838z
The synthesis and characterization of new Mn(I)- and Re(I)-centered organometallic complexes fashioned with 1,4-diazabutadiene (DAB) ligands is reported. Ten compounds of the type fac-(α-diimine)M(CO)3Br (M = Mn, Re) were obtained in moderate to excellent yield (35–80%) and high purity from the coordination of the five ligands with M(CO)5Br in refluxing ethanol. Despite the electronic similarity of DAB to 2,2′-bipyridyl, the complexes described herein were poor mediators of electrochemical CO2 conversion to CO, but provide insight into the role of redox-active ligands in catalysis. Additional characterization of the one-electron reduced rhenium compounds, relevant intermediates in CO2 reduction, by EPR and single-crystal X-ray analysis is described.
Co-reporter:Andrew C. Lane, Matthew V. Vollmer, Charles H. Laber, Doris Y. Melgarejo, Gina M. Chiarella, John P. Fackler Jr., Xinzheng Yang, Gary A. Baker, and Justin R. Walensky
Inorganic Chemistry 2014 Volume 53(Issue 21) pp:11357-11366
Publication Date(Web):October 15, 2014
DOI:10.1021/ic501694d
Co-reporter:Ellen M. Matson, Andrew T. Breshears, John J. Kiernicki, Brian S. Newell, Phillip E. Fanwick, Matthew P. Shores, Justin R. Walensky, and Suzanne C. Bart
Inorganic Chemistry 2014 Volume 53(Issue 24) pp:12977-12985
Publication Date(Web):November 21, 2014
DOI:10.1021/ic5020658
The trivalent uranium phenylchalcogenide series, Tp*2UEPh (Tp* = hydrotris(3,5-dimethylpyrazolyl)borate, E = O (1), S (2), Se (3), Te (4)), has been synthesized to investigate the nature of the U–E bond. All compounds have been characterized by 1H NMR, infrared and electronic absorption spectroscopies, and in the case of 4, X-ray crystallography. Compound 4 was also studied by SQUID magnetometry. Computational studies establish Mulliken spin densities for the uranium centers ranging from 3.005 to 3.027 (B3LYP), consistent for uranium–chalcogenide bonds that are primarily ionic in nature, with a small covalent contribution. The reactivity of 2–4 toward carbon disulfide was also investigated and showed reversible CS2 insertion into the U(III)–E bond, forming Tp*2U(κ2-S2CEPh) (E = S (5), Se (6), Te (7)). Compound 5 was characterized crystallographically.
Co-reporter:Lani A. Seaman, Justin R. Walensky, Guang Wu, and Trevor W. Hayton
Inorganic Chemistry 2013 Volume 52(Issue 7) pp:3556-3564
Publication Date(Web):June 20, 2012
DOI:10.1021/ic300867m
This Forum Article describes the pursuit of isolable homoleptic actinide alkyl complexes, starting with the pioneering work of Gilman during the Manhattan project. The initial reports in this area suggested that homoleptic uranium alkyls were too unstable to be isolated, but Wilkinson demonstrated that tractable uranium alkyls could be generated by purposeful “ate” complex formation, which serves to saturate the uranium coordination sphere and provide the complexes with greater kinetic stability. More recently, we reported the solid-state molecular structures of several homoleptic uranium alkyl complexes, including [Li(THF)4][U(CH2tBu)5], [Li(TMEDA)]2[UMe6], [K(THF)]3[K(THF)2][U(CH2Ph)6]2, and [Li(THF)4][U(CH2SiMe3)6], by employing Wilkinson’s strategy. Herein, we describe our attempts to extend this chemistry to thorium. The treatment of ThCl4(DME)2 with 5 equiv of LiCH2tBu or LiCH2SiMe3 at −25 °C in THF affords [Th(CH2tBu)5] (1) and [Li(DME)2][Th(CH2SiMe3)5 (2), respectively, in moderate yields. Similarly, the treatment of ThCl4(DME)2 with 6 equiv of K(CH2Ph) produces [K(THF)]2[Th(CH2Ph)6] (3), in good yield. Complexes 1–3 have been fully characterized, while the structures of 1 and 3 were confirmed by X-ray crystallography. Additionally, the electronic properties of 1 and 3 were explored by density functional theory.
