Co-reporter:Takashi Kippo, Kanako Hamaoka, Mitsuhiro Ueda, Takahide Fukuyama, and Ilhyong Ryu
Organic Letters October 6, 2017 Volume 19(Issue 19) pp:
Publication Date(Web):September 25, 2017
DOI:10.1021/acs.orglett.7b02471
A radical-chain addition of allyl bromides to aryl alkenes, vinyl ester, and vinyl phthalimide was studied in which elusive β-bromoalkyl radicals were trapped efficiently to give 5-bromo-1-pentenes in good to high yields (16 examples). A subsequent carbonylative radical cyclization with AIBN/Bu3SnH/CO was successful in giving the corresponding 3,5-disubstituted cyclohexanone derivatives in moderate yields. Synthesis of a piperidine ring was also successful by subsequent reaction with primary amine.
Co-reporter:Shuhei Sumino, Misae Uno, Takahide Fukuyama, Ilhyong Ryu, Makoto Matsuura, Akinori Yamamoto, and Yosuke Kishikawa
The Journal of Organic Chemistry May 19, 2017 Volume 82(Issue 10) pp:5469-5469
Publication Date(Web):May 5, 2017
DOI:10.1021/acs.joc.7b00609
Photoredox-catalyzed hydrodifluoroalkylation of alkenes proceeded smoothly in the presence of a Hantzsch ester as a hydrogen source under visible light irradiation. The reaction was also applicable to the hydrodifluoroalkylation of alkynes, and a continuous photo flow reaction was also successful.
Co-reporter:Takahide Fukuyama, Tomohiro Nishikawa, Keiichi Yamada, Davide Ravelli, Maurizio Fagnoni, and Ilhyong Ryu
Organic Letters December 1, 2017 Volume 19(Issue 23) pp:6436-6436
Publication Date(Web):November 20, 2017
DOI:10.1021/acs.orglett.7b03339
Tetrabutylammonium decatungstate (TBADT)-photocatalyzed C–H functionalization of alkylpyridines was investigated. Unlike alkylbenzene counterparts, alkylation of α-C–H bonds did not proceed for the reaction of 2- and 4-alkylpyridines and reluctantly proceeded for 3-alkylpyridines, which allow site-selective C(sp3)–H functionalization at nonbenzylic positions. The observed nonbenzylic site selectivities are rationalized by the polar inductive effects of pyridyl groups in the SH2 transition states. Consecutive γ-functionalization and α-bromofunctionalization were successfully carried out in selected cases.
Co-reporter:Matteo C. Quattrini;Saki Fujii;Keiichi Yamada;Takahide Fukuyama;Davide Ravelli;Maurizio Fagnoni
Chemical Communications 2017 vol. 53(Issue 15) pp:2335-2338
Publication Date(Web):2017/02/16
DOI:10.1039/C6CC09725A
A facile sunlight-induced derivatization of heteroaromatics via photocatalyzed C–H functionalization in amides, ethers, alkanes and aldehydes is described. Tetrabutylammonium decatungstate (TBADT) was used as the photocatalyst and allowed to carry out the process under mild conditions.
Co-reporter:Takahide Fukuyama;Md. Taifur Rahman;Hiroshi Mashima;Hideo Takahashi
Organic Chemistry Frontiers 2017 vol. 4(Issue 9) pp:1863-1866
Publication Date(Web):2017/08/22
DOI:10.1039/C7QO00331E
The ionic liquids bearing an aromatic vinylic C–H moiety are not innocent during Pd-catalyzed cross-coupling reactions of aryl halides, since palladium-catalyzed direct C–H arylation of thiazolium and imidazolium ionic liquids competes.
Co-reporter:Takahide Fukuyama, Yuki Fujita, Muhammad Abid Rashid, and Ilhyong Ryu
Organic Letters 2016 Volume 18(Issue 20) pp:5444-5446
Publication Date(Web):October 13, 2016
DOI:10.1021/acs.orglett.6b02727
A Cossy 5-exo-dig photocyclization of organohalides (X = Br, I) onto a C–C triple bond was studied using a flow photomicroreactor, which proceeded in a minute order of residence time. A deuterium labeling study supported the nonchain radical mechanism proposed by the Cossy group, in which a hydrogen source originates from a triethylamine cation radical. Scalable flow synthesis using a larger volume of flow reactor was also successful, providing 4 g of the product in high yield.
Co-reporter:Shuhei Sumino and Ilhyong Ryu
Organic Letters 2016 Volume 18(Issue 1) pp:52-55
Publication Date(Web):December 21, 2015
DOI:10.1021/acs.orglett.5b03238
The hydroalkylation of alkenes using alkyl iodides with Hantzsch ester as a hydrogen source occurred smoothly under a Pd/light system, in a novel, tin-free Giese reaction. A chemoselective reaction at C(sp3)–I in the presence of a C(sp2)–X (X = Br or I) bond was attained, which allowed for the stepwise functionalization of two types of C–X bonds in a one-pot procedure.
