Co-reporter:Soumyaditya Mula, Stéphane Frein, Virginie Russo, Gilles Ulrich, Raymond Ziessel, Joaquín Barberá, and Robert Deschenaux
Chemistry of Materials 2015 Volume 27(Issue 7) pp:2332
Publication Date(Web):March 23, 2015
DOI:10.1021/cm503577y
We have designed a series of modular and fluorescent poly(arylester) dendrimers functionalized with cyanobiphenyl subunits and fluorescent borondipyrromethene (Bodipy) dyes. The green emitter is a Bodipy with four methyl groups, and the Bodipy dye possessing extended conjugation with two methyl and two vinyl groups acts as a red emitter. The design element of these architectures relates to a secondary amide linkers interposed between the conventional Bodipy and the dendrons. The second- and third-generation dendrimers give rise to nematic and/or smectic A phases, whereas the first-generation dendrimers show smectic A and C phases or an unidentified mesophase. The novel materials are highly fluorescent in solution and in the as-obtained powders but not in the mesophase. Dilution of the dendritic dyes with the nonfluorescent acid dendron in the solid phase shifted the fluorescence to higher energy, and demonstrated the presence of aggregates in the solid state. Mixing the red and blue materials in a solid phase led to the observation of effective electronic energy transfer from the red dye to the blue one. Increasing the proportion of the red dye (energy donor) from 1 to 250 molar with respect to the blue dye (energy acceptor) resulted in the observation of residual emission of the red dye in the solid state mixture. Increasing the proportion from 1 to 1000 resulted in equal emission in the 540 to 760 nm range.
Co-reporter:A. Mirloup, Q. Huaulmé, N. Leclerc, P. Lévêque, T. Heiser, P. Retailleau and R. Ziessel
Chemical Communications 2015 vol. 51(Issue 79) pp:14742-14745
Publication Date(Web):29 Jul 2015
DOI:10.1039/C5CC05095B
The synthesis and characterization of bis(difluoroboryl)-1,2-bis((1H-pyrrol-2-yl)methylene)hydrazone functionalized with two lateral vinyl-thienyl modules and exhibiting strong absorption in the 400–800 nm window in thin films are reported. Bulk heterojunction solar cells assembled with these dyes and a fullerene derivative (PC71BM), using very small quantities of the additive diiodooctane, give a power conversion efficiency as high as 4.3% with short-circuit current values of 10.9 mA cm−2, an open-circuit voltage of 0.7 V and external quantum efficiencies higher than 70% over a broad range of wavelengths (580 to 720 nm).
Co-reporter:Adela Nano, Maria Pia Gullo, Barbara Ventura, Nicola Armaroli, Andrea Barbieri and Raymond Ziessel
Chemical Communications 2015 vol. 51(Issue 16) pp:3351-3354
Publication Date(Web):14 Jan 2015
DOI:10.1039/C4CC09832C
The preparation and the photophysical behaviour of novel julolidine derivatives designed for displaying excited state intramolecular proton transfer (ESIPT) are reported. These dyes exhibit panchromatic photoluminescence covering the whole visible spectral range, both in organic solvents and in the solid state.
Co-reporter:Quentin Huaulmé, Antoine Mirloup, Pascal Retailleau, and Raymond Ziessel
Organic Letters 2015 Volume 17(Issue 9) pp:2246-2249
Publication Date(Web):April 21, 2015
DOI:10.1021/acs.orglett.5b00858
BOPHY dyes bearing bromo (in 5,5′-position) and iodo (in 4,4′-position) were synthesized. Double Knoevenagel reactions allow the extension of conjugation, resulting in an absorption above 625 nm. Selective cross-coupling reactions promoted by palladium(0) and microwave irradiation allow linking of a perylene module. These dyes are highly fluorescent, and the intramolecular cascade energy transfer from the perylene moiety to the BOPHY framework is almost quantitative, providing large virtual Stoke shifts (>5100 cm–1).
Co-reporter:Alicia Jacquemet;Sra Rihn;Gilles Ulrich;Pierre-Yves Renard;Anthony Romieu
European Journal of Organic Chemistry 2015 Volume 2015( Issue 8) pp:1664-1669
Publication Date(Web):
DOI:10.1002/ejoc.201500047
Abstract
Derivatives of 2-(2′-hydroxy-5′-dimethylaminobenzyl)-4,6-dimethylamino-1,3,5-triazine were synthesized by an original multistep protocol that afforded, after quaternization of the N,N-dimethylaminobenzyl moiety, water-soluble fluorescent dyes. These fluorophores exhibited excited-state intramolecular proton transfer and large Stokes shifts (ΔSS > 10000 cm–1). The phenol residue was masked by an enzyme-labile protecting group based on self-immolative para-hydroxybenzyl alcohol. Lipases were used to unveil the fluorescence under physiological conditions (phosphate-buffered saline, pH 7.2) without aggregation or precipitation of the fluorescent dyes.
Co-reporter:Anthony Harriman, Patrycja Stachelek, Alexandra Sutter and Raymond Ziessel
Photochemical & Photobiological Sciences 2015 vol. 14(Issue 6) pp:1100-1109
Publication Date(Web):16 Apr 2015
DOI:10.1039/C5PP00021A
A molecular dyad, comprising two disparate extended boron dipyrromethene (BODIPY) units, has been identified as a potential component of artificial light-harvesting arrays. Highly efficient, intramolecular electronic energy transfer takes place under illumination but there is some competition from light-induced electron transfer along the molecular axis. The primary energy acceptor has a somewhat shortened excited-state lifetime and reduced emission quantum yield due to charge transfer from a terminal amine residue, the latter being required for the molecular system to operate in organic solar cells. Under continuous illumination with simulated solar light, the dyad undergoes very slow decomposition. In a protic solvent, both BODIPY units degrade at the same rate via an autocatalytic process. The products, one of which is a protonated analogue of the donor, degrade further by independent routes. In aprotic solvents or thin plastic films, the acceptor BODIPY dye absorbing at lowest energy undergoes photochemical degradation as above but the donor is much more stable under these conditions. At each stage of degradation, the molecule retains the ability to sensitize an amorphous silicon solar cell and the overall turnover number with respect to absorbed photons exceeds 10 million. The optical properties of the target compound nicely complement those of the solar cell and sensitization helps to avoid Staebler–Wronski photo-degradation.
Co-reporter:Dr. Jacek Dobkowski;Dr. Pawe&x142; Wnuk;Dr. Joanna Buczy&x144;ska;Maria Pszona;Gra&x17c;yna Orzanowska;Dr. Denis Frath;Dr. Gilles Ulrich;Dr. Julien Massue;Dr. Sra Mosquera-Vázquez; Eric Vauthey; Czes&x142;aw Radzewicz; Raymond Ziessel; Jacek Waluk
Chemistry - A European Journal 2015 Volume 21( Issue 3) pp:1312-1327
Publication Date(Web):
DOI:10.1002/chem.201404669
Abstract
Differently substituted anils (Schiff bases) and their boranil counterparts lacking the proton-transfer functionality have been studied using stationary and femtosecond time-resolved absorption, fluorescence, and IR techniques, combined with quantum mechanical modelling. Dual fluorescence observed in anils was attributed to excited state intramolecular proton transfer. The rate of this process varies upon changing solvent polarity. In the nitro-substituted anil, proton translocation is accompanied by intramolecular electron transfer coupled with twisting of the nitrophenyl group. The same type of structure is responsible for the emission of the corresponding boranil. A general model was proposed to explain different photophysical responses to different substitution patterns in anils and boranils. It is based on the analysis of changes in the lengths of CN and CC bonds linking the phenyl moieties. The model allows predicting the contributions of different channels that involve torsional dynamics to excited state depopulation.
Co-reporter:Sébastien Azizi, Gilles Ulrich, Maud Guglielmino, Stéphane le Calvé, Jerry P. Hagon, Anthony Harriman, and Raymond Ziessel
The Journal of Physical Chemistry A 2015 Volume 119(Issue 1) pp:39-49
Publication Date(Web):December 4, 2014
DOI:10.1021/jp5078246
It is noted that, for a small series of 3,5-diacetyl-1,4-dihydrolutidine (DDL) derivatives and the corresponding Hantzsch esters, the presence of methyl groups at the 2,6-positions serves to extinguish fluorescence in solution but not in the solid state. Emission is weakly activated and affected by changes in solvent polarity. The latter situation arises because the optical transition involves intramolecular charge transfer. Calculations, both semiempirical and DFT, indicate that, in all cases, rotation of the carbonyl function is facile and that the dihydropyridine ring is planar. These calculations also indicate that the 2,6-methyl groups do not affect the generic structure of the molecule. It is proposed that illumination increases the molecular dipole moment and pushes electron density toward the carbonyl oxygen atom. Proton transfer can now occur from one of the methyl groups, leading to formation of a relatively low-energy, neutral intermediate, followed by a second proton transfer step that forms the enol. Reaction profiles computed for the ground-state species indicate that this route is highly favored relative to hydrogen transfer from the 4-position. The barriers for light-induced proton transfer are greatly reduced relative to the ground-state process but such large-scale structural transformations are hindered in the solid state. A rigid analogue that cannot form an enol is highly emissive in solution, supporting the conclusion that proton transfer is in competition to fluorescence in solution.
Co-reporter:Elodie Heyer and Raymond Ziessel
The Journal of Organic Chemistry 2015 Volume 80(Issue 13) pp:6737-6753
Publication Date(Web):June 16, 2015
DOI:10.1021/acs.joc.5b00917
In the present account we describe unsymmetrical triads constructed from extended borondipyrromethene (BODIPY) dyes, diketopyrrolopyrrole (DPP) dyes, and electron donor fragments based on triarylamine. The assemblages are such that each module maintains its individual optical and redox properties. The use of phenyl-alkyne-phenyl or phenyl-alkyne-thienyl spacer units is favorable for weak electronic interaction between the modules. The step-by-step linking of each module using palladium-catalyzed cross-coupling reactions provides both mono- and disubstituted derivatives, the latter obtained by passing in particular through a pivotal monosubstituted DPP building block with a reactive bromo substituent. Thus, grafting of a second dye occurs in a controlled manner, providing the target triads in good yields. This protocol allows also the synthesis of key intermediates and dyads, which appear useful for the understanding of the electrochemical and spectroscopic properties. All the dyes exhibit redox and optical properties suitable for cascade energy transfer and photoinduced electron transfer processes in appropriate solvents.
Co-reporter:Chuanjiang Qin;Antoine Mirloup;Nicolas Leclerc;Ashraful Islam;Ahmed El-Shafei;Liyuan Han
Advanced Energy Materials 2014 Volume 4( Issue 11) pp:
Publication Date(Web):
DOI:10.1002/aenm.201400085
Co-reporter:Elodie Heyer, Pascal Retailleau, and Raymond Ziessel
Organic Letters 2014 Volume 16(Issue 9) pp:2330-2333
Publication Date(Web):April 10, 2014
DOI:10.1021/ol500572t
Both symmetrical and unsymmetrical α-fused dithienyl-BODIPY dyes have been prepared by oxidative ring closure induced by anhydrous FeCl3. Extension of the π-system in the fused BODIPY leads to a progressive shift to 579 and 665 nm respectively for the absorption wavelength maxima of the mono- and difused dyes relative to the unfused species (λabs = 502 nm). Linking such dyes to an NIR emitting module provides a panchromatic chromophore with a large absorption cross section in the visible range associated with efficient intramolecular cascade energy transfer.
Co-reporter:Catherine E. McCusker, Delphine Hablot, Raymond Ziessel, and Felix N. Castellano
Inorganic Chemistry 2014 Volume 53(Issue 23) pp:12564-12571
Publication Date(Web):November 13, 2014
DOI:10.1021/ic502169a
The synthesis, structural characterization, and excited-state dynamics of series of diketopyrrolopyrrole (DPP) bridged homodinuclear Ir(III) and heterodinuclear Ir(III)/Pt(II) complexes is described. Steady-state and time-resolved photoluminescence along with transient absorption measurements were used to probe the nature of the emissive and long-lived excited states. Upon excitation into the 1DPP ligand-localized excited state in the presence of coordinated Ir(III) or Pt(II) metal centers, the intersystem crossing is enhanced, leading to a quenching of the 1DPP fluorescence and the formation of the long-lived (τ ≈ 30–40 μs) 3DPP excited state in all instances.
Co-reporter:Francesco Nastasi, Fausto Puntoriero, Scolastica Serroni, Sebastiano Campagna, Jean-Hubert Olivier and Raymond Ziessel
Dalton Transactions 2014 vol. 43(Issue 47) pp:17647-17658
Publication Date(Web):02 Jul 2014
DOI:10.1039/C4DT01127A
A Bodipy species bearing an acetyl-acetonate (acac) group, 3, has been prepared from a blue absorbing borondipyrromethene core bearing gallate substituted paraffin chains. Compound 3 chelates a Pt(II) center having an orthometalated 2-phenyl-pyridine anion (ppy) as an additional ligand, giving rise to a new bichromophoric Pt(II)-Bodipy species, 1. The absorption spectra, redox behavior and photophysical properties of 1, 3 and of the neutral Pt(II) compound 2, containing ppy and an acac derivative as ligands, have been studied. Compounds 3 and 2 are used as models for the Bodipy-based and the metal-based subunits of 1, respectively. The 3LC emission of 2 is fully quenched in 1, whereas the Bodipy fluorescence is only weakly reduced in 1 compared to 3, indicating weak interaction between the subunits. Two different charge-separated (CS) states have a role in the intercomponent excited state decays of 1. Notably, whereas in all the previously investigated bichromophoric metal(polypyridine)-Bodipy compounds, the light absorbed by the metal-based unit leads to population of the lowest-energy triplet Bodipy-based level, in 1 it contributes with high efficiency (>99%) to the Bodipy fluorescence. An efficient and formally forbidden 3LC to 1Bodipy energy transfer occurring by Förster mechanism is, unprecedently, the dominant 3LC decay process in 1.
Co-reporter:Antoine Mirloup, Nicolas Leclerc, Sandra Rihn, Thomas Bura, Rony Bechara, Anne Hébraud, Patrick Lévêque, Thomas Heiser and Raymond Ziessel
New Journal of Chemistry 2014 vol. 38(Issue 8) pp:3644-3653
Publication Date(Web):16 May 2014
DOI:10.1039/C4NJ00294F
In this work we explore the synthesis of an extended 4,4-difluoro-4-bora-3a,4a-diaza-s-indacene dye (BODIPY) engineered from thiophene–benzothiadiazole–thiophene modules linked in the 3,5-substitution positions. We found that this highly soluble dye absorbs up to 800 nm in solution and up to 900 nm in thin films. An effective charge transfer absorption band was found at around 479 nm. The hybrid dye emits at 778 nm with a quantum yield of about 6%. Similar electrochemical and optical gaps were determined about 1.36 eV. When deposited in thin films the dye exhibits an ambipolar nature with well-balanced hole and electron mobilities. Bulk heterojunction solar cells based upon this dye blended with [6,6]phenylC61or71butyricacid methylester (PC61BM or PC71BM) provide a power conversion efficiency of about 1.26% upon a mild thermal annealing.
Co-reporter:Karima Benelhadj;Julien Massue;Pascal Retailleau;Siwar Chibani;Boris Le Guennic;Denis Jacquemin;Gilles Ulrich
European Journal of Organic Chemistry 2014 Volume 2014( Issue 32) pp:7156-7164
Publication Date(Web):
DOI:10.1002/ejoc.201402806
Abstract
This paper describes the synthesis of π-conjugated fluorophores based either on an anil or a benzoxazole scaffold incorporating a rigid 2-(6′-hydroxy-5′-benzofuryl) fragment. Their subsequent coordination to a BF2 motif led to highly luminescent BIII complexes called boranils or HBBO borate complexes, respectively. All the new compounds were characterized by NMR spectroscopy, mass spectrometry, and elemental analysis. The study of their optical properties in solution revealed two distinct photophysical behaviors depending on the substitution. Complexes bearing a strong electron-donating p-nBu2NC6H4 group displayed a marked internal charge transfer (ICT) leading to a pronounced solvatochromism with λem ranging from 507 to 663 nm and quantum yields of up to 87 %. Alternatively, borate complexes functionalized with p-tBuC6H4 or p-OMeC6H4 displayed fluorescence in the visible range that is not influenced by the nature of the solvent. Although the emission of the boranils was quenched in the solid state, HBBO borate complexes displayed intense fluorescence from 489 to 567 nm and quantum yields of up to 23 %. Finally, the excited states of a selection of borate complexes were modeled by using time-dependent DFT, which allowed calculation of the dipole moments of the dyes in their excited states.