Co-reporter:Andrew C. Behrle, Charles L. Barnes, Nikolas Kaltsoyannis, and Justin R. Walensky
Inorganic Chemistry 2013 Volume 52(Issue 18) pp:10623-10631
Publication Date(Web):August 29, 2013
DOI:10.1021/ic401642a
Homoleptic soft-donor actinide complexes of the general form An[E2PROR′]4 were synthesized from salt metathesis between ThCl4(DME)2 or UI4(1,4-dioxane)2 and M[E2PROR′], M = Na, K, to yield 2 (An = Th, E = S, R = 4-MeOC6H4, R′ = Me), 3 (An = Th, E = S, R = 4-MeOC6H4, R′ = tBu), 4 (An = U, E = S, R = 4-MeOC6H4, R′ = Me), 5 (An = Th, E = Se, R = C6H5, R′ = Me), and 6 (An = U, E = Se, R = C6H5, R′ = Me). In addition thorium and uranium thioselenophosphinate complexes 7 and 8 were produced from the reaction of ThCl4(DME)2 and UI4(1,4-dioxane)2 and Na[SSePPh2], respectively. All compounds were characterized using elemental analysis, 1H and 31P NMR, and IR spectroscopy, and the U(IV) compounds were also examined with UV–vis spectroscopy. The 77Se NMR spectrum of 5 reveals the first reported resonance with a Th–Se bond. The solid-state structures of 2, 5, 7, and 8 were determined by X-ray crystallography. The actinide–ligand bonding was examined using density functional theory calculations in conjunction with quantum theory of atoms-in-molecules analysis and shows slightly increased covalency in actinide–selenium bonds than actinide–sulfur.
Co-reporter:Agnes Mrutu;Charles L. Barnes;Suzanne C. Bart
European Journal of Inorganic Chemistry 2013 Volume 2013( Issue 22-23) pp:4050-4055
Publication Date(Web):
DOI:10.1002/ejic.201300390
Abstract
As the reactivity of transition-metal complexes with redox-active ligands has gained significant attention in recent years, we have begun expanding to the actinides. Here, we report the synthesis, characterization, and reactivity of (MesDABMe)2Th(THF) {1, MesDABMe = [MesNC(Me)=C(Me)NMes]2–, Mes = 2,4,6-trimethylphenyl, THF = tetrahydrofuran}, which cleaves the C–I bond in CH3I with subsequent Th–I and C–C bond formation to produce (MesDABMe)(MesDAB)ThI {2, MesDAB = [MesN=C(Me)C(Me)2NMes]–}. When 2 is crystallized in acetonitrile, the solvated adduct (MesDABMe)(MesDAB)ThI(NCCH3) (2-MeCN) is isolated. The reaction of 2 equiv. of 1 with Me3SiI produces (MesDABMe)2ThI (3) and Me3SiSiMe3. In 3, one MesDABMe ligand is now monoanionic and provides an electron to cleave the Si–I bond. This represents a rare example of the reactivity of thorium with a redox-active ligand.
Co-reporter:Ellen M. Matson;Mitchell D. Goshert;John J. Kiernicki;Brian S. Newell;Phillip E. Fanwick; Matthew P. Shores; Justin R. Walensky; Suzanne C. Bart
Chemistry - A European Journal 2013 Volume 19( Issue 48) pp:16176-16180
Publication Date(Web):
DOI:10.1002/chem.201303095
Co-reporter:Agnes Mrutu, Andrew C. Lane, Jonathan M. Drewett, Steven D. Yourstone, Charles L. Barnes, Christopher M. Halsey, Jason W. Cooley, Justin R. Walensky
Polyhedron 2013 Volume 54() pp:300-308
Publication Date(Web):30 April 2013
DOI:10.1016/j.poly.2013.02.011
The synthesis and spectroscopy of divalent first row transition metals bearing two monoanionic salicylaldiminate ligands is reported. The reaction of MnCl2, FeCl2, CoBr2, 1,2-(Ph2PCH2CH2PPh2)NiCl2, and CuCl2 with 2 equiv. of the alkali metal salt of [OC6H2tBu2C(H)N(C6H3Me2)]1− produces the corresponding M[OC6H2tBu2C(H)N(C6H3Me2)]2, M = Mn, Fe, Co, Ni, and Cu. Reaction of ZnEt2 with 2 equiv. of the protonated ligand affords Zn[OC6H2tBu2C(H)N(C6H3Me2)]2. The molecular structure of each complex has been analyzed using IR spectroscopy and X-ray crystallography while UV–Vis, CW-EPR, solution magnetic susceptibilities, and DFT calculations were also used to probe their electronic structure.The synthesis, structure, and spectroscopy of first row transition metal, Mn–Zn, complexes with two salicylaldiminate ligands are reported. The complexes exhibit non-covalent interactions which add stability to the tetrahedral geometry about the metal center instead of square planar which is observed if these interactions are absent.
Co-reporter:Skye Fortier ; Justin R. Walensky ; Guang Wu ;Trevor W. Hayton
Journal of the American Chemical Society 2011 Volume 133(Issue 18) pp:6894-6897
Publication Date(Web):April 13, 2011
DOI:10.1021/ja2001133
Addition of the Wittig reagent Ph3P═CH2 to the U(III) tris(amide) U(NR2)3 (R = SiMe3) generates a mixture of products from which the U(IV) complex U═CHPPh3(NR2)3 (2) can be obtained. Complex 2 features a short U═C bond and represents a rare example of a uranium carbene. In solution, 2 exists in equilibrium with the U(IV) metallacycle U(CH2SiMe2NR)(NR2)2 and free Ph3P═CH2. Measurement of this equilibrium as a function of temperature provides ΔHrxn = 11 kcal/mol and ΔSrxn = 31 eu. Additionally, the electronic structure of the U═C bond was investigated using DFT analysis.