Co-reporter:Takashi Kippo, Kanako Hamaoka, Mitsuhiro Ueda, Takahide Fukuyama, Ilhyong Ryu
Tetrahedron 2016 Volume 72(Issue 48) pp:7866-7874
Publication Date(Web):1 December 2016
DOI:10.1016/j.tet.2016.05.084
The full scope (30 examples) of the radical bromoallylation of alkynes using allyl bromides was studied. In this reaction, bromine radical adds to alkynes to form vinyl radicals, which then undergo an SH2′ reaction with allyl bromides to produce good yields of 1-bromo-1,4-dienes with the liberation of a bromine radical, which creates a radical chain. The sp2-carbon–bromine bond of the product dienes was further functionalized via cross-coupling and carbonylation reactions.
Co-reporter:Shuhei Sumino, Takahito Ui, Yuki Hamada, Takahide Fukuyama, and Ilhyong Ryu
Organic Letters 2015 Volume 17(Issue 20) pp:4952-4955
Publication Date(Web):October 6, 2015
DOI:10.1021/acs.orglett.5b02302
A carbonylative Mizoroki–Heck reaction using alkyl iodides was achieved with a Pd/photoirradiation system using DBU as a base. In this reaction, alkyl radicals were formed from alkyl iodides via single-electron transfer (SET) and then underwent a sequential addition to CO and alkenes to give β-keto radicals. It is proposed that DBU would abstract a proton α to carbonyl to form radical anions, giving α,β-unsaturated ketones via SET.
Co-reporter:Keiichi Yamada, Megumi Okada, Takahide Fukuyama, Davide Ravelli, Maurizio Fagnoni, and Ilhyong Ryu
Organic Letters 2015 Volume 17(Issue 5) pp:1292-1295
Publication Date(Web):February 18, 2015
DOI:10.1021/acs.orglett.5b00282
β- or γ-Site-selective C–H alkylation of aliphatic nitriles has been achieved using a decatungstate salt as the photocatalyst. The observed site selectivity was justified by a radical polar effect in transition states for hydrogen abstraction.
Co-reporter:Dr. Takuji Kawamoto;Aoi Sato ;Dr. Ilhyong Ryu
Chemistry - A European Journal 2015 Volume 21( Issue 42) pp:14764-14767
Publication Date(Web):
DOI:10.1002/chem.201503164
Abstract
Transition metal-catalyzed aminocarbonylation of aryl halides with CO and amines, pioneered by Heck and co-workers in the 1970s, is among the most commonly employed reactions to make aromatic amides. A catalyst-free aminocarbonylation of aryl iodides with CO and amines, which simply uses photoirradiation conditions by Xe-lamp, has now been developed. This methodology shows broad functional-group tolerance, including that of heteroaromatic amides. A hybrid radical/ionic chain mechanism, involving electron transfer from zwitterionic radical intermediates generated by nucleophilic attack of amines to aroyl radicals, is proposed.
Co-reporter:Megumi Okada, Keiichi Yamada, Takahide Fukuyama, Davide Ravelli, Maurizio Fagnoni, and Ilhyong Ryu
The Journal of Organic Chemistry 2015 Volume 80(Issue 18) pp:9365-9369
Publication Date(Web):August 28, 2015
DOI:10.1021/acs.joc.5b01850
A photocatalytic synthesis of homoallyl ketones was achieved via a one-pot procedure starting from a Norrish Type I reaction of cyclopentanones, followed by a decatungstate-catalyzed hydroacylation of electron-deficient olefins by the resulting 4-pentenals. The site-selective formyl H-abstraction in the second step can be explained by radical polar effects in the transition state.
Co-reporter:Shuhei Sumino, Akira Fusano, Takahide Fukuyama, and Ilhyong Ryu
Accounts of Chemical Research 2014 Volume 47(Issue 5) pp:1563
Publication Date(Web):April 8, 2014
DOI:10.1021/ar500035q
Numerous methods for transition metal catalyzed carbonylation reactions have been established. Examples that start from aryl, vinyl, allyl, and benzyl halides to give the corresponding carboxylic acid derivatives have all been well documented. In contrast, the corresponding alkyl halides often encounter difficulty. This is inherent to the relatively slow oxidative addition step onto the metal center and subsequent β-hydride elimination which causes isomerization of the alkyl metal species. Radical carbonylation reactions can override such problems of reactivity; however, carbonylation coupled to iodine atom transfer (atom transfer carbonylation), though useful, often suffers from a slow iodine atom transfer step that affects the outcome of the reaction. We found that atom transfer carbonylation of primary, secondary, and tertiary alkyl iodides was efficiently accelerated by the addition of a palladium catalyst under light irradiation. Stereochemical studies support a mechanistic pathway based on the synergic interplay of radical and Pd-catalyzed reaction steps which ultimately lead to an acylpalladium species. The radical/Pd-combined reaction system has a wide range of applications, including the synthesis of carboxylic acid esters, lactones, amides, lactams, and unsymmetrical ketones such as alkyl alkynyl and alkyl aryl ketones. The design of unique multicomponent carbonylation reactions involving vicinal C-functionalization of alkenes, double and triple carbonylation reactions, in tandem with radical cyclization reactions, has also been achieved. Thus, the radical/Pd-combined strategy provides a solution to a longstanding problem of reactivity involving the carbonylation of alkyl halides. This novel methodology expands the breadth and utility of carbonylation chemistry over either the original radical carbonylation reactions or metal-catalyzed carbonylation reactions.