Co-reporter:Alexandra Sutter, Pascal Retailleau, Wei-Ching Huang, Hao-Wu Lin and Raymond Ziessel
New Journal of Chemistry 2014 vol. 38(Issue 4) pp:1701-1710
Publication Date(Web):07 Feb 2014
DOI:10.1039/C3NJ01436C
This work explores the synthesis of extended dipyrromethene dyes engineered from thiophene-linked triphenylamine modules in order to favor pronounced charge transfer within the dyes and to enable their use in solar cells. We found that these soluble dyes absorb up to 760 nm in solution and 850 nm in thin films. An effective charge transfer absorption band was found at around 460 nm. In the crystal lattice, molecules are organized in layers separated by 3.50 Å with pronounced π–π stacking and S⋯S interactions. The electroactivity of the dyes indicates that electron injection in PC71BM is feasible. Bulk heterojunction solar cells based upon dyes 1 and 2 in an optimized device structure ITO/Ca/1 or 2:PC71BM/MoO3/Ag provide a power conversion efficiency of about 1.5% after thermal annealing.
Co-reporter:Julien Massue, Karima Benelhadj, Siwar Chibani, Boris Le Guennic, Denis Jacquemin, Pascal Retailleau, Gilles Ulrich, Raymond Ziessel
Tetrahedron Letters 2014 Volume 55(Issue 30) pp:4136-4140
Publication Date(Web):23 July 2014
DOI:10.1016/j.tetlet.2014.06.002
The multi-step synthesis, structural and optical properties of original luminescent borate complexes derived from 2-(2′-hydroxybenzofuran)benzoxazole (HBBO) are reported. Functionalization at position 3 of the benzofuran ring was readily achieved through an electrophilic cyclization key step followed by a Sonogashira cross-coupling reaction. The optical properties of the resulting boron difluoride dyes highlight different photophysical behaviors depending on the nature of the substitution at position 3 of the benzofuran core (tBu-phenylacetylene or NnBu2-phenylacetylene). The NnBu2-phenylacetylene moiety favors a sizeable intramolecular charge transfer as evidenced by a strong solvatochromism; a feature further confirmed by ab initio calculations.
Co-reporter:Quentin Huaulmé, Elsa Cece, Antoine Mirloup, Raymond Ziessel
Tetrahedron Letters 2014 Volume 55(Issue 35) pp:4953-4958
Publication Date(Web):27 August 2014
DOI:10.1016/j.tetlet.2014.07.026
The engineering of photoactive arrays built from a flat, functionalized triazatruxene (TAT) platform is described. The primary synthetic strategy involved the step by step connection of one, two or three bis(thienyl)diketopyrrolopyrrole (DPP) modules. Subsequent bromination of the pendent thiophene ring was not selective and provided a mixture of regioisomers. However, selective grafting of boron dipyrromethene (Bodipy) units via Pd-catalysed cross couplings enabled the construction of TAT/DPP/Bodipy arrays. As well, direct coupling of two green F-Bodipy units to dibromoTAT provided a substrate suitable for reaction with hydroxyl-propargyl-substituted red Bodipy dyes to give ready access to O-Bodipy linked multichromophoric systems. All the new dyes displayed strong absorption in the near-UV and visible region of the solar spectra (400–750 nm), with intramolecular cascade energy transfer enabling photon concentration and fluorescence at approximately 740 nm.
Co-reporter:Thomas Roland ; Elodie Heyer ; Li Liu ; Adrian Ruff ; Sabine Ludwigs ; Raymond Ziessel ;Stefan Haacke
The Journal of Physical Chemistry C 2014 Volume 118(Issue 42) pp:24290-24301
Publication Date(Web):September 22, 2014
DOI:10.1021/jp507474r
The photoreactions that occur in a molecular triad composed of derivatives of boron-dipyrromethene (BOD), diketopyrrolopyrrole (DPP), and of triphenylamine (TPA) specifically designed for the use as a possible donor material for organic solar cells are studied by ultrafast fluorescence and transient absorption spectroscopy with hyper spectral coverage (300–900 nm). While the latter is often sufficient to reveal the reaction scenario in mono- or bicomponent organic molecules, the overlap of ground state absorption and fluorescence spectra of the components present in the TPA-DPP-BOD triad requires a careful and accurate characterization of the excited state differential absorption spectra and kinetics of the individual isolated moieties. In addition, the absorption spectra of the TPA cation and the DPP and BOD anions are determined by in situ spectroelectrochemical experiments. Picosecond fluorescence demonstrates efficient excited state quenching of both DPP and BOD, with downhill energy transfer from TPA occurring in a subpicosecond time scale, leading to its complete excited state quenching. Additionally, an analysis of the multiexponential fluorescence decay indicates the existence of reactive (quenched) and nonreactive (radiative) subpopulations of DPP and BOD, most probably due to structural heterogeneities. In order to treat this complexity of multiple reaction pathways, the transient absorption data are analyzed by a combination of global fitting, providing decay-associated differential spectra (DADS), and a reconstitution of these DADS by linear combinations of the characteristic difference spectra of all molecular species and excited states. This allows us to disentangle the photoreactions that the TPA, DPP and BOD excited state subpopulations undergo on time scales ranging from 200 fs to a few ns, and to clarify the contribution of TPA as an electron donor in the formation of an intramolecular charge transfer (CT) state. DPP has a dual role as an ancillary light-harvesting unit capable of performing ultrafast energy transfer to BOD, and as an electron acceptor for the CT state. The latter has a remarkably long lifetime of 0.5 ns. We conclude that the triad could act as a very efficient electron-donor material in a blend with commonly used acceptors such as phenyl-C61-butyric acid methyl ester (PCBM).
Co-reporter:Arnaud Poirel, Antoinette De Nicola, and Raymond Ziessel
The Journal of Organic Chemistry 2014 Volume 79(Issue 23) pp:11463-11472
Publication Date(Web):November 10, 2014
DOI:10.1021/jo502068u
Fluorescent dithienyl-borondipyrromethene (BODIPY) dyes formylated in the β′-position (2b, 2c) have been treated with l-cysteine to provide thiazolidine derivatives. N-Protection of the thiazolidine unit by ethoxycarbonylation facilitated isolation of the two major diasteroisomers 6 and 7. These stereoisomers have been fully characterized by 1H NMR spectroscopy, allowing assignment of their stereochemistry as 2R,4R,aS and 2S,4R,aR, respectively. The optical properties of the thiazolidine dyes differ markedly in both absorption (λabs = 612 nm for 6 and 615 nm for 7) and emission (λem = 669 nm, ΦF = 62% for 6 and λem = 672 nm, ΦF = 19% for 7) from those of the BODIPY-carboxaldehydes 2b (λabs = 643 nm and λem = 719 nm, ΦF = 26%) and 2c (λabs = 636 nm and λem = 710 nm, ΦF = 36%). In a mixed solvent [phosphate buffer saline (PBS), pH = 7.4/ethanol 1:9], the fluorescence response of the dyes in the presence of l-cysteine is slow, but a ratiometric detection process in the therapeutic window (650 to 800 nm) is evident.
Co-reporter:Arnaud Poirel;Dr. Pascal Retailleau;Dr. Antoinette De Nicola;Dr. Raymond Ziessel
Chemistry - A European Journal 2014 Volume 20( Issue 5) pp:
Publication Date(Web):
DOI:10.1002/chem.201490014
Co-reporter:Dr. Denis Frath;Dr. Julien Massue;Dr. Gilles Ulrich;Dr. Raymond Ziessel
Angewandte Chemie International Edition 2014 Volume 53( Issue 9) pp:2290-2310
Publication Date(Web):
DOI:10.1002/anie.201305554
Abstract
Multidisciplinary research on novel organic luminescent dyes is propelled by potential applications in plastic electronics and biomedical sciences. The construction of sophisticated fluorescent dyes around a tetrahedral boron(III) center is a particular approach that has fueled the creativity of chemists. Success in this enterprise has been readily achieved with simple synthetic protocols, the products of which display unusual spectroscopic behavior. This account is a critical review of recent advances in the field of boron(III) complexes (excluding BODIPYs and acetylacetonate boron complexes) involving species displaying similar coordination features, and we outline their potential development in several disciplines.
Co-reporter:Dr. Gilles Ulrich;Dr. Alberto Barsella;Dr. Alex Boeglin;Dr. Songlin Niu;Dr. Raymond Ziessel
ChemPhysChem 2014 Volume 15( Issue 13) pp:2693-2700
Publication Date(Web):
DOI:10.1002/cphc.201402123
Abstract
A set of linear and dissymmetric BODIPY-bridged push–pull dyes are synthesized. The electron-donating substituents are anisole and dialkylanilino groups. The strongly electron-accepting moiety, a 1,1,4,4-tetracyanobuta-1,3-diene (TCBD) group, is obtained by insertion of an electron-rich ethyne into tetracyanoethylene. A nonlinear push–pull system is developed with a donor at the 5-position of the BODIPY core and the acceptor at the 2-position. All dyes are fully characterized and their electrochemical, linear and nonlinear optical properties are discussed. The linear optical properties of dialkylamino compounds show strong solvatochromic behavior and undergo drastic changes upon protonation. The strong push–pull systems are non-fluorescent and the TCBD-BODIPY dyes show diverse photochemistry and electrochemistry, with several reversible reduction waves for the tetracyanobutadiene moiety. The hyperpolarizability μβ of selected compounds is evaluated using the electric-field-induced second-harmonic generation technique. Two of the TCBD-BODIPY dyes show particularly high μβ (1.907 μm) values of 2050×10−48 and 5900×10−48 esu. In addition, one of these dyes shows a high NLO contrast upon protonation–deprotonation of the donor residue.
Co-reporter:Karima Benelhadj;Wenziz Muzuzu;Dr. Julien Massue;Dr. Pascal Retailleau;Dr. Azzam Charaf-Eddin;Dr. Adèle D. Laurent; Denis Jacquemin;Dr. Gilles Ulrich;Dr. Raymond Ziessel
Chemistry - A European Journal 2014 Volume 20( Issue 40) pp:12843-12857
Publication Date(Web):
DOI:10.1002/chem.201402717
Abstract
The synthesis, structural, and photophysical properties of a new series of original dyes based on 2-(2′-hydroxybenzofuran)benzoxazole (HBBO) is reported. Upon photoexcitation, these dyes exhibit intense dual fluorescence with contribution from the enol (E*) and the keto (K*) emission, with K* being formed through excited-state intramolecular proton transfer (ESIPT). We show that the ratio of emission intensity E*/K* can be fine-tuned by judiciously decorating the molecular core with electron-donating or -attracting substituents. Push–pull dyes 9 and 10 functionalized by a strong donor (nNBu2) and a strong acceptor group (CF3 and CN, respectively) exhibit intense dual emission, particularly in apolar solvents such as cyclohexane in which the maximum wavelength of the two bands is the more strongly separated. Moreover, all dyes exhibit strong solid-state dual emission in a KBr matrix and polymer films with enhanced quantum yields reaching up to 54 %. A wise selection of substituents led to white emission both in solution and in the solid state. Finally, these experimental results were analyzed by time-dependent density functional theory (TD-DFT) calculations, which confirm that, on the one hand, only E* and K* emission are present (no rotamer) and, on the other hand, the relative free energies of the two tautomers in the excited state guide the ratio of the E*/K* emission intensities.
La synthèse, les études structurales et photophysiques d’une nouvelle série de fluorophore basée sur une architecture 2-(2′-hydroxybenzofuran)benzoxazole (HBBO) sont décrites. Après photoexcitation, ces composés présentent une intense émission duale issue de l’émission simultanée des états excités des formes énol (E*) et kéto (K*). La forme K* est formée après un transfert de proton intramoléculaire dans l’état excité (ESIPT). Nous montrons que le ratio des intensités d’émission E*/K* peut être finement modulé par un choix judicieux de décoration sur le squelette moléculaire avec des substituents fortement donneur ou attracteur. Les colorants push–pull 9 et 10 fonctionnalisés par un donneur fort (nNBu2) sur la partie phénol et un accepteur fort (CF3 et CN respectivement) sur la partie benzoxazole présentent une émission duale intense, et ceci plus particulièrement dans des solvants apolaires tels que le cyclohexane, cas où la séparation entre les deux pics d’émission est la plus importante. Tous ces composés présentent en plus, une importante émission à l’état solide, que ce soit dans une matrice de KBr ou dans un film polymère, avec un rendement quantique de fluorescence allant jusqu’à 54 %. Une sélection pertinente de substituents permet d’obtenir une émission blanche en solution ou à l’état solide. Enfin, ces résultats expérimentaux ont été confirmés par TD-DFT (time-dependent density functional theory); ces études montrent non seulement que seules les émissions des espèces E* et K* sont présentes durant le processus (pas de rotamère), mais aussi que l’énergie libre relative des deux tautomères dans l’état excite guide le ratio des intensités d’émission E*/K*.
Co-reporter:Arnaud Poirel;Dr. Pascal Retailleau;Dr. Antoinette De Nicola;Dr. Raymond Ziessel
Chemistry - A European Journal 2014 Volume 20( Issue 5) pp:1252-1257
Publication Date(Web):
DOI:10.1002/chem.201303988
Abstract
Thesynthesis of three red-emitting and water-soluble thienyl-BODIPYs has beenachieved. The trimethyl(propargyl)ammonium group was chosen as a vector forwater solubility. One or two cationic arms were introduced either on the2-position of the thienyl unit or on the 4-position on the boron atom. Thesedyes have pronounced absorption around 600 nm and intense emission at 650 nmwith quantum yield of about 60% in water. Grafting of such BODIPYs via a flexible arm to BSA is veryefficient, allowing attachment of 1 to 30 labels in a controlled manner. Very strong fluorescence (quantum yield 56%)without aggregation of the dye at a low loading ratio (1:5 BSA/label) in PBSbuffer is measured.
Co-reporter:Arnaud Poirel;Dr. Pascal Retailleau;Dr. Antoinette DeNicola;Dr. Raymond Ziessel
Chemistry - A European Journal 2014 Volume 20( Issue 5) pp:
Publication Date(Web):
DOI:10.1002/chem.201304847
Abstract
Invited for the cover of this issue is the group of Raymond Ziessel at the University of Strasbourg. The image depicts the bright red fluorescence of a probe, both free in aqueous solution and when covalently linked to a model protein, such as BSA. Read the full text of the article at 10.1002/chem.201303988.
Co-reporter:Adela Nano;Dr. Raymond Ziessel;Patrycja Stachelek;Dr. Mohammed A. H. Alamiry; Dr. Anthony Harriman
ChemPhysChem 2014 Volume 15( Issue 1) pp:177-186
Publication Date(Web):
DOI:10.1002/cphc.201300805
Abstract
The photophysical properties of a prototypic donor–acceptor dyad, featuring a conventional boron dipyrromethene (Bodipy) dye linked to a dicyanovinyl unit through a meso-phenylene ring, have been recorded in weakly polar solvents. The absorption spectrum remains unperturbed relative to that of the parent Bodipy dye but the fluorescence is extensively quenched. At room temperature, the emission spectrum comprises roughly equal contributions from the regular π, π* excited-singlet state and from an exciplex formed by partial charge transfer from Bodipy to the dicyanovinyl residue. This mixture moves progressively in favor of the locally excited π, π* state on cooling and the exciplex is no longer seen in frozen media; the overall emission quantum yield changes dramatically near the freezing point of the solvent. The exciplex, which has a lifetime of approximately 1 ns at room temperature, can also be seen by transient absorption spectroscopy, in which it decays to form the locally excited triplet state. Under applied pressure (P<170 MPa), formation of the exciplex is somewhat hindered by restricted rotation around the semirigid linkage and again the emission profile shifts in favor of the π, π* excited state. At higher pressure (170<P<550 MPa), the molecule undergoes reversible distortion that has a small effect on the yield of π, π* emission but severely quenches exciplex fluorescence. In the limiting case, this high-pressure effect decreases the molar volume of the solute by approximately 25 cm3 and opens a new channel for nonradiative deactivation of the excited-state manifold.
