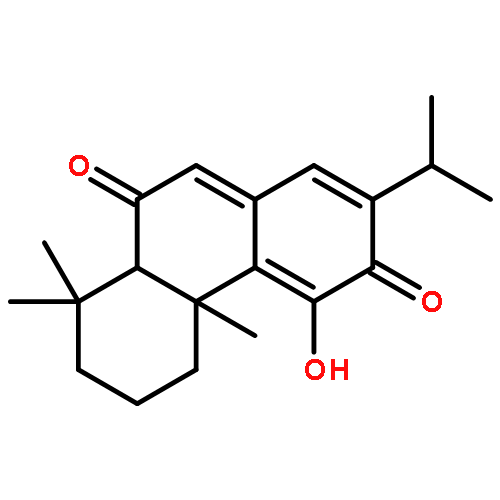
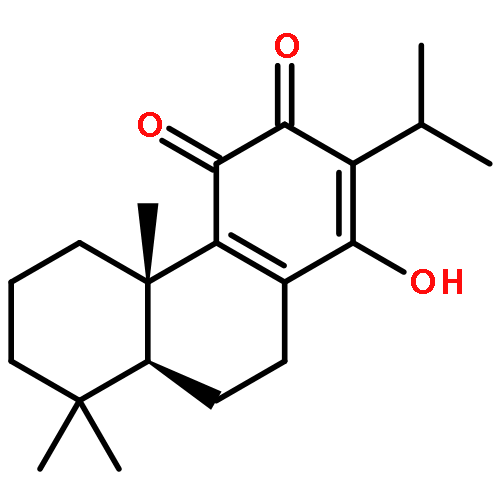
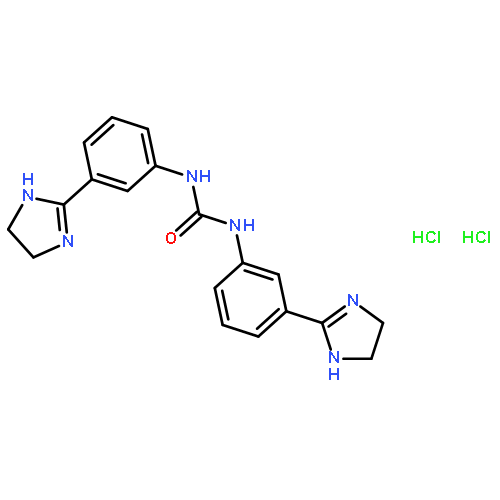
Co-reporter:Xiaohua Zhu, Abdelbasset A. Farahat, Meena Mattamana, April Joice, Trupti Pandharkar, Elizabeth Holt, Moloy Banerjee, Jamie L. Gragg, Laixing Hu, Arvind Kumar, Sihyung Yang, Michael Zhuo Wang, David W. Boykin, Karl A. Werbovetz
Bioorganic & Medicinal Chemistry Letters 2016 Volume 26(Issue 10) pp:2551-2556
Publication Date(Web):15 May 2016
DOI:10.1016/j.bmcl.2016.03.082
Arylimidamide (AIA) compounds containing two pyridylimidamide terminal groups (bis-AIAs) possess outstanding in vitro antileishmanial activity, and the frontrunner bis-AIA DB766 (2,5-bis[2-(2-isopropoxy)-4-(2-pyridylimino)aminophenyl]furan) is active in visceral leishmaniasis models when given orally. Eighteen compounds containing a single pyridylimidamide terminal group (mono-AIAs) were synthesized and evaluated for their antileishmanial potential. Six of these compounds exhibited sub-micromolar potency against both intracellular Leishmania donovani and Leishmania amazonensis amastigotes, and three of these compounds also displayed selectivity indexes of 25 or greater for the parasites compared to a J774 macrophage cell line. When given orally at a dose of 100 mg/kg/day for five days, compound 1b (N-(3-isopropoxy-4-(5-phenylfuran-2-yl)phenyl)picolinimidamide methanesulfonate) reduced liver parasitemia by 46% in L. donovani-infected mice. Mono-AIAs are thus a new class of candidate molecules for antileishmanial drug development.Figure optionsDownload full-size imageDownload as PowerPoint slide
Co-reporter:Xiaohua Zhu, Kurt S. Van Horn, Megan M. Barber, Sihyung Yang, Michael Zhuo Wang, Roman Manetsch, Karl A. Werbovetz
Bioorganic & Medicinal Chemistry 2015 Volume 23(Issue 16) pp:5182-5189
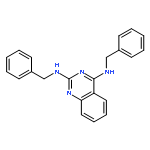
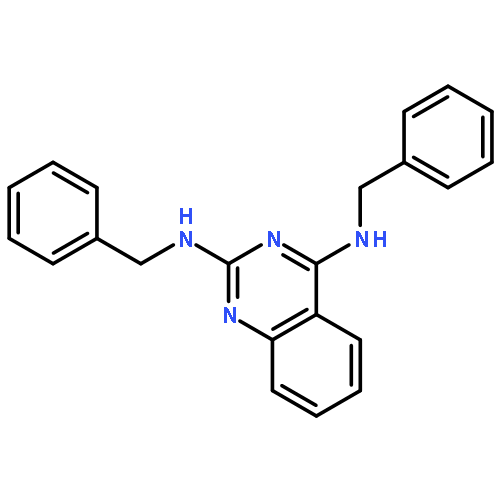
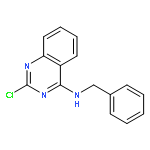