Co-reporter:Megumi Okada, Takahide Fukuyama, Keiichi Yamada, Ilhyong Ryu, Davide Ravelli and Maurizio Fagnoni
Chemical Science 2014 vol. 5(Issue 7) pp:2893-2898
Publication Date(Web):08 May 2014
DOI:10.1039/C4SC01072H
Sunlight-induced direct regioselective β-alkylation of cyclopentanones with electron-deficient alkenes was accomplished by using tetrabutylammonium decatungstate (TBADT) as the catalyst. The regiochemistry can be rationalized by a polar transition state for an SH2 reaction. In the presence of CO, the reaction gave the three-component β-acylation product in good yield.
Co-reporter:Takahide Fukuyama, Takenori Totoki and Ilhyong Ryu
Green Chemistry 2014 vol. 16(Issue 4) pp:2042-2050
Publication Date(Web):13 Dec 2013
DOI:10.1039/C3GC41789A
Microreactors have brought significant improvements to chemical synthesis and production because of their advantageous characteristics over batch reactors, which include highly efficient mixing, efficient heat and mass transfer ability, precise control of the residence time, large surface area-to-volume ratio and high operational safety. Microreactor technology has been found to be beneficial for gas–liquid biphasic reactions, for which the large interfacial area between the two phases is ensured. Carbonylation reactions with carbon monoxide, by which a wide range of carbonyl compounds can be prepared, deal with a variety of reactive species, such as organo transition metals, radicals, cations and anions. These reactions have long been carried out using a glass batch flask or a stainless-steel made autoclave, however, carbonylation reactions using a flow microreactor are now rapidly increasing in popularity. This review focuses on a new greener wave of carbonylation reactions using a flow microreactor.
Co-reporter:Takashi Kippo and Ilhyong Ryu
Chemical Communications 2014 vol. 50(Issue 45) pp:5993-5996
Publication Date(Web):16 Apr 2014
DOI:10.1039/C4CC01597E
A bromine-radical mediated three-component coupling reaction was effectively achieved by the use of allenes, electron-deficient alkenes, and allyl bromides and led to the synthesis of 2-bromo-1,7-dienes in good to high yields. This protocol was extended to the three-component process using alkylidenecyclopropane, which gave 2-bromo-1,8-diene along with alkylidenecyclopentane.
Co-reporter:Takuji Kawamoto, Aoi Sato, and Ilhyong Ryu
Organic Letters 2014 Volume 16(Issue 8) pp:2111-2113
Publication Date(Web):April 3, 2014
DOI:10.1021/ol500614q
Radical biaryl coupling of iodoarenes and benzene can be effectively promoted by tetrabutylammonium cyanoborohydride with air under photoirradiation conditions using a Xe lamp. The utility of this methodology is highlighted by its functional group tolerance and chemoselectivity.
Co-reporter:Takahide Fukuyama, Shinji Maetani, Kazusa Miyagawa, and Ilhyong Ryu
Organic Letters 2014 Volume 16(Issue 12) pp:3216-3219
Publication Date(Web):June 9, 2014
DOI:10.1021/ol5012407
An efficient approach to the synthesis of fluorenones via the rhodium-catalyzed intramolecular acylation of biarylcarboxylic acids was developed. Using this procedure, fluorenones with various substituents can be synthesized in good to high yields. This work marks the first recorded use of catalytic intramolecular acylation to synthesize fluorenones.
Co-reporter:Takahide Fukuyama, Takenori Totoki, and Ilhyong Ryu
Organic Letters 2014 Volume 16(Issue 21) pp:5632-5635
Publication Date(Web):October 16, 2014
DOI:10.1021/ol5026958
The generation of, and subsequent reactions with, 1-silyl-substituted organolithiums with CO was carried out using serially connected flow microreactors. The flow system proved to be quite useful for the carbonylation of silyl-substituted organolithiums under slightly pressurized conditions of CO, which was created conveniently by the use of a back-pressure regulator. This flow system, coupled with heating, accelerated the carbonylation reaction of 1-silyl-substituted organolithiums and allowed the stable silyl-substituted alkyllithium, 1,3-disilylallyllithium, which was not effective in a batch-flask reaction under a CO atmosphere, to participate in an efficient carbonylation reaction.