Co-reporter:Raymond Ziessel ; Gilles Ulrich ; Alexandre Haefele ;Anthony Harriman
Journal of the American Chemical Society 2013 Volume 135(Issue 30) pp:11330-11344
Publication Date(Web):July 3, 2013
DOI:10.1021/ja4049306
An artificial light-harvesting array, comprising 21 discrete chromophores arranged in a rational manner, has been synthesized and characterized fully. The design strategy follows a convergent approach that leads to a molecular-scale funnel, having an effective chromophore concentration of 0.6 M condensed into ca. 55 nm3, able to direct the excitation energy to a focal point. A cascade of electronic energy-transfer steps occurs from the rim to the focal point, with the rate slowing down as the exciton moves toward its ultimate target. Situated midway along each branch of the V-shaped array, two chromophoric relays differ only slightly in terms of their excitation energies, and this situation facilitates reverse energy transfer. Thus, the excitation energy becomes spread around the array, a situation reminiscent of a giant holding pattern for the photon that can sample many different chromophores before being trapped by the terminal acceptor. At high photon flux under conditions of relatively slow off-load to a device, such as a solar cell, electronic energy transfer encounters one or more barriers that hinder forward progress of the exciton and thereby delays arrival of the second photon. Preliminary studies have addressed the ability of the array to function as a sensitizer for amorphous silicon solar cells.
Co-reporter:Karima Benelhadj, Julien Massue, Pascal Retailleau, Gilles Ulrich, and Raymond Ziessel
Organic Letters 2013 Volume 15(Issue 12) pp:2918-2921
Publication Date(Web):June 6, 2013
DOI:10.1021/ol400849a
The synthesis, structural, and optical properties of a series of luminescent N-alkylated 2-(2′-hydroxyphenyl)benzimidazole (HBI) or N-arylated 9,10-phenanthroimidazole (HPI) borate complexes are described. The optical properties of these complexes as well as their corresponding ligands were evaluated in solution and the solid state. Efficient emission in the blue-green region was obtained with quantum yields up to 91% in CH2Cl2 and 27% in the solid state. These emissions originate from excited state intramolecular proton transfer (ESIPT) for the ligands and from a singlet excited state for the borate complexes.
Co-reporter:Denis Frath, Arnaud Poirel, Gilles Ulrich, Antoinette De Nicola and Raymond Ziessel
Chemical Communications 2013 vol. 49(Issue 43) pp:4908-4910
Publication Date(Web):10 Apr 2013
DOI:10.1039/C3CC41555D
Complexation of iminocoumarin derivatives with BF3·OEt2 provides novel N⁁N boron(III) dyes exhibiting high absorption coefficients and quantum yields as great as 81%. The excellent chemical stability of these dyes enables the grafting of Boranil or Bodipy units to give derivatives in which the Borico subunit can act either as an energy acceptor or as an antenna for a red emitting fluorophore.
Co-reporter:M. Teresa Indelli, Thomas Bura, and Raymond Ziessel
Inorganic Chemistry 2013 Volume 52(Issue 6) pp:2918-2926
Publication Date(Web):March 7, 2013
DOI:10.1021/ic302222q
Two iridium(III) complexes displaying for one a high HOMO–LUMO gap and for the other a weaker gap were linked in a controlled and logical manner to closo-p-carborane spacers. The bridging ligand is composed of 5-ethynyl-2,2′-bipyridine units, and the peripherical Ir-ligands are orthometalated 2′,4′-difluoro-2-phenylpyridine (dfppy) (λabs at 400 nm for the “Ir(dfppy)2(bpy′)”) for the energy donor fragment and dibenzo[a,c]phenazine (dbpz) (λabs at 525 nm for “Ir(dbpz)2(bpy′)”) for the energy acceptor fragment.Redox, spectroscopic, and photophysical properties for models and the donor–carborane–acceptor complex were determined. Efficient energy transfer from the “Ir(dfppy)2(bpy′)” moiety to the “Ir(dbpz)2(bpy′)” fragment is occurring with a rate constant of 3.3 × 108 s–1 despite weak electronic coupling through the inert p-carborane spacer. From flash photolysis experiments it is shown that, by excitation of the donor, a low lying triplet state localized on the acceptor bridging ligand side is formed which decays by conversion to the 3MLCT of the acceptor fragment which phosphoresces at 644 nm.
Co-reporter:Thomas Bura, Maria Pia Gullo, Barbara Ventura, Andrea Barbieri, and Raymond Ziessel
Inorganic Chemistry 2013 Volume 52(Issue 15) pp:8653-8664
Publication Date(Web):July 19, 2013
DOI:10.1021/ic400809a
Here we report on the synthesis, characterization, and photophysics of multichromophoric arrays based on a triptycene scaffold that acts as a bridging ligand for Ir(III) and Os(II) satellite active components. The triptycene scaffold not only furnishes a rigid star-shaped 3D displacement of the metallic units in space but also plays an active role in the energy cascade. The transition metal complexes have been designed in order to display an ideal cascade in their lowest excited state energy levels. For this purpose, a novel Ir(III) complex containing two dbpz (dibenzo[a,c]phenazine) ligands (Ir) has been synthesized. The key step in the synthesis of the array was the final cross-coupling between the mixed complex IrF–Os and Ir, providing the target heterotrinuclear complex IrF–Ir–Os. The photophysical properties of models confirmed the appropriate energy displacement of the single chosen active units, in the order triptycene > IrF > Ir > Os, and fast and efficient energy transfer processes leading to the final population of the Os-based triplet level have been evidenced. The reported arrays can be considered as efficient antenna systems with an absorption range extending up to 700 nm, where the triptycene bridging ligand provides both a structural and a photophysical function.
Co-reporter:Thomas Bura, Pascal Retailleau, Maria Teresa Indelli and Raymond Ziessel
Dalton Transactions 2013 vol. 42(Issue 13) pp:4544-4551
Publication Date(Web):07 Jan 2013
DOI:10.1039/C2DT32538A
New Ir(III) complexes involving N,C-chelating difluorophenyl-pyridine (dfppy) or dibenzo[a,c]phenazine (dbpz) ligands along with either N,N-bound 5-ethynyl-2,2′-bipyridine (e-bpy) or CO + Cl co-ligands have been obtained as [Ir(dfppy)2(e-bpy)]PF6, [Ir(dbpz)2(e-bpy)]PF6 and cis-[Ir(dbpz)2(Cl)(CO)]. A single-crystal X-ray diffraction study of cis-[Ir(dbpz)2(CO)Cl] has shown the Ir(III) centre to adopt a distorted octahedral coordination geometry with cis-CO/Cl and trans-N,N configurations. Pronounced π–π stacking interactions involving different dibenzo[a,c]phenazine units are evident. Electronic absorption and luminescence spectroscopy at 298 K and 77 K, along with cyclic voltammetry were used to study the three complexes. Excited state lifetimes varied from 1.4 to 2.9 μs at rt with quantum yields ranging from 10.2 to 0.7%. With 5-ethynyl-2,2′-bipyridine in the first coordination sphere, the lowest emitting state is that associated with the MLCT band, whereas in the bis-phenazine case the emission is ligand-centred. Importantly, the emission wavelength can be tuned from 552 to 640 nm by changing the conjugation length of the N,C ligand.
Co-reporter:Maria Pia Gullo, Julie Batcha Seneclauze, Barbara Ventura, Andrea Barbieri and Raymond Ziessel
Dalton Transactions 2013 vol. 42(Issue 48) pp:16818-16828
Publication Date(Web):01 Oct 2013
DOI:10.1039/C3DT52007B
This paper describes expeditious stepwise synthesis of polynuclear complexes based on heteroleptic iridium(III) and osmium(II) fragments linked to a central Pt(II) module via a spirobifluorene-bridge using a strategy based on the construction of preformed complexes. The luminescence features of the final multi-chromophoric array, i.e. a tetrad consisting of spirobifluorene-bridged Pt, Ir and Os complexes, have been studied by comparison with the features of reference complexes bearing two identical luminophores (Ir or Os) at the periphery. The 3MPtLCT and 3LC states of the Pt and spiro ligand undergo fast energy transfer into the 3MIrLCT or the 3MOsLCT state in the Pt–M2 (M = Ir or Os) arrays, whereas the 3LC and the 3MPtLCT states function as energy reservoirs for the metal excited states close in energy, resulting in a pronounced increase of the excited state lifetimes of these arrays. The tetrad efficiently works as an antenna system where the collected light energy is transferred to the Os unit acting as the final collector.
Co-reporter:Soumyaditya Mula, Delphine Hablot, Krishna K. Jagtap, Elodie Heyer and Raymond Ziessel
New Journal of Chemistry 2013 vol. 37(Issue 2) pp:303-308
Publication Date(Web):04 Sep 2012
DOI:10.1039/C2NJ40569E
We have devised new diketopyrrolopyrrole dyes soluble in polar solvents. The colors of the dyes have been changed by substituting the 3,6-phenyl residues for thiophene groups. The solubility in polar solvents has been improved by alkylation of a dimethylaminopropyne fragment with a 1,3-sultone giving rise to sulfobetaine moieties. These dyes exhibit strong and broad absorption in the visible range (488 nm for the phenyl-DPP and 578 nm for the thiophene-DPP) and intense fluorescence at, respectively, 554 nm (ΦF 0.36) and 609 nm (ΦF 0.19). The phenyl-DPP dye exhibits a maximum broad band lasing efficiency of 14.6% and a maximum narrow band lasing efficiency of 9.5% with a wide tunable range (554 nm to 616 nm) on excitation with a second harmonic of a Q-switched Nd:YAG (532 nm) laser in MeOH/H2O. The lasing efficiency remains unaffected for hours. The thiophene-DPP dye does not show any lasing action.
Co-reporter:Julien Massue;Gilles Ulrich
European Journal of Organic Chemistry 2013 Volume 2013( Issue 25) pp:5701-5709
Publication Date(Web):
DOI:10.1002/ejoc.201300616
Abstract
This article describes the multistep synthesis and photophysical properties of three highly fluorescent dyes based on the 2-(2′-hydroxyphenyl)benzoxazole (HBO) scaffold and their resulting chelation to a BF2 fragment. These dyes possess functionalization at the 3,5-positions of the phenol ring with an ethynyl-extended fragment bearing TMS, p-tBuC6H4, or p-NnBu2C6H4 groups. All of the new compounds were characterized by NMR spectroscopy, mass spectrometry, and elemental analysis. The optical properties of the HBO dyes reveal the presence of enol and keto bands as a result of a strong excited-state intramolecular proton transfer (ESIPT). This ESIPT process is highly dependent on the nature of the electronic substituents and the polarity of the solvent. Upon coordination to a BF2 fragment, typical singlet emission is observed with λem ranging from 439 to 553 nm and quantum yields from 0.03 to 0.36. If aromatic amines are involved, strong aggregates are observed that could be dissociated upon protonation of the lone electron pair.
Co-reporter:Julien Massue, Pascal Retailleau, Gilles Ulrich and Raymond Ziessel
New Journal of Chemistry 2013 vol. 37(Issue 4) pp:1224-1230
Publication Date(Web):30 Jan 2013
DOI:10.1039/C3NJ41052H
Triphenylborane (BPh3) has been successfully coordinated to five π-conjugated aromatic systems containing a N∧O bidentate 2-(2′-hydroxyphenyl)benzoxazole (HBO) ligand. Complexes 6–8 are directly substituted on the phenolic side of the HBO core while the structure of derivatives 9–10 includes a 4-dibutylaminophenyl module linked through an ethynyl fragment, at position 4 or 5. The crystal structure of complexes 8 and 10 reveals different solid-state molecular packing depending on the substitution, a herringbone molecular packing being observed for complex 8 while the dibutylamino fragment present in complex 10 is in favour of a lamellar structure. The optical properties are highly dependent on the nature and the position of substituents. Solvatochromic charge-transfer emissions are observed for substitution at position 3 or 5 while singlet emission is favoured when position 4 is functionalized. Solid-state fluorescence reveals that complex 8 possesses red-shifted emission when dispersed in a KBr matrix.
Co-reporter:Elodie Heyer, Raymond Ziessel
Tetrahedron Letters 2013 Volume 54(Issue 26) pp:3388-3393
Publication Date(Web):26 June 2013
DOI:10.1016/j.tetlet.2013.04.073
3-Bromodibenzo[g,p]chrysene has been synthesized in a one-pot reaction using phenyliodine(III) bis(trifluoroacetate) (PIFA) as an oxidant and a Lewis acid. The bromo function was easily converted to the cyano, alkyne, and formyl derivatives. Bodipy dyes have either been constructed from the chrysene–aldehyde or by cross coupling reactions with the chrysene ethynyl derivatives. The linking of diketopyrrolopyrrole (DPP) fragments was realized using the chrysene–ethynyl compound or with the in situ prepared chrysene–borolane derivatives. The novel chrysene dyes are highly fluorescent in apolar solvents and were used as input energy in intramolecular energy transfer processes with covalently linked DPP or Bodipy dyes. All mixed dyes are redox active and each module is clearly identified by its redox behavior. Little electronic interaction is evidenced whatever the nature of the dyes and spacer.
Co-reporter:Antoine Mirloup, Pascal Retailleau, Raymond Ziessel
Tetrahedron Letters 2013 Volume 54(Issue 33) pp:4456-4462
Publication Date(Web):14 August 2013
DOI:10.1016/j.tetlet.2013.06.039
The synthesis of photoactive arrays consisting of Bodipy scaffoldings is described. The synthetic strategy involved first the construction of functionalised modules, then the use of specific Knoevenagel reactions. Boron-protection is required to avoid self-condensation. Comparison of thienylBodipy dyes with their phenyl analogues showed them to be both less chemically stable and more weakly fluorescent. In the multichromophoric species, efficient cascade energy transfer occurs upon absorption in the highest energy unit. The redox activity of the mixed dyes is analysed in terms of the behaviour of specific reference compounds.
Co-reporter:Thomas Bura;Dr. Francesco Nastasi;Dr. Fausto Puntoriero;Dr. Sebastiano Campagna;Dr. Raymond Ziessel
Chemistry - A European Journal 2013 Volume 19( Issue 27) pp:8900-8912
Publication Date(Web):
DOI:10.1002/chem.201300413
Abstract
A series of new compounds in which various Bodipy dyes are grafted logically on triptycene rigid structures are synthesized and characterized, and their absorption spectra and photophysical properties are studied, also by pump-probe transient absorption spectroscopy. The studied compounds are: the mono-Bodipy species TA, TB, and TC (where A, B, and C identify different Bodipy subunits absorbing and emitting at different wavelengths), the multichromophore species TA3, which bears three identical A subunits, and the three multichromophoric species TAB, TBC, and TABC, all of them containing at least two different types of Bodipy subunits. The triptycene moiety plays the role of a rigid scaffold, keeping the various dyes at predetermined distances and allowing for a three-dimensional structural arrangement of the multichromophoric species. The absorption spectra of the multichromophoric Bodipy species are essentially additive, indicating that negligible inter-chromophoric interaction takes place at the ground state. Luminescence properties and transient absorption spectroscopy indicate that a very fast (on the picosecond time scale) and efficient photoinduced energy transfer occurs in all the multi-Bodipy species, with the lower-energy Bodipy subunits of each multi-Bodipy compounds playing the role of an electronic energy collector. In TAB, an energy transfer from the A-type Bodipy subunit to the B-type one takes place with a rate constant of 1.6×1010 s−1, whereas in TBC an energy transfer from the B-type Bodipy subunit to the C-type subunit is bi-exponential, exhibiting rate constants of 1.7×1011 and 1.9×1010 s−1; the possible presence of different conformers with different donor–acceptor distances in this bichromophoric species is proposed to cause the bi-exponential energy-transfer process. Interpretation of the intricate energy-transfer pathways occurring in TABC is made with the help of the processes identified in the bichromophoric compounds. In all cases, the measured energy-transfer rate constants agree with a Förster mechanism for the energy-transfer processes.