Publication Date(Web):15 August 2015
DOI:10.1016/j.bmc.2015.02.020
Visceral leishmaniasis is a neglected parasitic disease that has a high fatality rate in the absence of treatment. New drugs that are inexpensive, orally active, and effective could be useful tools in the fight against this disease. We previously showed that N2,N4-disubstituted quinazoline-2,4-diamines displayed low- to sub-micromolar potency against intracellular Leishmania, and lead compound N4-(furan-2-ylmethyl)-N2-isopropyl-7-methylquinazoline-2,4-diamine (4) exhibited modest efficacy in an acute murine model of visceral leishmaniasis. In the present work, thirty-one N2,N4-disubstituted quinazoline-2,4-diamines that had not previously been examined for their antileishmanial activity were evaluated for their potency and selectivity against Leishmania donovani, the causative parasite of visceral leishmaniasis. Quinazoline-2,4-diamines with aromatic substituents at both N2 and N4 exhibited potent in vitro antileishmanial activity but relatively low selectivity, while compounds substituted with small alkyl groups at either N2 or N4 generally showed lower antileishmanial potency but were less toxic to a murine macrophage cell line. Based on their in vitro antileishmanial potency, N4-benzyl-N2-(4-chlorobenzyl)quinazoline-2,4-diamine (15) and N2-benzyl-N4-isopropylquinazoline-2,4-diamine (40) were selected for in vivo evaluation of their pharmacokinetic and antileishmanial properties. While 15 displayed a longer plasma half-life and a greater area under the curve than 40, both compounds showed low efficacy in an acute murine visceral leishmaniasis model. Although the present study did not identify new quinazoline-2,4-diamines with promising in vivo efficacy, the reduced in vitro toxicity of derivatives bearing small alkyl groups at either N2 or N4 may provide clues for the design of safe and effective antileishmanial quinazolines.