Co-reporter:Takuji Kawamoto and Ilhyong Ryu
Organic & Biomolecular Chemistry 2014 vol. 12(Issue 48) pp:9733-9742
Publication Date(Web):01 Oct 2014
DOI:10.1039/C4OB01784F
Borohydrides are an important class of reagents in both organic and inorganic chemistry. Though popular as hydride-transfer reagents for reduction, since earlier work from the 1970s, borohydride reagents have also been known to serve as hydrogen-transfer reagents. In pursuit of greener tin hydride substitutes, recent progress has been made to mediate radical C–C bond forming reactions, including Giese reactions, radical carbonylation and addition to HCHO reactions, with borohydride reagents. This review article focuses on state-of-the-art borohydride based radical reactions, also covering earlier work, kinetics and some DFT calculations with respect to the hydrogen transfer mechanism.
Co-reporter:Dr. Yoshiyuki Manabe;Yuriko Kitawaki;Masahiro Nagasaki; Koichi Fukase; Hiroshi Matsubara;Yoshiko Hino; Takahide Fukuyama; Ilhyong Ryu
Chemistry - A European Journal 2014 Volume 20( Issue 40) pp:12750-12753
Publication Date(Web):
DOI:10.1002/chem.201402303
Abstract
The photobromination of CH bonds by using molecular bromine was reinvestigated under microfluidic conditions. The continuous-flow method suppressed the production of dibrominated compounds and effectively produced the desired monobrominated products with high selectivity. Rapid bromination of benzylic substrates containing a photoaffinity azide group was achieved without any decomposition.
Co-reporter:Takuji Kawamoto, Shohei Uehara, Hidefumi Hirao, Takahide Fukuyama, Hiroshi Matsubara, and Ilhyong Ryu
The Journal of Organic Chemistry 2014 Volume 79(Issue 9) pp:3999-4007
Publication Date(Web):April 9, 2014
DOI:10.1021/jo500464q
Cyanoborohydrides are efficient reagents in the reductive addition reactions of alkyl iodides and electron-deficient olefins. In contrast to using tin reagents, the reaction took place chemoselectively at the carbon–iodine bond but not at the carbon–bromine or carbon–chlorine bond. The reaction system was successfully applied to three-component reactions, including radical carbonylation. The rate constant for the hydrogen abstraction of a primary alkyl radical from tetrabutylammonium cyanoborohydride was estimated to be <1 × 104 M–1 s–1 at 25 °C by a kinetic competition method. This value is 3 orders of magnitude smaller than that of tributyltin hydride.
Co-reporter:Takahide Fukuyama ; Nao Nakashima ; Takuma Okada
Journal of the American Chemical Society 2013 Volume 135(Issue 3) pp:1006-1008
Publication Date(Web):January 11, 2013
DOI:10.1021/ja312654q
A free-radical-mediated [2 + 2 + 1] cycloaddition reaction comprising acetylenes, amidines, and CO was achieved by radical chain reaction to give five-membered α,β-unsaturated lactams in good yields. Both acyclic and cyclic amidines reacted with a variety of terminal acetylenes to afford monocyclic, bicyclic, and tricyclic lactams. We propose that vinyl radical carbonylation and nucleophilic addition of the amidine onto the resulting α-ketenyl radical give stable intermediates that are ready to undergo five-membered ring closure with elimination of tin radical.
Co-reporter:Ilhyong Ryu, Akihiro Tani, Takahide Fukuyama, Davide Ravelli, Sara Montanaro, and Maurizio Fagnoni
Organic Letters 2013 Volume 15(Issue 10) pp:2554-2557
Publication Date(Web):May 7, 2013
DOI:10.1021/ol401061v
Tetrabutylammonium decatungstate (TBADT) accelerated the addition of C–H bonds to the N═N double bond of diisopropyl azodicarboxylate (DIAD) under irradiation conditions. The photoinduced three-component coupling between cyclic alkanes, CO, and DIAD was also achieved to give the corresponding acyl hydrazides.
Co-reporter:Célia Brancour, Takahide Fukuyama, Yu Mukai, Troels Skrydstrup, and Ilhyong Ryu
Organic Letters 2013 Volume 15(Issue 11) pp:2794-2797
Publication Date(Web):May 22, 2013
DOI:10.1021/ol401092a
Carbonylation reactions, such as Heck, Sonogashira, and radical carbonylations, were successfully carried out in a “two-chamber reactor” where carbon monoxide was produced ex situ by the Morgan reaction (dehydration of formic acid by sulfuric acid). By a subsequent application in a microflow system using a “tube-in-tube” reactor where gas-permeable Teflon AF2400 was used as the inner tube, it is demonstrated that formic acid/sulfuric acid can be employed concomitantly with an amine base such as triethylamine in the Heck aminocarbonylation of aryl iodide.