Une nouvelle séries de sondes Bodipy ont été construites sur une plateforme triptycene T préalablement fonctionnalisée. L’approche synthétique a consistée à construire d’abord un triptycene modifié avec trois Bodipy identiques TA3 et à sélectivement transformer les unités existantes en des composés ayant des couleurs différentes (A, B, et C). De nombreux composés modèles ont ainsi pu être préparés tels que TA, TB, TC, TAB, TBC. La molécule cible TABC possède un ensemble de chromophores capable de transférer leur énergie sur la sonde de plus basse énergie et ses processus en cascade permettent de concentrer les photons sur un émetteur unique localisé à basse énergie. Le spectre d’absorption de TABC est une combinaison linéaire de l’absorption individuelle des quatre modules démontrant une faible interaction dans l’état fondamental. Les transferts d’énergie sont extrêmement rapides (picoseconde) et ont été étudiés par absorption transitoire. Plus précisément dans le modèle TAB le transfert d’énergie de A vers B a lieu en 60 ps tandis que le transfert de B vers C dans TBC se fait en 6 et 50 ps. La présence de différents conformères a été invoquée pour expliquer le déclin bi-exponentiel. L’interprétation des phénomènes de transfert d’énergie dans TABC a été faite à l’aide des composés modèles et donne une image réaliste de l’ensemble des processus impliqués lors d’une irradiation. L’ensemble de ces données sont en accord avec un transfert d’énergie par résonnance lié à une superposition spectrale de l’émission du donneur et de l’absorption de l’accepteur (théorie de Förster). Ces résultats montrent les progrès réalisables dans la concentration de photons et la monochromatisation de la lumière dans des molécules tridimensionnelles. Abstract in Italian: E’ stata sintetizzata e caratterizzata una nuova famiglia di antenne costituite da diversi frammenti cromoforici Bodipy ancorati su un core tripticenico. Sono stati studiati i loro spettri di assorbimento e le loro proprietà fotofisiche, anche tramite spettroscopia pump-probe di assorbimento transiente. Oltre ai sistemi multicromoforici TAB, TBC, e TABC (A, B, e C identificano differenti subunità Bodipy, che assorbono ed emettono a diversa energia) sono state preparate e studiate sia le specie modello monocromoforiche TA, TB e TC sia la specie multicromoforica TA3, contenente tre Bodipy identici. Il frammento tripticenico ha il ruolo fondamentale di permettere un arrangiamento topologicamente controllato e rigido, nelle tre dimensioni, dei diversi cromofori. Gli spettri di assorbimento delle specie multicromoforiche sono essenzialmente additivi, indicando che allo stato fondamentale l′interazione intercromoforica è trascurabile. Le proprietà di luminescenza e la spettroscopia di assorbimento transiente hanno messo in evidenza che in tutti i sistemi multicromoforici investigati si verificano processi di trasferimento di energia elettronica fotoindotti, particolamente efficenti e veloci (nella scala temporale dei ps) verso la subunità a più bassa energia (trappola di energia) di ciascun sistema multi-Bodipy. In particolare, nel sistema TAB il trasferimento di energia dalla subunità di tipo A a quella di tipo B avviene con una costante di velocità pari a 1.6×1010 s−1, mentre in TBC il trasferimento da B a C è biesponenziale, con costanti di velocità pari a 1.7×1011 e 1.9×1010 s−1. Tale comportamento potrebbe essere razionalizzato grazie alla presenza, per tale specie, di diversi conformeri con differenti distanze donatore - accettore. I risultati ottenuti per i sistemi bicromoforici hanno permesso di interpretare le complesse dinamiche di decadimento degli stati eccitati caratteristiche della specie multicromoforica TABC. Per tutte le specie investigate, i risultati ottenuti sperimentalmente sono in accordo con un meccanismo di trasferimento energetico fotoindotto di tipo Förster.
Co-reporter:Dr. Raymond Ziessel;Adela Nano;Elodie Heyer;Thomas Bura;Dr. Pascal Retailleau
Chemistry - A European Journal 2013 Volume 19( Issue 8) pp:2582-2588
Publication Date(Web):
DOI:10.1002/chem.201203121
Co-reporter:Dr. Julien Massue;Denis Frath;Pascal Retailleau;Dr. Gilles Ulrich;Dr. Raymond Ziessel
Chemistry - A European Journal 2013 Volume 19( Issue 17) pp:5375-5386
Publication Date(Web):
DOI:10.1002/chem.201203625
Abstract
A series of thirteen luminescent tetrahedral borate complexes based on the 2-(2′-hydroxyphenyl)benzoxazole (HBO) core is presented. Their synthesis includes the incorporation of an ethynyl fragment by Sonogashira cross-coupling reaction, with the goal of extending the conjugation and consequently redshifting their emission wavelength. Different regioisomers, substituted in the 3-, 4-, or 5-position of the phenolate side of the HBO core, were studied in order to compare their photophysical properties. The complexes were characterized by X-ray diffraction and NMR, UV/Vis, and emission spectroscopy in solution and in the solid state. In all cases, complexation to boron leads to a donor–acceptor character that impacts their photophysical properties. Complexes with a 3- or 5-substituted fragment display mild to pronounced internal charge transfer (ICT), a feature strengthened by the presence of p-dibutylaminophenylacetylene in the molecular structure, protonation of the nitrogen atom of which leads to a significant blueshift and an increase in quantum yield. On the contrary, when the ethynyl module is grafted on the 4-position, narrow, structured, symmetrical absorption/emission bands are observed. Moreover, the fact that protonation has little effect on the emission maximum wavelength reveals singlet excited-state decay. Solid-state emission properties reveal a redshift compared to solution, explained by tight packing of the π-conjugated systems and the high planarity of the dyes. Subsequent connection of these complexes to other photoactive subunits (BODIPY, Boranil) provides dyads in which efficient cascade energy transfer is observed.
Cet article présente une série de treize complexes de bore tétraédriques luminescents basés sur le motif 2-(2′-hydroxyphenyl)benzoxazole (HBO). Leur synthèse comprend l’incorporation d’un fragment ethynyle par une réaction de couplage croisé de Sonogashira dans le but d’étendre la conjugaison et ainsi déplacer leur longueur d’onde d’émission dans le rouge (basses longueur d’onde). Différents régioisomères, substitués en position 3, 4 ou 5 du côté phénol du groupement HBO ont été étudiés afin de comparer leurs propriétés spectroscopiques. Les complexes ont été caractérisés par RMN, diffraction des rayons X sur mono-crystal, spectroscopie UV-Visible et émission à la fois en solution et à l’état solide. Pour chaque composé, la complexation au bore entraine un caractère donneur-accepteur de la molécule, ce va qui fortement influer sur les propriétés photophysiques. Les complexes fonctionnalisés en position 3 et 5 présentent un transfert de charge interne (TCI) qui est amplifié par la présence du groupement dibutylaminophénylacetylène dans la structure des molécules; la protonation de l’atome d’azote par de l’HCl gazeux entraine un déplacement de la longueur d’onde d’émission vers le bleu (longueur d’onde plus élevée) et une augmentation du rendement quantique. A l’opposé, lorsque l’espaceur éthynyle est greffé en position 4, des bandes d’absorption/émission étroites, structurées et symétriques sont observées. Par ailleurs, la protonation n’a que peu d’effet sur le maximum de la longueur d’onde d’émission, mettant en évidence une décroissance de l’état excité de type singulet. L’étude des propriétés d’émission à l’état solide révèlent un déplacement vers le rouge en comparaison des propriétés en solution, qui est expliqué par les empilements importants des systèmes conjugués et la grande planéité des colorants. La connexion de ces complexes à d’autres sous-unités photoactives (BODIPY, Boranil) permet d’obtenir des dyades où un transfert d’énergie en cascade est observé.
Co-reporter:Adela Nano;Dr. Raymond Ziessel;Patrycya Stachelek;Dr. Anthony Harriman
Chemistry - A European Journal 2013 Volume 19( Issue 40) pp:13528-13537
Publication Date(Web):
DOI:10.1002/chem.201301045
Abstract
A small series of donor–acceptor molecular dyads has been synthesized and fully characterized. In each case, the acceptor is a dicyanovinyl unit and the donor is a boron dipyrromethene (BODIPY) dye equipped with a single styryl arm bearing a terminal amino group. In the absence of the acceptor, the BODIPY-based dyes are strongly fluorescent in the far-red region and the relaxed excited-singlet states possess significant charge-transfer character. As such, the emission maxima depend on both the solvent polarity and temperature. With the corresponding push–pull molecules, there is a low-energy charge-transfer state that can be observed by both absorption and emission spectroscopy. Here, charge-recombination fluorescence is weak and decays over a few hundred picoseconds or so to recover the ground state. Overall, these results permit evaluation of the factors affecting the probability of charge-recombination fluorescence in push–pull dyes. The photophysical studies are supported by cyclic voltammetry and DFT calculations.
Co-reporter:Dr. Denis Frath;Dr. James E. Yarnell;Dr. Gilles Ulrich; Felix N. Castellano;Dr. Raymond Ziessel
ChemPhysChem 2013 Volume 14( Issue 14) pp:3348-3354
Publication Date(Web):
DOI:10.1002/cphc.201300547
Abstract
New boron-dipyrromethene (BODIPY) dyes linked to viologen are prepared and their photophysical and electrochemical properties are investigated. Both synthesized molecules have similar electronic absorption spectra with the absorption maximum localized at 517 and 501 nm for dye 1 and dye 2, respectively. They exhibit well-defined redox behavior, highlighting the presence of BODIPY and viologen subunits, with little perturbation of the redox potential of both subunits with respect to the parent compounds. Both dyes are heavily quenched by photoinduced electron transfer from the BODIPY to the viologen subunit. The transient absorption technique demonstrates that dye 2 forms the viologen radical within a timeframe of 7.1 ps, and that the charge-separated species has a lifetime of 59 ps. Sustained irradiation of dye 2 in the presence of a tertiary amine allows for the accumulation of BODIPY–methyl-4,4′-bipyridinium (BODIPY–MV+), as observed by its characteristic absorption at 396 and 603 nm. However, dye 2 does not generate catalytic amounts of hydrogen under standard conditions.
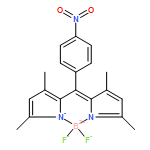
Co-reporter:Jean-Hubert Olivier ; Joaquín Barberá ; Effat Bahaidarah ; Anthony Harriman
Journal of the American Chemical Society 2012 Volume 134(Issue 14) pp:6100-6103
Publication Date(Web):March 28, 2012
DOI:10.1021/ja3007935
Red- and blue-absorbing boron dipyrromethene dyes, bearing opposite electronic charges, associate in solution to form a 1:2 complex having a stability constant of ca. 1017 M–2. The complex can be dismantled by addition of a large excess of tetra-N-butylammonium cations. The same complex displays liquid crystalline properties on heating from rt to above 150 °C, as characterized by various experimental techniques. Highly efficient electronic energy transfer from the red to the blue dye occurs in both the initial complex and the subsequent mesomorphic state.
Co-reporter:Thomas Bura ; Nicolas Leclerc ; Sadiara Fall ; Patrick Lévêque ; Thomas Heiser ; Pascal Retailleau ; Sandra Rihn ; Antoine Mirloup
Journal of the American Chemical Society 2012 Volume 134(Issue 42) pp:17404-17407
Publication Date(Web):October 5, 2012
DOI:10.1021/ja3072513
Green-absorbing dipyrromethene dyes engineered from bis-vinyl-thienyl modules are planar molecules, exhibiting strong absorption in the 713–724 nm range and displaying comparable electron and hole mobilities in thin films (maximum value 1 × 10–3 cm2/(V·s)). Bulk heterojunction solar cells assembled with these dyes and a fullerene derivative (PC61BM) at a low ratio give a power conversion efficiency as high as 4.7%, with short-circuit current values of 14.2 mA/cm2, open-circuit voltage of 0.7 V, and a broad external quantum efficiency ranging from 350 to 920 nm with a maximum value of 60%.
Co-reporter:Andrea Barbieri, Barbara Ventura, Raymond Ziessel
Coordination Chemistry Reviews 2012 Volume 256(15–16) pp:1732-1741
Publication Date(Web):August 2012
DOI:10.1016/j.ccr.2012.02.006
Transfer of excitation energy in natural systems is a fundamental process for light harvesting and propagation of information. The deep understanding of the energy transfer mechanisms and of the strategies for governing the directionality of the energy flow in artificial multichromophoric arrays is at the basis of their use in demanding fields such as solar energy conversion and optoelectronic applications. This review describes the recent activity of the authors in the synthesis and photophysical characterization of supramolecular arrays containing transition metal polypyridine complexes and organic units, with attention on the mechanisms and the pathways of the energy transfer processes that occur in the arrays. A topic of interest has been the exploitation of antenna effects as well as energy conduction in molecular wires. The design of the systems took into consideration several aspects that need to be balanced in the construction of efficient and useful arrays, in particular the spatial displacement of the units, their energy content distribution and the nature of the linkers that connect them. The latter point was of great relevance and the most recent studies afforded the preparation and the analysis of systems where the ligand is a large aromatic unit, that can both provide a structural role, a suitable electronic communication between the chromophores and an additional photoactive partnership.Graphical abstractHighlights► Energy transfer mechanisms in acetylide-linked multichromophoric systems. ► Overview of wire-like multichromophoric systems. ► Energy transfer dynamics in multichromophoric arrays based on a truxene scaffold.
Co-reporter:Julien Massue, Denis Frath, Gilles Ulrich, Pascal Retailleau, and Raymond Ziessel
Organic Letters 2012 Volume 14(Issue 1) pp:230-233
Publication Date(Web):December 16, 2011
DOI:10.1021/ol203014e
Complexation of boron trifluoride by a series of electron donor/acceptor substituted 2-(2′-hydroxy phenyl)benzoxazole (HBO) derivatives yields luminescent B(III) complexes with an emission wavelength ranging from 385 to 425 nm in dichloromethane or toluene. Appropriate chemical functionalization of these new dyes allows connection to different photoactive subunits (Boranil, BODIPY), endowing an efficient cascade energy transfer.
Co-reporter:Denis Frath, Sébastien Azizi, Gilles Ulrich, and Raymond Ziessel
Organic Letters 2012 Volume 14(Issue 18) pp:4774-4777
Publication Date(Web):September 7, 2012
DOI:10.1021/ol3020573
A Boranil fluorophore bearing a nitro-phenyl group has been selectively reduced to its anilino form and then successfully converted to amide, imine, urea, and thiourea derivatives which are fluorescent dyes. Its isolated isothiocyanate intermediate derivative was used in a model labeling experiment with Bovine Serum Albumin (BSA). The purified labeled-BSA exhibits strong luminescence (Φf = 47%) in a phosphate buffer at pH = 7.4.
Co-reporter:Arnaud Poirel, Antoinette De Nicola, and Raymond Ziessel
Organic Letters 2012 Volume 14(Issue 22) pp:5696-5699
Publication Date(Web):October 31, 2012
DOI:10.1021/ol302710z
The synthesis of unsymmetrical 3,5-dioligothienyl-BODIPY derivatives and their optical and redox properties are reported. The key step is the monobromination of the 2,6-dimethyl-3,5-dithienyl-BODIPY at the α position of the thiophene moiety. The additional thiophene modules are attached by palladium-catalyzed cross-coupling reactions. Increasing the number of modules on each side of the BODIPY core progressively shifts the absorption to 677 nm and the emission to 769 nm.