Co-reporter:Andrew C. Behrle; Andrew Kerridge
Inorganic Chemistry () pp:
Publication Date(Web):December 4, 2015
DOI:10.1021/acs.inorgchem.5b01342
We report a comparison of the molecular and electronic structures of dithio- and diselenophosphinate, (E2PR2)1– (E = S, Se; R = iPr, tBu), with thorium(IV) and uranium(IV) complexes. For the thorium dithiophosphinate complexes, reaction of ThCl4(DME)2 with 4 equiv of KS2PR2 (R = iPr, tBu) produced the homoleptic complexes, Th(S2PiPr2)4 (1S-Th-iPr) and Th(S2PtBu2)4 (2S-Th-tBu). The diselenophosphinate complexes were synthesized in a similar manner using KSe2PR2 to produce Th(Se2PiPr2)4 (1Se-Th-iPr) and Th(Se2PtBu2)4 (2Se-Th-tBu). U(S2PiPr2)4, 1S-U-iPr, could be made directly from UCl4 and 4 equiv of KS2PiPr2. With (Se2PiPr2)1–, using UCl4 and 3 or 4 equiv of KSe2PiPr2 yielded the monochloride product U(Se2PiPr2)3Cl (3Se-UiPr-Cl), but using UI4(1,4-dioxane)2 produced the homoleptic U(Se2PiPr2)4 (1Se-U-iPr). Similarly, the reaction of UCl4 with 4 equiv of KS2PtBu2 yielded U(S2PtBu2)4 (2S-U-tBu), whereas the reaction with KSe2PtBu2 resulted in the formation of U(Se2PtBu2)3Cl (4Se-UtBu-Cl). Using UI4(1,4-dioxane)2 and 4 equiv of KSe2PtBu2 with UCl4 in acetonitrile yielded U(Se2PtBu2)4 (2Se-U-tBu). Transmetalation reactions were investigated with complex 2Se-U-tBu and various CuX (X = Br, I) salts to yield U(Se2PtBu2)3X (6Se-UtBu-Br and 7Se-UtBu-I) and 0.25 equiv of [Cu(Se2PtBu2)]4 (8Se-Cu-tBu). Additionally, 2Se-U-tBu underwent transmetalation reactions with Hg2F2 and ZnCl2 to yield U(Se2PtBu2)3F (6) and U(Se2PtBu2)3Cl (4Se-UtBu-Cl), respectively. The molecular structures were analyzed using 1H, 13C, 31P, and 77Se NMR and IR spectroscopy and structurally characterized using X-ray crystallography. Using the QTAIM approach, the electronic structure of all homoleptic complexes was probed, showing slightly more covalent bonding character in actinide–selenium bonds over actinide–sulfur bonds.
Co-reporter:Andrew C. Behrle, Jessica R. Levin, Jee Eon Kim, Jonathan M. Drewett, Charles L. Barnes, Eric J. Schelter and Justin R. Walensky
Dalton Transactions 2015 - vol. 44(Issue 6) pp:NaN2702-2702
Publication Date(Web):2014/09/11
DOI:10.1039/C4DT01798F
We report M(IV) M = Ti, Zr, Hf, Ce, and Th, complexes of a selenium bis(phenolate) ligand, 2,2′-selenobis(4,6-di-tert-butylphenol), (H2ArOSeO), 1. Reaction of Ti(NEt2)4 with two equivalents of 1 affords Ti(ArOSeO)2, 2. Salt metathesis of ZrCl4 and HfCl4 with two equivalents of Na2ArOSeO produces Zr(ArOSeO)2(THF), 3, and Hf(ArOSeO)2(THF), 4, respectively. Protonolysis of ThCl[N(SiMe3)2]3 with two equivalents of 1 yields Th(ArOSeO)2(THF)2, 5. Salt metathesis of Ce(OTf)3 and two equivalents of Na2ArOSeO produces [Na(THF)3][Ce(ArOSeO)2], which was oxidized in situ using 0.5 equivalents of I2 to yield the diamagnetic Ce(IV) product, Ce(ArOSeO)2(THF)2, 6. Addition of 2,2′-bipyridyl to 6 forms Ce(ArOSeO)2(bipy), 6a. Each diamagnetic complex was characterized using 1H, 13C, and 77Se NMR and IR spectroscopy and the structures of 2–6a were established with X-ray crystallography. Electrochemical measurements using cyclic voltammetry on complexes 2, 5, and 6 are also reported.