Co-reporter:Shinji Maetani, Takahide Fukuyama, and Ilhyong Ryu
Organic Letters 2013 Volume 15(Issue 11) pp:2754-2757
Publication Date(Web):May 16, 2013
DOI:10.1021/ol4010905
Rhodium-catalyzed intramolecular C–H arylation of 2-aryloxybenzoic acids proceeded accompanied by decarbonylation to give dibenzofuran derivatives in high yields. The present reaction is widely applicable to substrates bearing various functionalities.
Co-reporter:Shuhei Sumino, Akira Fusano, and Ilhyong Ryu
Organic Letters 2013 Volume 15(Issue 11) pp:2826-2829
Publication Date(Web):May 22, 2013
DOI:10.1021/ol4011536
Atom-transfer radical (ATR) reactions of alkenes with R–X usually give products having new C–C and C–X bonds at the adjacent carbons. However, when the reaction was carried out under irradiation using a low-pressure Hg lamp, addition/reduction products were obtained in good yield. Hydrogen bromide, formed by H-abstraction of a bromine radical from alkenes, is likely to play a key role in the reductive ATR reaction.
Co-reporter:Shuhei Sumino, Takahito Ui, and Ilhyong Ryu
Organic Letters 2013 Volume 15(Issue 12) pp:3142-3145
Publication Date(Web):June 3, 2013
DOI:10.1021/ol401363t
Alkyl aryl ketones were synthesized by the carbonylative cross-coupling reaction of alkyl iodides and arylboronic acids under combined Pd/light conditions. In this reaction, it is likely that an acylpalladium species would be formed via carbonylation of the alkyl radical, which would then undergo transmetalation of an arylboronic acid to give the corresponding acyl(aryl)palladium species, ready to undergo reductive elimination to yield the alkyl aryl ketone.
Co-reporter:Takashi Kuwahara, Takahide Fukuyama and Ilhyong Ryu
RSC Advances 2013 vol. 3(Issue 33) pp:13702-13704
Publication Date(Web):24 Jun 2013
DOI:10.1039/C3RA42834F
The α-alkylation reaction of acetamides with primary alcohols to afford the corresponding amides was accomplished effectively using RuHCl(CO)(PPh3)3 as a catalyst, nitrogen tridentate ligand L1 as an additive, and KOtBu as a base. While the addition of bpy was effective only for benzylic alcohols, L1 affected the alkylation reaction when both benzylic and non-benzylic type alcohols were used.
Co-reporter:Takashi Kippo ; Kanako Hamaoka
Journal of the American Chemical Society 2012 Volume 135(Issue 2) pp:632-635
Publication Date(Web):December 26, 2012
DOI:10.1021/ja311821h
Bromine radical-mediated cyclopropylcarbinyl-homoallyl rearrangement of alkylidenecyclopropanes was effectively accomplished by C–C bond formation with allylic bromides, which led to the syntheses of 2-bromo-1,6-dienes. A three-component coupling reaction comprising alkylidenecyclopropanes, allylic bromides, and carbon monoxide also proceeded well to give 2-bromo-1,7-dien-5-ones in good yield.
Co-reporter:Shinji Maetani, Takahide Fukuyama, Nobuyoshi Suzuki, Daisuke Ishihara and Ilhyong Ryu
Chemical Communications 2012 vol. 48(Issue 19) pp:2552-2554
Publication Date(Web):10 Jan 2012
DOI:10.1039/C2CC18093F
The catalytic decarbonylation reaction of aliphatic carboxylic acids can be carried out in the presence of an iron complex, and it proceeds smoothly to give α-olefins with high selectivity.
Co-reporter:Takashi Kuwahara, Takahide Fukuyama, and Ilhyong Ryu
Organic Letters 2012 Volume 14(Issue 18) pp:4703-4705
Publication Date(Web):August 29, 2012
DOI:10.1021/ol302145a
The α-alkylation reaction of ketones with primary alcohols to give α-alkylated ketones was achieved using RuHCl(CO)(PPh3)3 as a catalyst in the presence of Cs2CO3 as a base. This reaction proceeds via an aldol condensation of ketones with aldehydes, formed via transfer dehydrogenation of alcohols, to give α,β-unsaturated ketones, which then undergo transfer hydrogenation with primary alcohols to give α-alkylated ketones and aldehydes, the latter of which participate in the next catalytic cycle. While the reaction of aliphatic primary alcohols was sluggish compared with that of benzylic alcohols, a catalytic amount of 1,10-phenanthroline was found to promote the alkylation dramatically.