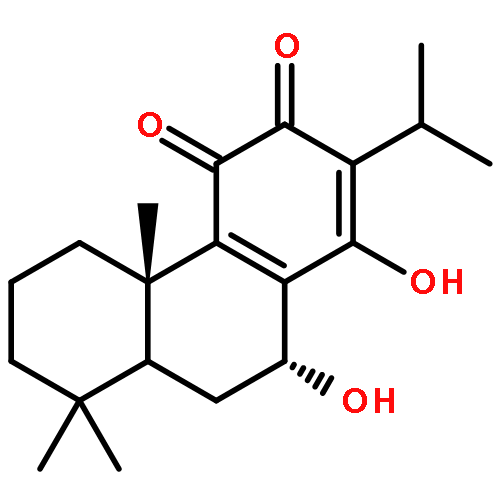
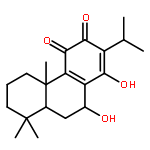
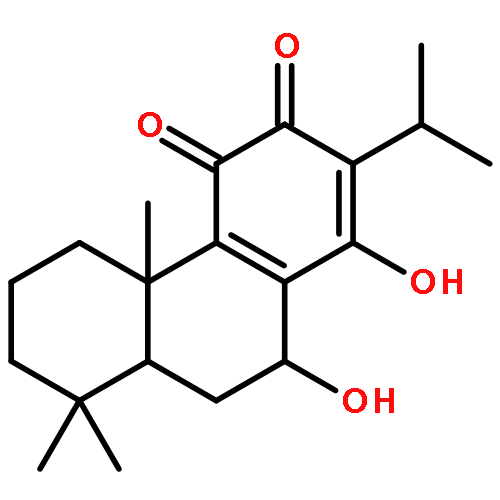
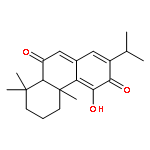
Co-reporter:Molla Endeshaw, Xiaohua Zhu, Shanshan He, Trupti Pandharkar, Emily Cason, Kiran V. Mahasenan, Hitesh Agarwal, Chenglong Li, Manoj Munde, W. David Wilson, Mark Bahar, Raymond W. Doskotch, A. Douglas Kinghorn, Marcel Kaiser, Reto Brun, Mark E. Drew, and Karl A. Werbovetz
Journal of Natural Products 2013 Volume 76(Issue 3) pp:311-315
Publication Date(Web):November 20, 2012
DOI:10.1021/np300638f
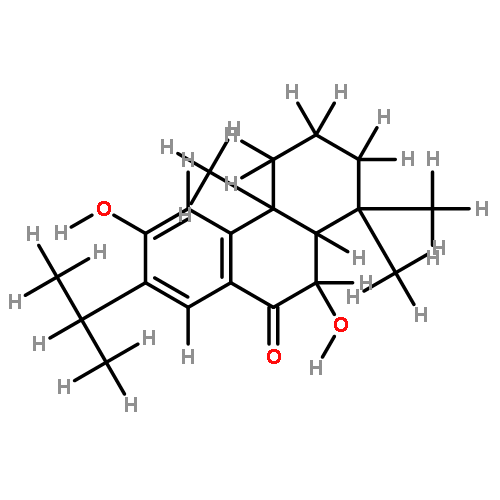
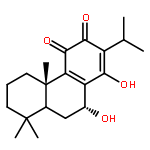
Semisynthetic 8,8-dialkyldihydroberberines (8,8-DDBs) were found to possess mid- to low-nanomolar potency against Plasmodium falciparum blood-stage parasites, Leishmania donovani intracellular amastigotes, and Trypanosoma brucei brucei bloodstream forms. For example, 8,8-diethyldihydroberberine chloride (5b) exhibited in vitro IC50 values of 77, 100, and 5.3 nM against these three parasites, respectively. In turn, two 8,8-dialkylcanadines, obtained by reduction of the corresponding 8,8-DDBs, were much less potent against these parasites in vitro. While the natural product berberine is a weak DNA binder, the 8,8-DDBs displayed no affinity for DNA, as assessed by changes in the melting temperature of poly(dA·dT) DNA. Selected 8,8-DDBs showed efficacy in mouse models of visceral leishmaniasis and African trypanosomiasis, with 8,8-dimethyldihydroberberine chloride (5a) reducing liver parasitemia by 46% in L. donovani-infected BALB/c mice when given at an intraperitoneal dose of 10 mg/kg/day for five days. The 8,8-DDBs may thus serve as leads for discovering new antimalarial, antileishmanial, and antitrypanosomal drug candidates.