Co-reporter:Barbara Ventura ; Andrea Barbieri ; Alessandra Degli Esposti ; Julie Batcha Seneclauze
Inorganic Chemistry 2012 Volume 51(Issue 5) pp:2832-2840
Publication Date(Web):February 22, 2012
DOI:10.1021/ic201903g
The synthesis, characterization, photophysics, and time-dependent density functional theory (TD-DFT) calculations of spirobifluorene-bipyridine based iridium(III), osmium(II), and mixed Ir/Os complexes are presented. The preparation of the reference and mixed complexes proceeded step-by-step and microwave irradiation facilitated the complexation of osmium. The absorption of the target heterobimetallic derivative, Ir-L-Os, is described by linear combination of half of the absorption spectra of the homobimetallic analogues, Ir-L-Ir and Os-L-Os, due to the occurrence of mixed ligand and metal based transitions when the spirobifluorene-(bpy)2 bridging ligand L is linked to the metal, confirming a negligible interaction between the substituted metallic chromophores. TD-DFT calculations on monometallic, homo- and hetero-bimetallic complexes fully disentangled the origin of the absorption features. Noticeably, in the mixed Ir-L-Os complex an almost quantitative energy transfer from the 3Ir to the 3Os MLCT state is occurring, with a rate constant of 4.1 × 108 s–1 and nearly exclusively via a Dexter-type mechanism mediated by the orbitals of the spiroconjugated ligand. This result, together with the outcomes of the TD-DFT calculations, supports the existence of spiroconjugation and evidences the interesting role of this kind of bridge in the energy transfer dynamics of the arrays. In all the complexes, moreover, the ligand fluorescence is heavily quenched by energy transfer processes toward the metallic appended units; the rate constant is estimated in the order of 1010 s–1 for Ir-L-Os and higher than 1012 s–1 for the other complexes. In the heterometallic array, both at room temperature and at 77 K, all photons are thus funneled to the emissive Os 3MLCT state, which acts as energy trap for the antenna cascade.
Co-reporter:Stéphane Diring, Barbara Ventura, Andrea Barbieri and Raymond Ziessel
Dalton Transactions 2012 vol. 41(Issue 42) pp:13090-13096
Publication Date(Web):02 Apr 2012
DOI:10.1039/C2DT30385J
The synthesis, photophysical characterization and energy-transfer features of a series of hybrid truxene derivatives peripherally decorated with inorganic Os-containing polypyridine units and organic Bodipy dyes are reported. The photoactive terminal units are coupled to the central truxene scaffold by rigid ethynyl linkers in a star-shaped arrangement. The absorption range widely covers the UV-Vis spectrum and the Os 3MLCT or the Bodipy triplet act as final collectors of the absorbed energy.
Co-reporter:Matthieu Starck and Raymond Ziessel
Dalton Transactions 2012 vol. 41(Issue 43) pp:13298-13307
Publication Date(Web):17 Jul 2012
DOI:10.1039/C2DT31181J
A series of lanthanide complexes has been prepared from ligands constructed from a bis-pyrazolyl-pyridine core bearing various chelating arms with anionic mixed carboxylate/phosphonate or phosphonate substituents. These ligands form particularly stable complexes with Eu(III) and Tb(III) which display outstanding spectroscopic properties, with excited state lifetimes ranging from 2.6 to 3.2 ms and quantum yields in the 16 to 48% range in water or phosphate buffer. The complexes are significantly more stable than those of analogous ligands bearing only carboxylate groups. Some of the new ligands have a central and flexible pendent link suitable for bio-labelling.
Co-reporter:Raymond Ziessel, Alexandre Steffen, Matthieu Starck
Tetrahedron Letters 2012 Volume 53(Issue 29) pp:3713-3716
Publication Date(Web):18 July 2012
DOI:10.1016/j.tetlet.2012.04.105
Several new ligands constructed from a pyridine-ethynylnaphthalene platform have been engineered with a chelating pocket comprising bisdimethylaminocarboxylate/phosphonate or bisdimethylaminophosphonate units. Selective hydrolysis provides pockets with four to six anionic carboxylate or phosphate functions suitable for lanthanide complexation. Linkage at the opposite side of a flexible hanging arm either via a triple bond or through an amide tether offers the possibility of further use of these ligands for bioconjugation. Protocols to link the dedicated pockets via hydrophilic flexible chain to Biotin are also detailed.A family of naphthalene-pyridine based ligands were synthesized stepwise from glyphosate and amino-di(methylenediethylphosphite). The linker is a flexible chain bearing a terminal ester or a Biotin fragment.
Co-reporter:Raymond Ziessel, Alexandra Sutter, Matthieu Starck
Tetrahedron Letters 2012 Volume 53(Issue 29) pp:3717-3721
Publication Date(Web):18 July 2012
DOI:10.1016/j.tetlet.2012.04.106
A series of ligands has been constructed from a central bis-pyrazolyl–pyridine core and various deprotonable chelating pockets based on mixed carboxylate/phosphate or phosphate anionic functions and a central flexible pendent arm for subsequent grafting to biomaterials. For some of these ligands, the sequence of reactions incorporates an additional step introducing a substituted diethynylphenyl residue to increase significantly their solubility in polar organic solvents. The terbium(III) complexes of some of these ligands display outstanding spectroscopic properties with lifetimes ranging from 2.7 to 3.2 ms and quantum yields from 16% to 26% in water.A variety of pyrazolyl–pyridine ligands adequately functionalized by carboxylate/phosphonate/phosphate functions form very stable lanthanide complexes and the terbium display outstanding spectroscopic properties.
Co-reporter:Arnaud Poirel, Antoinette De Nicola, Pascal Retailleau, and Raymond Ziessel
The Journal of Organic Chemistry 2012 Volume 77(Issue 17) pp:7512-7525
Publication Date(Web):July 28, 2012
DOI:10.1021/jo301300b
We report the synthesis of BODIPYs with unsubstituted 1,7,8-positions and their dimerization by oxidative coupling with phenyliodine(III)-bis(trifluoroacetate) (PIFA). This dimerization was achieved for BODIPYs substituted in the 3,5-positions with either methyl or thienyl groups. The position and the type of the linkage in the resulting dimers depended on the nature of the substituent. The 3,5-dimethyl-BODIPY dyes were linked either via direct 1,1′-pyrrole–pyrrole coupling or via a 1,3′-methylene bridge. The 3,5-dithienyl-BODIPY dyes provided, in excellent yields, unique compounds linked exclusively via the α-thienyl positions. All dyes were unreactive in the 8-position. Electrochemical and spectroscopic measurements on the monomers and dimers provided evidence of interactions between the two halves of the dimers. Thus, oxidation and reduction potentials were split by up to 210 mV, and modest excitonic coupling and an internal charge transfer were observed in some cases.
Co-reporter:Dr. Song-lin Niu;Cédrik Massif;Dr. Gilles Ulrich; Pierre-Yves Renard;Dr. Anthony Romieu;Dr. Raymond Ziessel
Chemistry - A European Journal 2012 Volume 18( Issue 23) pp:7229-7242
Publication Date(Web):
DOI:10.1002/chem.201103613
Abstract
A series of water-soluble red-emitting distyryl-borondipyrromethene (BODIPY) dyes were designed and synthesized by using three complementary approaches aimed at introducing water-solubilizing groups on opposite faces of the fluorescent core to reduce or completely suppress self-aggregation. An additional carboxylic acid functional group was introduced at the pseudo-meso position of the BODIPY scaffold for conjugation to amine-containing biomolecules/biopolymers. The optical properties of these dyes were evaluated under simulated physiological conditions (i.e., phosphate-buffered saline (PBS), pH 7.5) or in pure water. The emission wavelength (λmax) of these labels was found in the 640–660 nm range with quantum yields from modest to unprecedentedly high values (4 to 38 %). The bioconjugation of these distyryl-BODIPY dyes with bovine serum albumin (BSA) and the monoclonal antibody (mAb) 12A5 was successfully performed under mild aqueous conditions.
De nouveaux colorants fluorescents à architecture distyrylborondipyrromethene (BODIPY), émettant dans le rouge et solubles dans l’eau, ont été préparés en utilisant trois méthodes efficaces de fonctionnalisation chimique qui permettent l’introduction de groupements hydrophiles (motifs pseudo-PEG et sulfonate) sur les parties nord et sud de leur squelette conjugué, et ce afin de réduire ou de supprimer totalement l’agrégation en solution aqueuse. La solubilité dans l’eau des fluorophores ainsi obtenus est élevée et leurs propriétés optiques en milieu physiologique remarquables et comparables à celles déterminées dans les solvants organiques pour les composés hydrophobes parents. De plus, la présence d’une fonction acide carboxylique en position méso de ces composés distyryl-BODIPY a permis le marquage covalent de protéines d’intérêt (albumine de sérum bovin et anticorps monoclonaux 12A5) via la formation de liens amide dans des conditions biocompatibles.
Co-reporter:Delphine Hablot;Alexra Sutter;Dr. Pascal Retailleau;Dr. Raymond Ziessel
Chemistry - A European Journal 2012 Volume 18( Issue 7) pp:1890-1895
Publication Date(Web):
DOI:10.1002/chem.201103307
Co-reporter:Delphine Hablot;Dr. Ashraful Islam;Liyuan Han;Dr. Raymond Ziessel
ChemPlusChem 2012 Volume 77( Issue 6) pp:462-469
Publication Date(Web):
DOI:10.1002/cplu.201200059
Abstract
Novel metal-free and fluorescent dipyrrolopyrrole (DPP) dyes consisting of a central bis(lactam)-bearing pendent benzyl or branched hydrocarbon groups as solubilizing fragments and two orthogonal side arms (dimethylaminopropyne and benzoate anion) have been designed and synthesized. The UV/Vis absorption spectra recorded in THF are dominated by intense low-energy π–π* absorptions centered at 488 nm or 602 nm, respectively, for the phenyl- or thiophene-based DPP dyes. The fluorescence spectra also display broad bands at 555 nm (ΦF=0.57 and τF=4.6 ns) for the phenyl- and at 624 nm (ΦF=0.31 and τF=3.5 ns) for the thiophene-based molecular structures. Under standard global AM 1.5G irradiation a maximum photon-to-electron conversion efficiency of 2.54 % was achieved with dye-sensitized solar cells based on nanocrystalline TiO2 (Jsc=7.5 mA cm−2, Voc=0.49 V, and FF=0.70) for the phenyl-based DPP dye and 1.89 % (Jsc=7.1 mA cm−2, Voc=0.41 V, and FF=0.65) for the thiophene-based DPP dye.
Co-reporter:Gilles Ulrich, Raymond Ziessel, and Alexandre Haefele
The Journal of Organic Chemistry 2012 Volume 77(Issue 9) pp:4298-4311
Publication Date(Web):April 12, 2012
DOI:10.1021/jo3002408
An efficient protocol for the direct synthesis of 3-substituted and 3,5-disubstituted BODIPY derivatives via electrophilic attack with NBS was developed. Various substituents like ethers, sugar, hydroxyl, thiophene, sulfur, azide, tertiary amines, alkyne, vinyl, or phosphonate groups were obtained in moderate to excellent yields. The amine-substituted derivatives display unusual spectroscopic and electrochemical properties which were analyzed in solution in the presence of HCl. The diethylamino-substituted derivative has a proton association constant of log β = 4.7, and the disubstituted derivative has two association constants of log β = 6.2 and 12.1 in ethanol. In both cases, the quenching of the fluorescence is explained by photoinduced electron transfer from the tertiary amine to the Bodipy excited state.
Co-reporter:Gilles Ulrich, Alexandre Haefele, Pascal Retailleau, and Raymond Ziessel
The Journal of Organic Chemistry 2012 Volume 77(Issue 11) pp:5036-5048
Publication Date(Web):May 2, 2012
DOI:10.1021/jo300477p
A methodological study is presented dealing with carbopalladation reactions on BODIPY dyes bearing aryl–halogen functions. Using this technique, several ester and amide groups were efficiently introduced on the dyes. These changes do not affect the optical properties of the dyes and thus allow the construction of new BODIPY-based functional dyes with carboxylic anchoring groups or peptide links.
Co-reporter:Sandra Rihn, Pascal Retailleau, Antoinette De Nicola, Gilles Ulrich, and Raymond Ziessel
The Journal of Organic Chemistry 2012 Volume 77(Issue 20) pp:8851-8863
Publication Date(Web):July 26, 2012
DOI:10.1021/jo301059u
Derivatives of isomeric 2-(hydroxytolyl)-4,6-dimethylamino-1,3,5-triazines have been synthesized in high yields in a controlled manner using a multistep reaction sequence. Iodination of either 2-(1′-hydroxy-6′-methylphen-2′-yl)- or 2-(1′-hydroxy-4′-methylphen-2′-yl)-4,6-dimethylamino-1,3,5-triazine with ICl provides species differing in the positioning of the iodo group relative to the hydroxyl which readily undergo Suzuki, Sonogashira, and Heck reactions under Pd(0) catalysis. Thus, thienyl, bisthienyl, and 3,4-ethylenedioxythienyl groups have been directly grafted, while unsubstituted polycyclic aromatics such as pyrene and perylene have been linked via alkyne bridges, as have ethynyldifluoroborondipyrromethane (BODIPY) dyes prepared in situ. The presence of a hydrogen bond in the ground state involving the hydroxyl substituent has been established by proton NMR and several X-ray structure determinations. All of the new dyes with a simple substituent (phenyl, thienyl) exhibited a pronounced green fluorescence resulting from an intramolecular proton transfer in the excited state (ESIPT) which produces a large Stokes shift (>10 000 cm–1). With other dyes, the fluorescence of the keto form responsible for the ESIPT process could be used as the input energy in efficient intramolecular energy transfer processes. Replacing perylene with pyrene allowed reversal of the direction of energy transfer from the polyaromatic module to the keto form.
Co-reporter:Julien Iehl ; Jean-François Nierengarten ; Anthony Harriman ; Thomas Bura
Journal of the American Chemical Society 2011 Volume 134(Issue 2) pp:988-998
Publication Date(Web):December 8, 2011
DOI:10.1021/ja206894z
A sophisticated model of the natural light-harvesting antenna has been devised by decorating a C60 hexa-adduct with ten yellow and two blue boron dipyrromethene (Bodipy) dyes in such a way that the dyes retain their individuality and assist solubility of the fullerene. Unusually, the fullerene core is a poor electron acceptor and does not enter into light-induced electron-transfer reactions with the appended dyes, but ineffective electronic energy transfer from the excited-state dye to the C60 residue competes with fluorescence from the yellow dye. Intraparticle electronic energy transfer from yellow to blue dyes can be followed by steady-state and time-resolved fluorescence spectroscopy and by excitation spectra for isolated C60 nanoparticles dissolved in dioxane at 293 K and at 77 K. The decorated particles can be loaded into polymer films by spin coating from solution. In the dried film, efficient energy transfer occurs such that photons absorbed by the yellow dye are emitted by the blue dye. Films can also be prepared to contain C60 nanoparticles loaded with the yellow Bodipy dye but lacking the blue dye and, under these circumstances, electronic energy migration occurs between yellow dyes appended to the same nanoparticle and, at higher loading, to dye molecules on nearby particles. Doping these latter polymer films with the mixed-dye nanoparticle coalesces these multifarious processes in a single system. Thus, long-range energy migration occurs among yellow dyes attached to different particles before trapping at a blue dye. In this respect, the film resembles the natural photosynthetic light-harvesting complexes, albeit at much reduced efficacy. The decorated nanoparticles sensitize amorphous silicon photocells.
Co-reporter:Denis Frath, Sébastien Azizi, Gilles Ulrich, Pascal Retailleau, and Raymond Ziessel
Organic Letters 2011 Volume 13(Issue 13) pp:3414-3417
Publication Date(Web):June 7, 2011
DOI:10.1021/ol2011665
Complexation of a large variety of Anils (aniline-imines) with boron(III) precursors provides stable Boranils, some of which have been structurally characterized. Analysis of their optical properties reveals that the fluorescence stems from an intraligand charge transfer (ILCT) state with the best quantum yields reaching 90%. Chemistry on the Boranils allows grafting of photoactive modules acting as energy antennae for borondipyrromethene (Bodipy) and subphtalocyanine (SubPc) fluorophores.
Co-reporter:Jean-Hubert Olivier, Jack Harrowfield and Raymond Ziessel
Chemical Communications 2011 vol. 47(Issue 40) pp:11176-11188
Publication Date(Web):25 Jul 2011
DOI:10.1039/C1CC12771C
Synthetic routes for the construction of 3-substituted 2,4-pentadionate ligands are broadly surveyed. They involve sequential alkylation and arylation by numerous methods, including those based on reactions of coordinated ligands, and can provide access to various rationally designed ligands. Applications of such ligands in the synthesis of multichromophoric complexes are illustrated in some detail. Incorporation of 3-substituted 2,4-pentanedione units into mesomorphic and macromolecular structures is considered in relation to structural control of energy and electron transfer processes in photoactive systems. Aspects of the general supramolecular chemistry of complexes of 3-substituted-2,4-pentadionate ligands are briefly discussed to illustrate the utility of such species within the full range of 1,3-dionate chemistry.