Co-reporter: Takahide Fukuyama;Yuko Ohta;Dr. Célia Brancour;Kazusa Miyagawa; Ilhyong Ryu;Dr. Anne-Lise Dhimane; Louis Fensterbank; Max Malacria
Chemistry - A European Journal 2012 Volume 18( Issue 23) pp:7243-7247
Publication Date(Web):
DOI:10.1002/chem.201200045
Abstract
We have developed novel Rh-catalyzed [n+1]-type cycloadditions of 1,4-enyne esters, which involve an acyloxy migration as a key step. The efficient preparation of functionalized resorcinols, including biaryl derivatives, from readily available 1,4-enyne esters and CO was achieved by Rh-catalyzed [5+1] cycloaddition accompanied by 1,2-acyloxy migration. When enyne esters had an internal alkyne moiety, the reaction proceeded by a [4+1]-type cycloaddition involving 1,3-acyloxy migration, leading to cyclopentenones.
Co-reporter:Dr. Akira Fusano;Shuhei Sumino;Satoshi Nishitani;Takaya Inouye;Keisuke Morimoto;Dr. Takahide Fukuyama ; Ilhyong Ryu
Chemistry - A European Journal 2012 Volume 18( Issue 30) pp:9415-9422
Publication Date(Web):
DOI:10.1002/chem.201200752
Abstract
The atom-transfer carbonylation reaction of various alkyl iodides thereby leading to carboxylic acid esters was effectively accelerated by the addition of transition-metal catalysts under photoirradiation conditions. By using a combined Pd/hν reaction system, vicinal C-functionalization of alkenes was attained in which α-substituted iodoalkanes, alkenes, carbon monoxide, and alcohols were coupled to give functionalized esters. When alkenyl alcohols were used as acceptor alkenes, three-component coupling reactions, which were accompanied by intramolecular esterification, proceeded to give lactones. Pd-dimer complex [Pd2(CNMe)6][PF6]2, which is known to undergo homolysis under photoirradiation conditions, worked quite well as a catalyst in these three- or four-component coupling reactions. In this metal/radical hybrid system, both Pd radicals and acyl radicals are key players and a stereochemical study confirmed the carbonylation step proceeded through a radical carbonylation mechanism.
Co-reporter:Takuji Kawamoto ; Takahide Fukuyama
Journal of the American Chemical Society 2011 Volume 134(Issue 2) pp:875-877
Publication Date(Web):December 20, 2011
DOI:10.1021/ja210585n
Hydroxymethylation of alkyl halides was achieved using paraformaldehyde as a radical C1 synthon in the presence of tetrabutylammonium cyanoborohydride as a hydrogen source. The reaction proceeds via a radical chain mechanism involving an alkyl radical addition to formaldehyde to form an alkoxy radical, which abstracts hydrogen from a hydroborate anion.
Co-reporter:Ilhyong Ryu, Takahide Fukuyama, Mami Tojino, Yoshitaka Uenoyama, Yuka Yonamine, Nozomi Terasoma and Hiroshi Matsubara
Organic & Biomolecular Chemistry 2011 vol. 9(Issue 10) pp:3780-3786
Publication Date(Web):23 Mar 2011
DOI:10.1039/C1OB05145H
Tin hydride mediated radical carbonylation and cyclization reaction was investigated using a variety of ω-alkynyl amines as substrates. In this reaction α-methylene and α-stannylmethylene lactams having five to eight membered rings were obtained as principal products. In cases where the nitrogen has a substituent capable of giving stable radicals, such as an α-phenethyl group, the lactam ring formation again took place with extrusion of an α-phenethyl radical. Coupled with the subsequent protodestannylation procedure (TMSCl plus MeOH), these reactions provide a useful entry to α-methylene lactams with incorporation of CO as a lactam carbonyl group. In cases where the amines do not have a substituent acting as a radical leaving group, a reaction course involving a 1,4-H shift is chosen so as to liberate tin radicals ultimately. Thus the proposed mechanism involves (i) nucleophilic attack of amine nitrogen onto a carbonyl group of α,β-unsaturated acyl radicals/α-ketenyl radicals via lone pair–π* interaction, which leads to zwitterionic radical species, (ii) the subsequent proton shift from N to O to give hydroxyallyl radicals, (iii) 1,4-hydrogen shift from O to C, and (iv) β-scission to give lactams with liberation of tin radicals. DFT calculations reveal that the 1,4-hydrogen shifts, the key step of the reaction mechanism, can proceed under usual reaction conditions. On the other hand, an SHi type reaction to give lactams may be the result of the β-scission of the similar zwitterionic radical intermediates. DFT calculations also predict that an SHi type reaction would result when the intermediate has a good (radical) leaving group such as a phenethyl group.