Co-reporter:Carolyn S. Reid, Abdelbasset A. Farahat, Xiaohua Zhu, Trupti Pandharkar, David W. Boykin, Karl A. Werbovetz
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 22) pp:6806-6810
Publication Date(Web):15 November 2012
DOI:10.1016/j.bmcl.2012.06.037
Analogs of the lead antileishmanial bis-arylimidamide DB766 were prepared that possess unsymmetrical substitutions on the diphenylfuran linker, and an additional compound was synthesized that contains isopropoxy groups meta to the central furan. These agents all displayed nanomolar in vitro potency against intracellular Leishmania with selectivity indexes >100 compared to J774 macrophages. While the unsymmetrical analogs were toxic to mice when given ip at 30 mg/kg/day, the compound bearing the meta isopropoxy groups was well tolerated by mice and showed activity in a murine model of visceral leishmaniasis when administered ip at 30 mg/kg/day for five days.
Co-reporter:Carolyn S. Reid, Donald A. Patrick, Shanshan He, Jean Fotie, Kokku Premalatha, Richard R. Tidwell, Michael Zhuo Wang, Qiang Liu, Pavel Gershkovich, Kishor M. Wasan, Tanja Wenzler, Reto Brun, Karl A. Werbovetz
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 1) pp:513-523
Publication Date(Web):1 January 2011
DOI:10.1016/j.bmc.2010.11.003
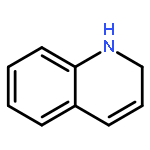
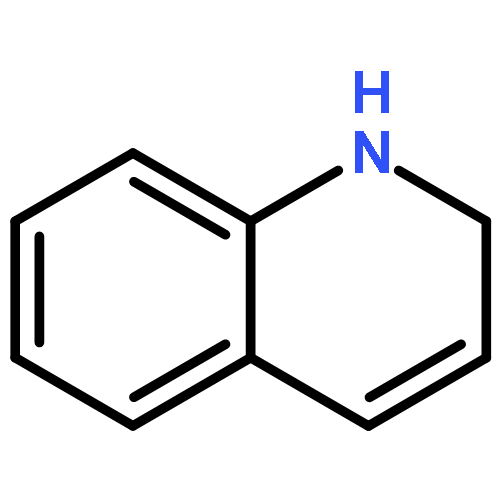
Analogs of the trypanocidal lead compound 1-benzyl-1,2-dihydro-2,2,4-trimethylquinolin-6-yl acetate were prepared to extend the structure–activity relationship in this series of molecules, improve the in vivo antitrypanosomal activity of the lead, and determine whether ester prodrugs are needed to overcome the instability of the dihydroquinolin-6-ols. Two of the most active compounds identified in this study were 1-benzyl-1,2-dihydro-2,2,4-trimethylquinolin-6-ol hydrochloride and 1-(2-methoxy)benzyl-1,2-dihydro-2,2,4-trimethylquinolin-6-ol hydrochloride. These stable solids possessed low nanomolar IC50 values against Trypanosoma brucei rhodesiense STIB900 in vitro and provided cures in an early treatment acute mouse model of African trypanosomiasis when given ip at 50 mg/kg/day for four consecutive days.