Co-reporter:Fabian Spaenig ; Jean-Hubert Olivier ; Valentina Prusakova ; Pascal Retailleau ; Raymond Ziessel ;Felix N. Castellano
Inorganic Chemistry 2011 Volume 50(Issue 21) pp:10859-10871
Publication Date(Web):September 2, 2011
DOI:10.1021/ic201397v
The synthesis, complete structural characterization, electrochemistry, and excited-state dynamics of a series of four bis-heteroleptic iridium(III) charge-transfer complexes composed of a single acac-functionalized and two ortho-metalated 2-phenylpyridine ligands. The formed iodophenyl complex (2) was used as a metallosynthon to introduce extended-core ethynyltolyl (3), ethynylpyrene (4), and ethynylperylene (5) residues into these structures projecting from the acac ancillary ligand. Static and dynamic photoluminescence along with ultrafast and conventional transient absorption measurements in conjunction with cyclic voltammetry were employed to elucidate the nature of the intramolecular energy-transfer processes occurring in the excited states of polychromophores 4 and 5 and are directly compared with those of model complexes 2 and 3. Upon charge-transfer excitation of these molecules, the long-lived triplet-state metal-to-ligand charge-transfer (3MLCT)-based photoluminescence readily observed in 2 and 3 (τ = 1 μs) is nearly quantitatively quenched, resulting from production of the associated triplet intraligand (3IL) excited states in 4 and 5 through intramolecular triplet–triplet energy transfer. The respective formation of the extended-core 3*pyrenyl and 3*perylenyl-localized excited states in 4 and 5 is confirmed by their ultrafast excited-state evolution, which ultimately generates features associated with these 3IL excited states and their greatly extended excited-state lifetimes with respect to the parent complexes 2 and 3.
Co-reporter:Franck Camerel, Bertrand Donnio and Raymond Ziessel
Soft Matter 2011 vol. 7(Issue 2) pp:412-428
Publication Date(Web):22 Oct 2010
DOI:10.1039/C0SM00547A
Two novel families of highly functionalized molecules consisting of a central carbazole or naphthalene core carrying actionable acid functions and linked to gallate platforms equipped with three long alkoxy chains have been rationally designed. The presence of amide tethers and chelating rigid fragments generate a new class of luminescent mesomorphic materials. Platforms bearing long paraffin chains (n = 12 and 16) self-organize into columnar liquid crystalline phases in which the columns are arranged according to a 2D oblique, rectangular or hexagonal lattice symmetry as deduced from powder XRD experiments. FT-IR studies confirm that the common driving force for aggregation and micro-segregation in the mesophase is due to the occurrence of a tight intermolecular H-bonded network. Spectroscopic measurements show that these platforms are luminescent in solution and in the solid state. Due to the synthetic availability of carbazole and naphthalene rigid cores and the simplicity by which these platforms can be graphed, this methodology represents a practical alternative to the production of fluorescent and mesomorphic materials.
Co-reporter:Song Lin Niu, Cédrik Massif, Gilles Ulrich, Raymond Ziessel, Pierre-Yves Renard and Anthony Romieu
Organic & Biomolecular Chemistry 2011 vol. 9(Issue 1) pp:66-69
Publication Date(Web):18 Nov 2010
DOI:10.1039/C0OB00693A
The synthesis and preliminary bio-conjugation studies of a novel water-soluble red-emitting di-styryl BODIPY dye are disclosed. Aggregation behaviour of this compound under physiological conditions was suppressed by specific introduction of a di-sulfonated peptide-based linker at the mesophenyl substituent, sultonated styryl arms and short polyethyleneglycol chains at the boron center. Thus, a good quantum yield of 22% in PBS for this red-emitting BODIPY was obtained. Introduction of an activated ester function enabled successful bio-conjugation to monoclonal antibodies and proteins.
Co-reporter:Thomas Bura, Delphine Hablot, Raymond Ziessel
Tetrahedron Letters 2011 Volume 52(Issue 18) pp:2370-2374
Publication Date(Web):4 May 2011
DOI:10.1016/j.tetlet.2011.02.094
Several new extended fluorescent dyes have been prepared by stepwise functionalisation of the methyl groups located in the 1, 3, 5, and 7 substitution positions of Bodipy dyes. In one case, a ferrocene residue has been connected, giving rise to severe fluorescence quenching and a rich redox behavior. The stepwise syntheses were largely based on Knoevenagel reactions allowing the attachment of three different modules in a tetravinyl configuration. Most of these dyes exhibit reversible formation of radical cations and radical anions, as well as an irreversible formation of dications and dianions. Spectroscopic examination of the dyes under various conditions including the presence of acid leads to the conclusion that the delocalization is more effective in the 3,5-substitution positions than in the 1,7-positions. These novel dyes have absorption and emission wavelengths spanning the ranges of 573–718 nm and 585–778 nm, respectively.Conjugated Bodipy dyes constructed with various functionalized benzaldehydes have been prepared and display interesting redox and spectroscopic properties depending on their environment.
Co-reporter:Songlin Niu, Gilles Ulrich, Pascal Retailleau, Raymond Ziessel
Tetrahedron Letters 2011 Volume 52(Issue 38) pp:4848-4853
Publication Date(Web):21 September 2011
DOI:10.1016/j.tetlet.2011.07.028
A series of BODIPY bridged push–pull chromophores were prepared by a sequence of reactions involving: i) cross-coupling reactions promoted by palladium complexes; ii) Knoevenagel condensation leading to dicyano derivatives; iii) [2+2] cycloaddition. The last reaction is regioselective, providing mono-derivatives with anisole substituents. With dimethylaminophenyl donor groups, double addition is feasible, providing highly colored dyes displaying remarkable electrochemical properties.A family of push–pull chromophores were synthesized by a stepwise series of reactions involving cross-coupling reactions, Knoevenagel condensation and [2+2] cycloaddition.
Co-reporter:Dr. Jean-Hubert Olivier;Josiane Widmaier;Dr. Raymond Ziessel
Chemistry - A European Journal 2011 Volume 17( Issue 42) pp:11709-11714
Publication Date(Web):
DOI:10.1002/chem.201101407
Co-reporter:Delphine Hablot;Dr. Anthony Harriman;Dr. Raymond Ziessel
Angewandte Chemie International Edition 2011 Volume 50( Issue 34) pp:7833-7836
Publication Date(Web):
DOI:10.1002/anie.201102065
Co-reporter:Jean-Hubert Olivier;Dr. Franck Camerel ; Raymond Ziessel
Chemistry - A European Journal 2011 Volume 17( Issue 33) pp:9113-9122
Publication Date(Web):
DOI:10.1002/chem.201100296
Abstract
While acetylacetone (acacH) derivatives are, upon deprotonation, ubiquitous ligands in coordination chemistry, their potential to form stable ionic liquids has not been studied so far. Here we describe a straightforward synthesis of novel trifluoroacetylacetone-functionalised imidazolium salts. These salts are built from an imidazolium ring substituted on one side with a flexible chain of fixed length carrying a terminal acacH group and on the opposite side a paraffin chain of various lengths. By changing the length of these flexible chains (n=4, 8, 12) and the nature of the counter-anions (PF6−, BF4−, NTf2−), room-temperature ionic liquids were produced. Their application for the extraction of lanthanide salts (Eu, Tb) from dilute aqueous solution has been investigated. The presence of a strong UV absorber (imidazolium ring, λabs=290 nm) allows photosensitisation of the EuIII and TbIII luminescence by efficient energy transfer, and thus extraction of these two lanthanides can be followed by fluorescence techniques. It appears that loading of the ionic liquids onto silica particles pre-treated with a dilute aqueous solution of NaOH is the most efficient way to obtain fast and almost quantitative (>99.9 %) extraction of the metal ions as their diketonato complexes. The procedure is reproducible and the loaded SiO2 particles can be simply treated with acid to strip the metal ions and regenerate the adsorbed (protonated) extractant.
Co-reporter:Gilles Ulrich, Sébastien Goeb, Antoinette De Nicola, Pascal Retailleau, and Raymond Ziessel
The Journal of Organic Chemistry 2011 Volume 76(Issue 11) pp:4489-4505
Publication Date(Web):April 18, 2011
DOI:10.1021/jo200246q
A general method for the synthesis of difluorobora-diisoindolomethene dyes with phenyl, p-anisole, or ethyl-thiophene substituents has been developed. The nature of the substituents allows modulation of the fluorescence from 650 to 780 nm. Replacement of the fluoro ligands by ethynyl-aryl or ethyl residues is facile using Grignard reagents. Several X-ray molecular structures have been determined, allowing establishment of structure–fluorescence relationships. When the steric crowding around the boron center is severe, the aromatic substituents α to the diisoindolomethene nitrogens are twisted out of coplanarity, and hypsochromic shifts are observed in the absorption and emission spectra. This shift reached 91 nm with ethyl substituents compared to fluoro groups. When ethynyl linkers are used, the core remains flat, and a bathochromic shift is observed. All the fluorophores exhibit relatively high quantum yields for emitters in the 650–800 nm region. When perylene or pyrene residues are connected to the dyes, almost quantitative energy transfer from them to the dye core occurs, providing large virtual Stokes shifts spanning from 8000 to 13 000 cm–1 depending on the nature of the dye. All the dyes are redox active, providing the Bodipy radical cation and anion in a reversible manner. Stepwise reduction or oxidation to the dication and dianion is feasible at higher potentials. We contend that the present work paves the way for the development of a new generation of stable, functionalized luminophores for bioanalytical applications.
Co-reporter:Alexandre Haefele, Chantal Zedde, Pascal Retailleau, Gilles Ulrich and Raymond Ziessel
Organic Letters 2010 Volume 12(Issue 8) pp:1672-1675
Publication Date(Web):March 17, 2010
DOI:10.1021/ol100109j
A boradiazaindacene (BODIPY) fluorophore with a chirality held on the central boron has been synthesized and the racemate resolved. Dissymetrization of the BODIPY core was obtained by oxidation of the 3-methyl group to the corresponding carboxaldehyde. A hydrogen bond between the aldehyde proton and the fluorine on the boron atom was evidenced by both 1H NMR and X-ray diffraction. Chiral high-performance liquid chromatography as well as circular dichroism confirm the persistence of both enantiomers.
Co-reporter:Jean-Hubert Olivier, Alexandre Haefele, Pascal Retailleau and Raymond Ziessel
Organic Letters 2010 Volume 12(Issue 3) pp:408-411
Publication Date(Web):December 28, 2009
DOI:10.1021/ol902386u
New, acetylacetone-linked borondipyrromethene (BODIPY) dyes were readily obtained from BODIPY cores by various protocols involving direct grafting with acetylacetone or cross-coupling from a preorganized phenylacacH derivative bearing either an iodo or an ethynyl function. Facile anchoring on TiO2 powder is obtained and scrutinized by FT-IR spectroscopy.
Co-reporter:Aaron A. Rachford ; Raymond Ziessel ; Thomas Bura ; Pascal Retailleau ;Felix N. Castellano
Inorganic Chemistry 2010 Volume 49(Issue 8) pp:3730-3736
Publication Date(Web):March 19, 2010
DOI:10.1021/ic901996u
The synthesis, structural characterization, electrochemistry, and molecular photophysics of [Ir(ppy)2(bpy-C≡C-Bodipy)](PF6), where ppy is 2-phenylpyridine and bpy-C≡C-Bodipy is 5-ethynyl-2,2′-bipyridine-8-phenyl-1,3,5,7-tetramethyl-4,4-bis(2,5-dioxaoct-7-ynyl)-4-bora-3a,4a-diaza-s-indacene (4), is presented. Static and dynamic photoluminescence and absorption measurements in conjunction with cyclic voltammetry were employed to elucidate the nature of the intramolecular energy transfer processes occurring in the excited state of the title chromophore. Parallel studies were performed on appropriate model chromophores (2 and 3) intended to represent the photophysics of the isolated molecular subunits, that is, triplet metal-to-ligand-charge-transfer (3MLCT) and triplet Bodipy intraligand (3IL) excited states, respectively. Upon charge transfer excitation of the title chromophore, the 3MLCT based phosphorescence readily observed in 2 (Φem = 0.027, τ = 243 ns) is quantitatively quenched resulting from production of the 3Bodipy excited state through intramolecular triplet−triplet energy transfer. The formation of the 3Bodipy-localized excited state is confirmed by features in the transient absorption difference spectrum, extended excited-state lifetime (τ = 25 μs), as well the observation of 3IL Bodipy-based phosphorescence detected at 730 nm at 77 K. The low temperature Bodipy phosphorescence is readily produced in 4 as a result of the internal Ir(III) heavy atom.
Co-reporter:Barbara Ventura ; Andrea Barbieri ; Francesco Barigelletti ; Stéphane Diring
Inorganic Chemistry 2010 Volume 49(Issue 18) pp:8333-8346
Publication Date(Web):August 17, 2010
DOI:10.1021/ic1008413
A rigid star-shaped tetrachromophoric trimetallic complex engineered from a 5,5′,10,10′,15,15′-hexabutyltruxene platform functionalized in the 2,7,12 positions with three different metal centers, namely, a terpyridine-Pt(II) ethynylene unit and Ru(II) and Os (II) bipyridine centers, was synthesized in a controlled fashion and characterized by 1H NMR and mass spectrometry. The protocol was devised in such a way that key mono and dinuclear model complexes and two reference truxene ligands could also be prepared. Room temperature (RT) optical absorption and RT and 77 K luminescence studies were performed on the truxene ligands, the trimetallic species, the various mono- and binuclear complexes and precursors lacking the truxene fragment; RT nanosecond transient absorption measurements were also carried out in particular cases. The electronic properties of the Ru and Os subunits in the arrays were found to be unaffected by the presence of the truxene core whereas direct linking of the Pt subunit to the truxene via the σ-alkyne bond markedly influences the spectroscopic behavior of the Pt center. Remarkably the truxene phosphorescence was clearly established in the two ligands (lifetime of 4.3 s for the mono ethynyl-bipy substituted truxene and 17.5 ms for the bis ethynyl-bipy substituted truxene) and also detected in the Pt-containing complexes PtL′ (model Pt-truxene) and Pt−Os (Pt-truxene-Os dyad) at low temperature. This is attributed to the closeness in energy of the Pt 3CT level and the truxene triplet at low temperature and to the spin−orbit coupling induced by the Pt heavy atom. Transient absorbance measurements evidenced the population of the Pt-based triplet in the Pt-truxene mononuclear complex PtL′ at room-temperature. For the trimetallic complex, where the various centers exhibit an energy gradient for the local excited levels, and following an approach based on the use of selected excitation of the components, an initial energy transfer was found to occur from the central truxene unit toward the peripheral Pt, Ru, and Os metal-based centers. Subsequent Pt-based and Ru-based excited state depletion contributes to the final sensitization of the low-lying Os triplet excited state; the excited state dynamics for these multicascade processes are examined in detail.
Co-reporter:Francesco Nastasi, Fausto Puntoriero, Sebastiano Campagna, Jean-Hubert Olivier and Raymond Ziessel
Physical Chemistry Chemical Physics 2010 vol. 12(Issue 27) pp:7392-7402
Publication Date(Web):09 Jun 2010
DOI:10.1039/C003789C
By tailoring the chemistry of the 4,4-difluoro-4-bora-3a,4a-diaza-s-indacene (bodipy) core in order to change its spectroscopic properties, we linked some modified bodipy dyes to Pt(II)-terpyridine centers thereby preparing three novel hybrid complexes. In one of such species, two pyrene subunits are also present. The absorption spectra and luminescence properties (both at room temperature in acetonitrile solution and at 77 K in butyronitrile rigid matrix) of the new hybrid complexes and of their bodipy parent species have been investigated, and compared with those of previously studied compounds. Absorption spectra showed that the different chromophoric subunits roughly keep their own features in the multicomponent systems, indicating the supramolecular nature of the compounds. Efficient photoinduced intercomponent energy transfer takes place in the various hybrid Pt(II)-terpyridine-bodipy compounds, by taking advantage of different mechanisms: at room temperature in fluid solution very efficient energy transfer from the 3MLCT state(s) of the Pt residue to the lowest-lying 3bodipy is mediated by a charge transfer state via a two-step electron transfer process. Direct energy transfer from 3MLCT state(s) to the 3bodipy states takes place at 77 K, most likely via the Dexter mechanism. When two pyrene fragments are grafted to the boron, cascade energy transfer from the pyrene to the 1bodipy also occurs by the Förster mechanism. For one of the new compounds, 3bodipy emission with a maximum at 738 nm is also observed at cryogenic temperature.