Co-reporter: Ilhyong Ryu;Akihiro Tani;Dr. Takahide Fukuyama;Davide Ravelli; Maurizio Fagnoni; Angelo Albini
Angewandte Chemie International Edition 2011 Volume 50( Issue 8) pp:1869-1872
Publication Date(Web):
DOI:10.1002/anie.201004854
Co-reporter:Shinji Maetani, Takahide Fukuyama, Nobuyoshi Suzuki, Daisuke Ishihara, and Ilhyong Ryu
Organometallics 2011 Volume 30(Issue 6) pp:1389-1394
Publication Date(Web):February 22, 2011
DOI:10.1021/om1009268
Vaska’s complex, IrCl(CO)(PPh3)2, when combined with KI as an additive, served as an excellent catalyst for the decarbonylation of long-chain aliphatic carboxylic acids to give internal alkenes with high selectivity. On combination with KI and Ac2O as additives under controlled temperatures, decarbonylation proceeded to give terminal alkenes with high selectivity.
Co-reporter: Ilhyong Ryu;Akihiro Tani;Dr. Takahide Fukuyama;Davide Ravelli; Maurizio Fagnoni; Angelo Albini
Angewandte Chemie 2011 Volume 123( Issue 8) pp:1909-1912
Publication Date(Web):
DOI:10.1002/ange.201004854
Co-reporter:Aurélien Denichoux, Takahide Fukuyama, Takashi Doi, Jiro Horiguchi and Ilhyong Ryu
Organic Letters 2010 Volume 12(Issue 1) pp:1-3
Publication Date(Web):November 25, 2009
DOI:10.1021/ol902289r
The cross-coupling reaction of α,β-unsaturated aldehydes with primary alcohols to give 2-hydroxymethyl ketones was achieved using RuHCl(CO)(PPh3)3 as a catalyst. This atom-economical reaction is likely to proceed via the hydroruthenation of α,β-unsaturated aldehydes followed by an aldol reaction of the resultant enolates with aldehydes to give α-formylated ketones, which undergo transfer hydrogenation with primary alcohols leading to α-hydroxymethyl ketones. The reduction step can generate aldehydes, participating in the next catalytic cycle.
Co-reporter:Takashi Kippo, Takahide Fukuyama, and Ilhyong Ryu
Organic Letters 2010 Volume 12(Issue 18) pp:4006-4009
Publication Date(Web):August 16, 2010
DOI:10.1021/ol1016096
The free-radical-mediated bromoallylation of acetylenes proceeded efficiently in the presence of V-65 (2,2-azobis(2,4-dimethylvaleronitrile)) as the radical initiator. The regioselective reaction, which yields 1-bromo-2-substituted 1,4-dienes, is complementary to the Pd-catalyzed bromoallylation reaction previously reported by Kaneda. The products of the free-radical-mediated bromoallylation of acetylenes could be converted into a variety of substituted dienes by subsequent Pd-catalyzed reactions.
Co-reporter:Shoji Kobayashi, Takuji Kawamoto, Shohei Uehara, Takahide Fukuyama and Ilhyong Ryu
Organic Letters 2010 Volume 12(Issue 7) pp:1548-1551
Publication Date(Web):March 3, 2010
DOI:10.1021/ol1002847
Tin-free radical/ionic hydroxymethylation of secondary and tertiary alkyl iodides proceeded efficiently in the presence of tetrabutylammonium borohydride as the hydrogen source under atmospheric pressure of CO in conjunction with photoirradiation using black light. Two possible mechanisms were proposed, both of which involve hybrid radical/ionic processes.
Co-reporter:Akira Fusano, Takahide Fukuyama, Satoshi Nishitani, Takaya Inouye and Ilhyong Ryu
Organic Letters 2010 Volume 12(Issue 10) pp:2410-2413
Publication Date(Web):April 21, 2010
DOI:10.1021/ol1007668
Under photoirradiation conditions using xenon light, in the presence of a catalytic amount of PdCl2(PPh3)2 with triethylamine as a base, a three-component coupling reaction of iodoalkanes, carbon monoxide, and terminal alkynes proceeded to give alkyl alkynyl ketones in good yields.
Co-reporter:Inga C. Wienhöfer, Armido Studer, Md. Taifur Rahman, Takahide Fukuyama and Ilhyong Ryu
Organic Letters 2009 Volume 11(Issue 11) pp:2457-2460
Publication Date(Web):May 7, 2009
DOI:10.1021/ol900713d
Highly efficient thermal radical carboaminoxylations of various olefins by using the novel alkoxyamine A to give adducts of type B are described. It is reported that these radical addition reactions can be performed in a microflow reaction system. As compared to conventional batch reaction setup, significantly higher yields are obtained by running carboaminoxylations using the microflow system under analogous conditions.