Co-reporter:Jean Fotie ; Marcel Kaiser ; Dawn A. Delfín ; Joshua Manley ; Carolyn S. Reid ; Jean-Marc Paris ; Tanja Wenzler ; Louis Maes ; Kiran V. Mahasenan ; Chenglong Li ;Karl A. Werbovetz
Journal of Medicinal Chemistry 2010 Volume 53(Issue 3) pp:966-982
Publication Date(Web):January 4, 2010
DOI:10.1021/jm900723w
The current chemotherapy for second stage human African trypanosomiasis is unsatisfactory. A synthetic optimization study based on the lead antitrypanosomal compound 1,2-dihydro-2,2,4-trimethylquinolin-6-yl 3,5-dimethoxybenzoate (TDR20364, 1a) was undertaken in an attempt to discover new trypanocides with potent in vivo activity. While 6-ether derivatives were less active than the lead compound, several N1-substituted derivatives displayed nanomolar IC50 values against T. b. rhodesiense STIB900 in vitro, with selectivity indexes up to >18000. 1-Benzyl-1,2-dihydro-2,2,4-trimethylquinolin-6-yl acetate (10a) displayed an IC50 value of 0.014 μM against these parasites and a selectivity index of 1700. Intraperitoneal administration of 10a at 50 (mg/kg)/day for 4 days caused a promising prolongation of lifespan in T. b. brucei STIB795-infected mice (>14 days vs 7.75 days for untreated controls). Reactive oxygen species were produced when T. b. brucei were exposed to 10a in vitro, implicating oxidative stress in the trypanocidal mode of action of these 1,2-dihydroquinoline derivatives.
Co-reporter:Molla M. Endeshaw, Catherine Li, Jessica de Leon, Ni Yao, Kirk Latibeaudiere, Kokku Premalatha, Naomi Morrissette, Karl A. Werbovetz
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 17) pp:5179-5183
Publication Date(Web):1 September 2010
DOI:10.1016/j.bmcl.2010.07.003
The synthesis and evaluation of 20 dinitroanilines and related compounds against the obligate intracellular parasite Toxoplasmagondii is reported. Using in vitro cultures of parasites in human fibroblasts, we determined that most of these compounds selectively disrupted Toxoplasma microtubules, and several displayed sub-micromolar potency against the parasite. The most potent compound was N1,N1-dipropyl-2,6-dinitro-4-(trifluoromethyl)-1,3-benzenediamine (18b), which displayed an IC50 value of 36 nM against intracellular T. gondii. Based on these data and another recent report [Ma, C.; Tran, J.; Gu, F.; Ochoa, R.; Li, C.; Sept, D.; Werbovetz, K.; Morrissette, N. Antimicrob. AgentsChemother. 2010, 54, 1453], an antimitotic structure–activity relationship for dinitroanilines versus Toxoplasma is presented.
Co-reporter:Laixing Hu, Reem K. Arafa, Mohamed A. Ismail, Tanja Wenzler, Reto Brun, Manoj Munde, W. David Wilson, Sandra Nzimiro, Serene Samyesudhas, Karl A. Werbovetz, David W. Boykin
Bioorganic & Medicinal Chemistry Letters 2008 Volume 18(Issue 1) pp:247-251
Publication Date(Web):1 January 2008
DOI:10.1016/j.bmcl.2007.10.091
Eighteen diamidino azaterphenyls and analogues were evaluated as anti-leishmanials; nine of the compounds gave IC50 values less than 1 μM, five exhibited values less than 0.40 μM, and two gave values less than 0.10 μM in a Leishmania donovani axenic amastigote assay. The activity of the diamidines strongly depends on the ring N-atom location relative to the amidine groups and correlates with DNA affinity. Transmission electron microscopy studies showed a dramatic dilation of the mitochondrion and evidence of disintegration of the kinetoplast of the amastigotes.