Co-reporter:Pokpong Rungthanaphatsophon, Charles L. Barnes and Justin R. Walensky
Dalton Transactions 2016 - vol. 45(Issue 36) pp:NaN14276-14276
Publication Date(Web):2016/08/18
DOI:10.1039/C6DT02709A
The coordination chemistry of copper has interest due to its use in biological systems as well as for photochemical and medicinal properties. We report the coordination chemistry of copper(I) complexes using terphenyl-based dithiocarboxylate, thiolate, and selenolate ligands. The number of metal ions of the resulting complexes can be tuned by varying the steric properties of the terphenyl ligands, changing the starting material, as well as adding PEt3. In addition, the steric crowding of the terphenyl ligand leads to varying reactivity. For example, while the reaction of carbon disulfide with [Cu(2,6-(Ph)2C6H3)]2 results in an insertion into the copper–carbon bond, no reaction occurs with [Cu(2,4,6-(Mes)2C6H3)], Mes = 2,4,6-Me3C6H2, or [Et3PCu(2,4,6-(Mes)2C6H2)2C6H3]. The synthesis and characterization of new copper(I) complexes using NMR and IR spectroscopy, as well as X-ray crystallography is described.
Co-reporter:Andrew C. Behrle, Alexander J. Myers, Pokpong Rungthanaphatsophon, Wayne W. Lukens, Charles L. Barnes and Justin R. Walensky
Chemical Communications 2016 - vol. 52(Issue 100) pp:NaN14375-14375
Publication Date(Web):2016/11/16
DOI:10.1039/C6CC08105C
The synthesis and characterisation of a rare U(III) alkyl complex, U[η4-Me2NC(H)C6H5]3, using the dimethylbenzylamine (DMBA) ligand has been accomplished. While attempting to prepare the U(IV) compound, reduction to the U(III) complex occurred. In the analogous Th(IV) system, C–H bond activation of a methyl group of one dimethylamine was observed yielding Th[η4-Me2NC(H)C6H5]2[η5-(CH2)MeNC(H)C6H5] with a dianionic DMBA ligand. The utility of these complexes as starting materials has been analyzed using a bulky dithiocarboxylate ligand to yield tetravalent actinide species.
Co-reporter:Andrew C. Behrle and Justin R. Walensky
Dalton Transactions 2016 - vol. 45(Issue 24) pp:NaN10049-10049
Publication Date(Web):2016/04/28
DOI:10.1039/C6DT00776G
The reactivity of thorium–phosphido and thorium–arsenido bonds was probed using tert-butyl isocyanide, tBuNC. Reaction of (C5Me5)2Th[E(H)R]2, E = P, As; R = 2,4,6-iPr3C6H2, 2,4,6-Me3C6H2 with tBuNC affords the first phosphaazaallene and arsaazaallene moieties with an f-element.














![Phenol, 2,4-bis(1,1-dimethylethyl)-6-[(8-quinolinylimino)methyl]-](http://img.cochemist.com/ccimg/252100/252048-83-2.png)
![Phenol, 2,4-bis(1,1-dimethylethyl)-6-[(8-quinolinylimino)methyl]-](http://img.cochemist.com/ccimg/252100/252048-83-2_b.png)


![Phenol, 2-[[[2,6-bis(1-methylethyl)phenyl]imino]methyl]-4,6-bis(1,1-dimethylethyl)-](/data/chemimg/135600/210882-24-9.png)
![Phenol, 2-[[[2,6-bis(1-methylethyl)phenyl]imino]methyl]-4,6-bis(1,1-dimethylethyl)-](/data/chemimg/135600/210882-24-9_b.png)


![Phenol, 2-[[[2,6-bis(1-methylethyl)phenyl]imino]methyl]-](http://img.cochemist.com/ccimg/187700/187605-57-8.png)
![Phenol, 2-[[[2,6-bis(1-methylethyl)phenyl]imino]methyl]-](http://img.cochemist.com/ccimg/187700/187605-57-8_b.png)
![Phosphine, [2,4,6-tris(1-methylethyl)phenyl]-](http://img.cochemist.com/ccimg/112500/112438-22-9.png)
![Phosphine, [2,4,6-tris(1-methylethyl)phenyl]-](http://img.cochemist.com/ccimg/112500/112438-22-9_b.png)


![Thorium,dichlorobis[(1,2,3,4,5-h)-1,2,3,4,5-pentamethyl-2,4-cyclopentadien-1-yl]-](http://img.cochemist.com/ccimg/67600/67506-88-1.png)
![Thorium,dichlorobis[(1,2,3,4,5-h)-1,2,3,4,5-pentamethyl-2,4-cyclopentadien-1-yl]-](http://img.cochemist.com/ccimg/67600/67506-88-1_b.png)