Co-reporter:Thomas Bura, Raymond Ziessel
Tetrahedron Letters 2010 Volume 51(Issue 21) pp:2875-2879
Publication Date(Web):26 May 2010
DOI:10.1016/j.tetlet.2010.03.095
Several new fluorophores have been prepared by grafting boradiazaindacene (red absorbing) and/or styrylboradiazaindacene (blue absorbing) units as terminal energy acceptors onto a fluorene-derived platform. In one case, an amino-bis(bipyridine) pocket has been attached to enable strong binding of transition metal ions. The stepwise syntheses were largely based on Pd-catalysed cross-coupling reactions. The electrochemistry of the dyes has been analysed by reference to the properties of the various synthetic intermediates, protonation of the tertiary amine site present in the bis(bipyridine) species enabling processes involving this centre to be distinguished from those associated with the boradiazaindacene (Bodipy) unit.Mixed Bodipy dyes appended to a soluble fluorene platform have been prepared, one Bodipy was constructed with a bis-bipyridine complexation pocket.
Co-reporter:Thomas Bura;Pascal Retailleau Dr. Dr.
Angewandte Chemie 2010 Volume 122( Issue 37) pp:6809-6813
Publication Date(Web):
DOI:10.1002/ange.201003206
Co-reporter:Dr. Raymond Ziessel;Sra Rihn;Dr. Anthony Harriman
Chemistry - A European Journal 2010 Volume 16( Issue 39) pp:11942-11953
Publication Date(Web):
DOI:10.1002/chem.201001142
Abstract
Synthetic strategies have been devised that allow the rational design and isolation of highly coloured boron dipyrromethene (BODIPY) dyes that absorb across much of the visible region. Each dye has an aryl polycycle (usually pyrene or perylene) connected to the central BODIPY core through a conjugated tether at the 3,5-positions. Both mono- and difunctionalised derivatives are accessible, in certain cases containing both pyrene and perylene residues. For all new compounds, the photophysical properties have been recorded in solution at ambient temperature and in a glassy matrix at 77 K. The presence of the aryl polycycle(s) affects the absorption and emission maxima of the BODIPY nucleus, thereby confirming that these units are coupled electronically. Indeed, the band maxima and oscillator strengths depend on the conjugation length of the entire molecule, whereas there is no sign of fluorescence from the polycycle. As a consequence, the radiative rate constant tends to increase with each added appendage. The nature of the linkage (styryl, ethenyl, or ethynyl) also exerts an effect on the photophysical properties and, in particular, the absorption spectrum is perturbed in the region of the aryl polycycle. The perylene-containing BODIPY derivatives absorb over a wide spectral range and emit in the far-red region in almost quantitative yield. A notable exception to this generic behaviour is provided by the anthracenyl derivative, which exhibits charge-transfer absorption and emission spectra in weakly polar media at ambient temperature. Regular BODIPY-like behaviour is restored in a glassy matrix at 77 K. Overall, these new dyes represent an important addition to the range of strongly absorbing and emitting reagents that could be used as solar concentrators.
Co-reporter:Stéphane Diring Dr. ;Francesco Barigelletti Dr.;Andrea Barbieri Dr.;Barbara Ventura Dr.
Chemistry - A European Journal 2010 Volume 16( Issue 30) pp:9226-9236
Publication Date(Web):
DOI:10.1002/chem.200903305
Abstract
We report on the synthesis, optical properties and energy-transfer features of a series of transition-metal-containing complexes and dyads, based on a pre-organised truxene scaffold. In this series, the [Ru(bpy)3]2+ and [Os(bpy)3]2+ photoactive terminals are coupled to the bridging aromatic truxene core through rigid ethynyl linkers. The photoinduced energy-transfer processes taking place from the Ru- to the Os-based levels, and from the truxene bridging ligand to the terminal-metal chromophores are studied and the pathways and mechanisms are discussed. The photoinduced energy-transfer process observed in the dyad proceeds rapidly through: i) an efficient 1L1Os direct energy transfer followed by intersystem crossing to 3Os, and ii) a fast 1L1Ru energy-transfer step and subsequent intersystem crossing to 3Ru followed by a 3Ru3Os energy-transfer process. The first 1L1Os and 1L1Ru steps are controlled by a dipole–dipole interaction (Förster mechanism), whereas the subsequent 3Ru3Os step proceeds by means of a long-range (≈24 Å) through-bond mediated Dexter mechanism, facilitated by the conjugation along the bpy-truxene-bpy molecular axis.
Co-reporter:Jean-Hubert Olivier;Franck Camerel Dr.;Gilles Ulrich Dr.;Joaquín Barberá Dr. Dr.
Chemistry - A European Journal 2010 Volume 16( Issue 24) pp:7134-7142
Publication Date(Web):
DOI:10.1002/chem.201000339
Abstract
A series of modular mesogenic salts based on the combination of anionic 4,4-difluoro-4-bora-3a,4a-diaza-s-indacene (F-BODIPY) 2,6-disulfonate dyes and trialkoxybenzyl-functionalised imidazolium cations has been designed and synthesised. Each salt contains a rigid dianionic BODIPY core associated with two imidazolium cations functionalised by 1,2,3-trialkoxybenzyl (alkyl=n-C8, n-C12 or n-C16) units or, in one case, with imidazolium cations functionalised by a trialkylgallate (3,4,5-trialkoxybenzoate) unit in which the 3,5-dialkyl groups are terminated with a polymerisable acrylate entity. All these compounds were highly fluorescent in solution with quantum yields ranging from 54 to 62 %. In the solid state, the width of the emission band observed at around 650 nm is a clear signature of aggregation. With the trialkoxybenzylimidazolium cations, polarised optical microscopy (POM) and X-ray scattering experiments showed that columnar mesophases were formed. Differential scanning calorimetry (DSC) studies confirmed the mesomorphic behaviour from room temperature to about 130 °C for salts with alkyl chains containing 8, 12 and 16 carbon atoms. The strong luminescence of the BODIPY unit was maintained in the mesophase and fluorescence measurements confirmed the presence of J aggregates in all cases. The salt containing the gallate-functionalised imidazolium cations showed no mesomorphism but the acrylate terminal units could be used to engender photoinitiated polymerisation thereby allowing the material to be immobilised on glass plates. The polymerisation process was followed by FTIR spectroscopy and the fixed and patterned films were highly fluorescent with a solid-state emission close to that of the complex in the solid state.
Co-reporter:Jean-Hubert Olivier;Franck Camerel Dr.;Gilles Ulrich Dr.;Joaquín Barberá Dr. Dr.
Chemistry - A European Journal 2010 Volume 16( Issue 24) pp:
Publication Date(Web):
DOI:10.1002/chem.201090114
Co-reporter:Fausto Puntoriero;Francesco Nastasi;Sebastiano Campagna;Thomas Bura
Chemistry - A European Journal 2010 Volume 16( Issue 29) pp:8832-8845
Publication Date(Web):
DOI:10.1002/chem.201000466
Abstract
A series of novel multichromophoric, luminescent compounds has been prepared, and their absorption spectra, luminescence properties (both at 77 K in rigid matrix and at 298 K in fluid solution), and photoinduced intercomponent energy-transfer processes have been studied. The series contains two new multichromophoric systems 1 and 2, each one containing two different boron–dipyrromethene (Bodipy) subunits and one bridging fluorene species, and two fluorene–Bodipy bichromophoric species, 6 and 7. Three monochromophoric compounds, 3, 4, and 5, used as precursors in the synthetic process, were also fully characterized. The absorption spectra of the multichromophoric compounds are roughly the summation of the absorption spectra of their individual components, thus demonstrating the supramolecular nature of the assemblies. Luminescence studies show that quantitative energy transfer occurs in 6 and 7 from the fluorene chromophore to the Bodipy dyes. Luminescence studies, complemented by transient-absorption spectroscopy studies, also indicate that efficient inter-Bodipy energy transfer across the rigid fluorene spacer takes place in 1 and 2, with rate constants, evaluated by several experimental methods, between 2.0 and 7.0×109 s−1. Such an inter-Bodipy energy transfer appears to be governed by the Förster mechanism. By taking advantage of the presence of various protonable sites in the substituents of the lower-energy Bodipy subunit of 1 and 2, the effect of protonation on the energy-transfer rates has also been investigated. The results suggest that control of energy-transfer rate and efficiency of inter-Bodipy energy transfer in this type of systems can be achieved by an external, reversible input.
Co-reporter:Soumyaditya Mula, Kristopher Elliott, Anthony Harriman, and Raymond Ziessel
The Journal of Physical Chemistry A 2010 Volume 114(Issue 39) pp:10515-10522
Publication Date(Web):September 13, 2010
DOI:10.1021/jp106626v
We have designed and synthesized a series of modular, dual-color dyes comprising a conventional boron dipyrromethene (Bodipy) dye, as a yellow emitter, and a Bodipy dye possessing extended conjugation that functions as a red emitter. A flexible tether of variable length, built from ethylene glycol residues, connects the terminal dyes. A critical design element of this type of dyad relates to a secondary amine linkage interposed between the conventional Bodipy and the tether. Cyclic voltammetry shows both Bodipy dyes to be electroactive and indicates that the secondary amine is quite easily oxidized. The ensuing fluorescence quenching is best explained in terms of the rapid formation of an intermediate charge-transfer state. In fact, exciplex-type emission is observed in weakly polar solvents and over a critical temperature range. In the dual-color dyes, direct excitation of the yellow emitter results in the appearance of red fluorescence, indicating that the exciplex is likely involved in the energy-transfer event, and provides for a virtual Stokes shift of 5000 cm−1. Replacing the red emitter with a higher energy absorber (namely, pyrene) facilitates the collection of near-UV light and extends the virtual Stokes shift to 8000 cm−1. Modulation of the efficacy of intramolecular energy transfer is achieved by preorganization of the connector in the presence of certain cations. This latter behavior, which is fully reversible, corresponds to an artificial allosteric effect.
Co-reporter:Thomas Bura;Pascal Retailleau Dr. Dr.
Angewandte Chemie International Edition 2010 Volume 49( Issue 37) pp:6659-6663
Publication Date(Web):
DOI:10.1002/anie.201003206
Co-reporter:Anthony Harriman ; Laura J. Mallon ; Kristopher J. Elliot ; Alexandre Haefele ; Gilles Ulrich
Journal of the American Chemical Society 2009 Volume 131(Issue 37) pp:13375-13386
Publication Date(Web):August 28, 2009
DOI:10.1021/ja9038856
A series of donor−spacer−acceptor triads has been synthesized and fully characterized. Both donor and acceptor units are built from boron dipyrromethene (BODIPY) dyes but they differ in their respective conjugation lengths, and thereby offer quite disparate optical properties. The spacer units comprise an oligomer of 1,4-phenylene-diethynylene repeat units and allow the boron−boron separation distance to be varied progressively from 18 to 38 Å. A notable feature of this series is that each subunit can be selectively excited with monochromatic light. Highly efficacious electronic energy transfer (EET) occurs from the first-excited singlet state localized on the conventional BODIPY dye to its counterpart resident on the expanded BODIPY-based nucleus, but the rate constant follows a nonlinear evolution with separation distance. Overall, the rate of EET falls by only a factor of 4-fold on moving from the shortest to the longest spacer. This shallow length dependence is a consequence of the energy gap between donor and spacer units becoming smaller as the molecular length increases. Interestingly, a simple relationship exists between the measured electronic resistance of the spacer unit and the Huang−Rhys factor determined by emission spectroscopy. Both parameters relate to the effective conjugation length. Direct illumination of the spacer unit leads to EET to both terminals, followed by EET from conventional BODIPY to the expanded version. In each case, EET to the expanded dye involves initial population of the second-singlet excited state, whereas transfer from spacer to the conventional BODIPY dye populates the S2 state for shorter lengths but the S1 state for the longer analogues. The rate of EET from spacer to conventional BODIPY dye, as measured for the corresponding molecular dyads, is extremely fast (>1011 s−1) and scales with the spectral overlap integral. The relative partitioning of EET from the spacer to each terminal is somewhat sensitive to the molecular length, with the propensity to populate the conventional BODIPY dye changing from 65% for N = 0 to 45% for N = 2. The most likely explanation for this behavior can be traced to the disparate spectral overlap integrals for the two dyes. These systems have been complemented by a molecular tetrad in which pyrene residues replace the fluorine atoms present on the conventional BODIPY-based dye. Here, rapid EET occurs from pyrene to the BODIPY dye and is followed by slower, long-range EET to the opposite terminal. Such materials are seen as highly attractive solar concentrators when dispersed in transparent plastic media and used under conditions where both inter- and intramolecular EET operate.
Co-reporter:Stéphane Frein, Franck Camerel, Raymond Ziessel, Joaquín Barberá and Robert Deschenaux
Chemistry of Materials 2009 Volume 21(Issue 17) pp:3950
Publication Date(Web):August 14, 2009
DOI:10.1021/cm9008078
Fluorescent mesomorphic materials have been synthesized by grafting difluoro-bora-diaza-s-indacene (F-Bodipy) onto first-, second-, and third-generation liquid-crystalline poly(aryl ester) dendrons functionalized with cyanobiphenyl units. The second- and third-generation dendrimers give rise to smectic A phases; the first-generation dendrimer shows a nematic phase and an unidentified phase. The supramolecular organization within the smectic A phase could be established from X-ray experiments: the dendritic core is oriented approximately parallel to the layer planes and the mesogenic units are oriented perpendicularly above and below the dendritic core; interdigitation occurs between neighboring layers. All the materials are highly fluorescent both in solution and in the mesophases. For the first-generation dendrimer, the formation of J-aggregates was detected. The higher-generation dendrons prevented the formation of aggregates. Therefore, the dendrons play the role of liquid-crystalline promoters and protective shells.
Co-reporter:Maria L. Muro, Stéphane Diring, Xianghuai Wang, Raymond Ziessel and Felix N. Castellano
Inorganic Chemistry 2009 Volume 48(Issue 24) pp:11533-11542
Publication Date(Web):November 13, 2009
DOI:10.1021/ic901036a
The synthesis, structural characterization, photoluminescence, and excited state absorption properties of a series of platinum(II) terpyridyl complexes bearing a bipyridyl acetylide subunit are presented. The [tBu3tpyPtC≡Cbpy]+ (1) complex displays a broad and structureless emission profile at room temperature (RT), a lifetime of 5.8 μs, and transient absorption (TA) difference spectra characteristic of a charge transfer (CT) excited state. Upon coordination of Fe2+ to 1, producing tetranuclear 2, the CT emission was quantitatively quenched presumably through the low-lying iron-based ligand field states present. Surprisingly, the addition of Zn2+ to solutions of 1 produces a higher energy emissive state with a substantially longer excited state lifetime of 16.1 μs. The combined spectroscopic data measured for the zinc titration product (3) suggests that the overall excited state is dominated by a CT manifold, albeit at higher energy relative to 1. The photophysics of a bis-phosphine complex bearing two trans-disposed bpy-acetylide subunits (4) produced a model chromophore possessing an intraligand triplet excited state with a lifetime of 26 μs at RT. The bipyridyl analogue of 1, tBu2bpyPt(C≡Cbpy)2 (5), was also prepared and its photophysics are consistent with a lowest CT parentage at RT. The 77 K emission spectra measured for complexes 1, 3, 4, and 5 are all consistent with a triplet bpy-acetylide localized excited state; the E00 energies vary over a modest 344 cm−1 across the series. However, the shorter 77 K excited state lifetimes observed for 1, 3, and 5 in comparison to 4 suggests that the energetically proximate CT state in the former compounds significantly influences excited state decay at low temperature.