Co-reporter:Tesmol G. George, Jayaseharan Johnsamuel, Dawn A. Delfín, Adam Yakovich, Mitali Mukherjee, Mitch A. Phelps, James T. Dalton, Dan L. Sackett, Marcel Kaiser, Reto Brun, Karl A. Werbovetz
Bioorganic & Medicinal Chemistry 2006 Volume 14(Issue 16) pp:5699-5710
Publication Date(Web):15 August 2006
DOI:10.1016/j.bmc.2006.04.017
N1-Phenyl-3,5-dinitro-N4,N4-di-n-propylsulfanilamide (1) and N1-phenyl-3,5-dinitro-N4,N4-di-n-butylsulfanilamide (2) show potent in vitro antimitotic activity against kinetoplastid parasites but display poor in vivo activity. Seventeen new dinitroaniline sulfonamide and eleven new benzamide analogs of these leads are reported here. Nine of the sulfonamides display in vitro IC50 values under 500 nM against African trypanosomes, and the most active antikinetoplastid compounds also inhibit the in vitro assembly of purified leishmanial tubulin with potencies similar to that of 2. While several of the potent compounds are rapidly degraded by rat liver S9 fractions in vitro, N1-(3-hydroxy)phenyl-3,5-dinitro-N4,N4-di-n-butylsulfanilamide (21) displays an IC50 value of 260 nM against African trypanosomes in vitro and is more stable than 2 in the in vitro metabolism assay.Seventeen new dinitroaniline sulfonamides and eleven dinitroaniline benzamides are reported. Nine of the sulfonamides display in vitro IC50 values under 500 nM against African trypanosomes.
Co-reporter:Denise M Cottrell, Jeffrey Capers, Manar M Salem, Kate DeLuca-Fradley, Simon L Croft, Karl A Werbovetz
Bioorganic & Medicinal Chemistry 2004 Volume 12(Issue 11) pp:2815-2824
Publication Date(Web):1 June 2004
DOI:10.1016/j.bmc.2004.03.054
A series of 5-thiocyanatomethyl- and 5-alkyl-3-aryl-1,2,4-oxadiazoles were synthesized and evaluated for their activity against kinetoplastid parasites. Formation of the oxadiazole ring was accomplished through the reaction of benzamidoximes with acyl chlorides, while the thiocyanate group was inserted by reacting the appropriate 5-halomethyl oxadiazole with ammonium thiocyanate. The thiocyanate-containing compounds possessed low micromolar activity against Leishmania donovani and Trypanosoma brucei, while the 5-alkyl oxadiazoles were less active against these parasites. 3-(4-Chlorophenyl)-5-(thiocyanatomethyl)-1,2,4-oxadiazole (compound 4b) displayed modest selectivity for L. donovani axenic amastigote-like parasites over J774 macrophages, PC3 prostate cancer cells, and Vero cells (6.4-fold, 3.8-fold, and 9.1-fold, respectively), while 3-(3,4-dichlorophenyl)-5-(thiocyanatomethyl)-1,2,4-oxadiazole (compound 4h) showed 30-fold selectivity against Vero cells but was not selective against PC3 cells. In a murine model of visceral leishmaniasis, compound 4b decreased liver parasitemia caused by L. donovani by 48% when given in five daily i.v. doses at 5 mg/kg and by 61% when administered orally for 5 days at 50 mg/kg. These results indicate that aromatic thiocyanates hold promise for the treatment of leishmanial infections if the selectivity of these compounds can be improved.A series of 3-aryl-5-thiocyanatomethyl-1,2,4-oxadiazoles was synthesized and tested for activity against Leishmania and African trypanosomes. Compounds 4b (R1 = Cl, R2 = R3 = H) and 4h (R1 = R2 = Cl, R3 = H) were the most active in vitro, displaying IC50 values of 4.5 and 2.3 μM against L. donovani amastigote-like forms, respectively. Compound 4b reduced liver parasite burdens in a murine model of visceral leishmaniasis by 61% at an oral dose of 50 mg/kg/day for 5 days.


![4-QUINAZOLINAMINE, 2-CHLORO-N-[(4-METHOXYPHENYL)METHYL]-](http://img.cochemist.com/ccimg/512900/512803-45-1.png)
![4-QUINAZOLINAMINE, 2-CHLORO-N-[(4-METHOXYPHENYL)METHYL]-](http://img.cochemist.com/ccimg/512900/512803-45-1_b.png)