Co-reporter:Soumyaditya Mula, Gilles Ulrich, Raymond Ziessel
Tetrahedron Letters 2009 50(46) pp: 6383-6388
Publication Date(Web):
DOI:10.1016/j.tetlet.2009.08.091
Co-reporter:Jean-Hubert Olivier;Franck Camerel Dr.;Joaquín Barberá Dr.;Pascal Retailleau Dr. Dr.
Chemistry - A European Journal 2009 Volume 15( Issue 33) pp:8163-8174
Publication Date(Web):
DOI:10.1002/chem.200900721
Abstract
We have designed and synthesised a series of modular, mesogenic complexes based on anthracene-2,6-disulfonate and trialkoxybenzyl-functionalised imidazolium cations. Each complex contains a central, rigid, dianionic anthracene core and two flexible monocations bearing paraffin chains anchored on imidazolium rings. Anthracene-2,6-disulfonate can be crystallised with various simple alkylammonium ions and, in the case of +N(CH3)2(C16H33)2, a crystal structure determination has shown that the long paraffinic chains are intercalated between the anthracene moieties. The dianion forms columnar mesophases with trialkoxybenzylimidazolium cations, as identified by polarising optical microscopy and X-ray scattering measurements. Differential scanning calorimetry studies confirmed mesomorphic behaviour from room temperature to about 200 °C for alkyl chains containing 8, 12 and 16 carbon atoms. The strong luminescence of anthracene is maintained in the mesophase and fluorescence measurements confirmed the presence of J aggregates in all cases. The new functional materials described herein provide an easy access to stable and luminescent mesomorphic materials engineered by an ionic self-assembly process.
Une nouvelle série de complexes mésogènes obtenus par association électrostatique entre l’anthracène-2,6-disulfonate et des imidazoliums fonctionnalisés a été synthétisée. Les propriétés optiques et d’auto-organisation de ces complexes, constitués d’un noyau rigide central dianionique d’anthracène et de deux cations flexibles à cœur imidazolium, ont été étudiées. Associé à des dérivés d’ammoniums portant de longues chaînes carbonées, l’anthracène-2,6-disulfonate peut être cristallisé et la structure obtenue avec la cation +N(CH3)2(C16H33)2 montre qu’il n’y a pas de ségrégation entre les chaînes grasses et le cœur rigide du dérivé d’anthracène. Par contre, associé à des imidazoliums fonctionnalisés par des dérivés tris-alkoxybenzyl, une microségrégation peut être engendrée et des mésophases colonnaires ont pu être observées et identifiées par POM ainsi que par diffraction aux rayons X. Des études par calorimétrie différentielle à balayage (DSC) confirme le comportement mésomorphe des complexes sur une large gamme de température s’étalant de 20°C à 200°C avec des chaînes alkyles à 8, 12, 16 atomes de carbone. Les propriétés de luminescence de l’anthracène sont maintenues au sein des mésophases et des études par spectroscopie d’émission confirment la présence d’agrégat de type J entre les fragments aromatiques. Ces nouveaux complexes fonctionnels, obtenus par auto-assemblage par voie électrostatique, fournissent un accès rapide et simple à des matériaux mésomorphes et luminescents. Abstract in Spanish: Hemos diseñado y sintetizado una serie de complejos mesógenos modulares basados en antraceno-2,6-disulfonato y cationes imidazolio funcionalizados con tris(alcoxi)bencilo. Todos los complejos contienen un núcleo central rígido dianiónico de antraceno y dos monocationes flexibles que portan cadenas parafínicas ancladas en los anillos de imidazol. El antraceno-2,6-disulfonato puede cristalizarse con varios iones alquilamonio simples y, en el caso de +N(CH3)2(C16H33)2, la determinación de la estructura cristalina ha mostrado que las cadenas parafínicas largas están intercaladas entre las unidades de antraceno. Con los cationes tris(alcoxi)bencilimidazolio, el dianión forma mesofases columnares identificadas por medidas de microscopía óptica con luz polarizada y difracción de rayos X. Los estudios de DSC confirman el comportamiento mesomorfo desde temperatura ambiente hasta 200ºC para cadenas alquílicas que contienen 8, 12 y 16 átomos de carbono. La fuerte luminiscencia del antraceno se mantiene en la mesofase y las medidas de fluorescencia confirman la presencia de agregados J en todos los casos. Los nuevos materiales funcionales descritos aquí representan un fácil acceso a materiales mesomorfos luminiscentes estables generados por un proceso de auto-ensamblaje iónico.
Co-reporter:Francesco Nastasi, Fausto Puntoriero, Sebastiano Campagna, Jean-Hubert Olivier and Raymond Ziessel
Physical Chemistry Chemical Physics 2010 - vol. 12(Issue 27) pp:NaN7402-7402
Publication Date(Web):2010/06/09
DOI:10.1039/C003789C
By tailoring the chemistry of the 4,4-difluoro-4-bora-3a,4a-diaza-s-indacene (bodipy) core in order to change its spectroscopic properties, we linked some modified bodipy dyes to Pt(II)-terpyridine centers thereby preparing three novel hybrid complexes. In one of such species, two pyrene subunits are also present. The absorption spectra and luminescence properties (both at room temperature in acetonitrile solution and at 77 K in butyronitrile rigid matrix) of the new hybrid complexes and of their bodipy parent species have been investigated, and compared with those of previously studied compounds. Absorption spectra showed that the different chromophoric subunits roughly keep their own features in the multicomponent systems, indicating the supramolecular nature of the compounds. Efficient photoinduced intercomponent energy transfer takes place in the various hybrid Pt(II)-terpyridine-bodipy compounds, by taking advantage of different mechanisms: at room temperature in fluid solution very efficient energy transfer from the 3MLCT state(s) of the Pt residue to the lowest-lying 3bodipy is mediated by a charge transfer state via a two-step electron transfer process. Direct energy transfer from 3MLCT state(s) to the 3bodipy states takes place at 77 K, most likely via the Dexter mechanism. When two pyrene fragments are grafted to the boron, cascade energy transfer from the pyrene to the 1bodipy also occurs by the Förster mechanism. For one of the new compounds, 3bodipy emission with a maximum at 738 nm is also observed at cryogenic temperature.
Co-reporter:Francesco Nastasi, Fausto Puntoriero, Scolastica Serroni, Sebastiano Campagna, Jean-Hubert Olivier and Raymond Ziessel
Dalton Transactions 2014 - vol. 43(Issue 47) pp:NaN17658-17658
Publication Date(Web):2014/07/02
DOI:10.1039/C4DT01127A
A Bodipy species bearing an acetyl-acetonate (acac) group, 3, has been prepared from a blue absorbing borondipyrromethene core bearing gallate substituted paraffin chains. Compound 3 chelates a Pt(II) center having an orthometalated 2-phenyl-pyridine anion (ppy) as an additional ligand, giving rise to a new bichromophoric Pt(II)-Bodipy species, 1. The absorption spectra, redox behavior and photophysical properties of 1, 3 and of the neutral Pt(II) compound 2, containing ppy and an acac derivative as ligands, have been studied. Compounds 3 and 2 are used as models for the Bodipy-based and the metal-based subunits of 1, respectively. The 3LC emission of 2 is fully quenched in 1, whereas the Bodipy fluorescence is only weakly reduced in 1 compared to 3, indicating weak interaction between the subunits. Two different charge-separated (CS) states have a role in the intercomponent excited state decays of 1. Notably, whereas in all the previously investigated bichromophoric metal(polypyridine)-Bodipy compounds, the light absorbed by the metal-based unit leads to population of the lowest-energy triplet Bodipy-based level, in 1 it contributes with high efficiency (>99%) to the Bodipy fluorescence. An efficient and formally forbidden 3LC to 1Bodipy energy transfer occurring by Förster mechanism is, unprecedently, the dominant 3LC decay process in 1.
Co-reporter:Matthieu Starck and Raymond Ziessel
Dalton Transactions 2012 - vol. 41(Issue 43) pp:NaN13307-13307
Publication Date(Web):2012/07/17
DOI:10.1039/C2DT31181J
A series of lanthanide complexes has been prepared from ligands constructed from a bis-pyrazolyl-pyridine core bearing various chelating arms with anionic mixed carboxylate/phosphonate or phosphonate substituents. These ligands form particularly stable complexes with Eu(III) and Tb(III) which display outstanding spectroscopic properties, with excited state lifetimes ranging from 2.6 to 3.2 ms and quantum yields in the 16 to 48% range in water or phosphate buffer. The complexes are significantly more stable than those of analogous ligands bearing only carboxylate groups. Some of the new ligands have a central and flexible pendent link suitable for bio-labelling.
Co-reporter:Stéphane Diring, Barbara Ventura, Andrea Barbieri and Raymond Ziessel
Dalton Transactions 2012 - vol. 41(Issue 42) pp:NaN13096-13096
Publication Date(Web):2012/04/02
DOI:10.1039/C2DT30385J
The synthesis, photophysical characterization and energy-transfer features of a series of hybrid truxene derivatives peripherally decorated with inorganic Os-containing polypyridine units and organic Bodipy dyes are reported. The photoactive terminal units are coupled to the central truxene scaffold by rigid ethynyl linkers in a star-shaped arrangement. The absorption range widely covers the UV-Vis spectrum and the Os 3MLCT or the Bodipy triplet act as final collectors of the absorbed energy.
Co-reporter:Maria Pia Gullo, Julie Batcha Seneclauze, Barbara Ventura, Andrea Barbieri and Raymond Ziessel
Dalton Transactions 2013 - vol. 42(Issue 48) pp:NaN16828-16828
Publication Date(Web):2013/10/01
DOI:10.1039/C3DT52007B
This paper describes expeditious stepwise synthesis of polynuclear complexes based on heteroleptic iridium(III) and osmium(II) fragments linked to a central Pt(II) module via a spirobifluorene-bridge using a strategy based on the construction of preformed complexes. The luminescence features of the final multi-chromophoric array, i.e. a tetrad consisting of spirobifluorene-bridged Pt, Ir and Os complexes, have been studied by comparison with the features of reference complexes bearing two identical luminophores (Ir or Os) at the periphery. The 3MPtLCT and 3LC states of the Pt and spiro ligand undergo fast energy transfer into the 3MIrLCT or the 3MOsLCT state in the Pt–M2 (M = Ir or Os) arrays, whereas the 3LC and the 3MPtLCT states function as energy reservoirs for the metal excited states close in energy, resulting in a pronounced increase of the excited state lifetimes of these arrays. The tetrad efficiently works as an antenna system where the collected light energy is transferred to the Os unit acting as the final collector.
Co-reporter:Adela Nano, Maria Pia Gullo, Barbara Ventura, Nicola Armaroli, Andrea Barbieri and Raymond Ziessel
Chemical Communications 2015 - vol. 51(Issue 16) pp:NaN3354-3354
Publication Date(Web):2015/01/14
DOI:10.1039/C4CC09832C
The preparation and the photophysical behaviour of novel julolidine derivatives designed for displaying excited state intramolecular proton transfer (ESIPT) are reported. These dyes exhibit panchromatic photoluminescence covering the whole visible spectral range, both in organic solvents and in the solid state.
Co-reporter:A. Mirloup, Q. Huaulmé, N. Leclerc, P. Lévêque, T. Heiser, P. Retailleau and R. Ziessel
Chemical Communications 2015 - vol. 51(Issue 79) pp:NaN14745-14745
Publication Date(Web):2015/07/29
DOI:10.1039/C5CC05095B
The synthesis and characterization of bis(difluoroboryl)-1,2-bis((1H-pyrrol-2-yl)methylene)hydrazone functionalized with two lateral vinyl-thienyl modules and exhibiting strong absorption in the 400–800 nm window in thin films are reported. Bulk heterojunction solar cells assembled with these dyes and a fullerene derivative (PC71BM), using very small quantities of the additive diiodooctane, give a power conversion efficiency as high as 4.3% with short-circuit current values of 10.9 mA cm−2, an open-circuit voltage of 0.7 V and external quantum efficiencies higher than 70% over a broad range of wavelengths (580 to 720 nm).
Co-reporter:Denis Frath, Arnaud Poirel, Gilles Ulrich, Antoinette De Nicola and Raymond Ziessel
Chemical Communications 2013 - vol. 49(Issue 43) pp:NaN4910-4910
Publication Date(Web):2013/04/10
DOI:10.1039/C3CC41555D
Complexation of iminocoumarin derivatives with BF3·OEt2 provides novel N⁁N boron(III) dyes exhibiting high absorption coefficients and quantum yields as great as 81%. The excellent chemical stability of these dyes enables the grafting of Boranil or Bodipy units to give derivatives in which the Borico subunit can act either as an energy acceptor or as an antenna for a red emitting fluorophore.
Co-reporter:Thomas Bura, Pascal Retailleau, Maria Teresa Indelli and Raymond Ziessel
Dalton Transactions 2013 - vol. 42(Issue 13) pp:NaN4551-4551
Publication Date(Web):2013/01/07
DOI:10.1039/C2DT32538A
New Ir(III) complexes involving N,C-chelating difluorophenyl-pyridine (dfppy) or dibenzo[a,c]phenazine (dbpz) ligands along with either N,N-bound 5-ethynyl-2,2′-bipyridine (e-bpy) or CO + Cl co-ligands have been obtained as [Ir(dfppy)2(e-bpy)]PF6, [Ir(dbpz)2(e-bpy)]PF6 and cis-[Ir(dbpz)2(Cl)(CO)]. A single-crystal X-ray diffraction study of cis-[Ir(dbpz)2(CO)Cl] has shown the Ir(III) centre to adopt a distorted octahedral coordination geometry with cis-CO/Cl and trans-N,N configurations. Pronounced π–π stacking interactions involving different dibenzo[a,c]phenazine units are evident. Electronic absorption and luminescence spectroscopy at 298 K and 77 K, along with cyclic voltammetry were used to study the three complexes. Excited state lifetimes varied from 1.4 to 2.9 μs at rt with quantum yields ranging from 10.2 to 0.7%. With 5-ethynyl-2,2′-bipyridine in the first coordination sphere, the lowest emitting state is that associated with the MLCT band, whereas in the bis-phenazine case the emission is ligand-centred. Importantly, the emission wavelength can be tuned from 552 to 640 nm by changing the conjugation length of the N,C ligand.
Co-reporter:Jean-Hubert Olivier, Jack Harrowfield and Raymond Ziessel
Chemical Communications 2011 - vol. 47(Issue 40) pp:NaN11188-11188
Publication Date(Web):2011/07/25
DOI:10.1039/C1CC12771C
Synthetic routes for the construction of 3-substituted 2,4-pentadionate ligands are broadly surveyed. They involve sequential alkylation and arylation by numerous methods, including those based on reactions of coordinated ligands, and can provide access to various rationally designed ligands. Applications of such ligands in the synthesis of multichromophoric complexes are illustrated in some detail. Incorporation of 3-substituted 2,4-pentanedione units into mesomorphic and macromolecular structures is considered in relation to structural control of energy and electron transfer processes in photoactive systems. Aspects of the general supramolecular chemistry of complexes of 3-substituted-2,4-pentadionate ligands are briefly discussed to illustrate the utility of such species within the full range of 1,3-dionate chemistry.
Co-reporter:Song Lin Niu, Cédrik Massif, Gilles Ulrich, Raymond Ziessel, Pierre-Yves Renard and Anthony Romieu
Organic & Biomolecular Chemistry 2011 - vol. 9(Issue 1) pp:NaN69-69
Publication Date(Web):2010/11/18
DOI:10.1039/C0OB00693A
The synthesis and preliminary bio-conjugation studies of a novel water-soluble red-emitting di-styryl BODIPY dye are disclosed. Aggregation behaviour of this compound under physiological conditions was suppressed by specific introduction of a di-sulfonated peptide-based linker at the mesophenyl substituent, sultonated styryl arms and short polyethyleneglycol chains at the boron center. Thus, a good quantum yield of 22% in PBS for this red-emitting BODIPY was obtained. Introduction of an activated ester function enabled successful bio-conjugation to monoclonal antibodies and proteins.