Co-reporter:Sohei Omura, Takahide Fukuyama, Yuji Murakami, Hiromi Okamoto and Ilhyong Ryu
Chemical Communications 2009 (Issue 44) pp:6741-6743
Publication Date(Web):19 Oct 2009
DOI:10.1039/B912850F
Lactonization of dialdehydes and keto aldehydes was effectively catalyzed by RuHCl(CO)(PPh3)3. A cascade lactonization sequence accompanied by cross-coupling with enones was also attained.
Co-reporter:Atsushi Sugimoto, Takahide Fukuyama, Yukihito Sumino, Makoto Takagi, Ilhyong Ryu
Tetrahedron 2009 65(8) pp: 1593-1598
Publication Date(Web):
DOI:10.1016/j.tet.2008.12.063
Co-reporter:Takahide Fukuyama;Takashi Kippo
Research on Chemical Intermediates 2009 Volume 35( Issue 8-9) pp:
Publication Date(Web):2009 November
DOI:10.1007/s11164-009-0069-x
The reaction of allyl bromide with phenylacetylene catalyzed by Pd/Al2O3 pellets and Pd/C powder was investigated. Bromoallylation of phenylacetylene took place to give a regioisomeric mixture of 1-bromo-substituted 1,4-dienes. The bromoallylation of phenylacetylene was successfully applied to a continuous flow synthesis using a stainless steel made column packed with Pd/Al2O3 pellets.
Co-reporter:Ilhyong Ryu;Hiroshi Matsubara;Hiroyuki Nakamura;Dennis P. Curran
The Chemical Record 2008 Volume 8( Issue 6) pp:351-363
Publication Date(Web):
DOI:10.1002/tcr.20161
Abstract
The phase-vanishing (PV) method is based on spontaneous reaction controlled by diffusion of reagents into fluorous media, such as perfluorohexanes (FC-72) and polyperfluoroethers. Thus, the original PV reaction utilizes a triphasic test tube method composed of a bottom reagent phase, a middle fluorous phase, and a top substrate phase. In such a triphasic system, the fluorous phase acts as a liquid membrane to transport the bottom reagents to the top organic phase containing substrates. In the end, the bottom layer disappears and two phases remain. Since the first demonstration of the PV method by bromination of alkenes with molecular bromine, a number of applications have been developed thus far. These include halogenation of alcohols with SOBr2 and PBr3, demethylation of methoxyarenes with BBr3, cyclopropanation of alkenes by CH2I2-AlEt3, and Friedel–Crafts acylation of aromatic compounds with SnCl4. A fluorous triphasic U-tube method is effective for chlorination of alcohols based on lighter (less dense) reagents such as SOCl2 and PCl3. A system using a solution containing reagents as a bottom phase is useful for oxidation with m-CPBA, which may be defined as a new category for the “extractive PV” method. Recent advances include a “quadraphasic” PV method, in which an aqueous “scavenger” phase is added to the original triphasic PV method to remove acidic by-products. © 2008 The Japan Chemical Journal Forum and Wiley Periodicals, Inc. Chem Rec 8: 351–363; 2008: Published online in Wiley InterScience (www.interscience.wiley.com) DOI 10.1002/tcr.20161
Co-reporter:Takahide Fukuyama Dr.;Takashi Doi Dr.;Satoshi Minamino;Sohei Omura Dr.
Angewandte Chemie International Edition 2007 Volume 46(Issue 29) pp:
Publication Date(Web):20 JUN 2007
DOI:10.1002/anie.200701005
It all adds up: Straightforward access to 2-alkyl-substituted 1,3-diketones is provided by a regioselective addition of aldehydes to enones catalyzed by the ruthenium hydride catalyst [RuHCl(CO)(PPh3)3] (see scheme). The reaction involves a hydrometalation of the enone to form a metal enolate, a cross-aldol reaction to form an alkoxymetal species, and a subsequent β-metal hydride elimination.
Co-reporter:Takahide Fukuyama Dr.;Takashi Doi Dr.;Satoshi Minamino;Sohei Omura Dr.
Angewandte Chemie 2007 Volume 119(Issue 29) pp:
Publication Date(Web):20 JUN 2007
DOI:10.1002/ange.200701005
Alles zusammen: Die durch den Rutheniumhydridkomplex [RuHCl(CO)(PPh3)3] katalysierte regioselektive Addition von Aldehyden an Enone eröffnet einen einfachen Zugang zu 2-Alkyl-substituierten 1,3-Diketonen (siehe Schema). Die Reaktion besteht aus der Hydrometallierung des Enons zu einem Metallenolat, einer gekreuzten Aldolreaktion, die eine Alkoxymetallspezies liefert, und einer β-Metallhydrideliminierung.
Co-reporter:Md. Taifur Rahman, Takahide Fukuyama, Naoya Kamata, Masaaki Sato and Ilhyong Ryu
Chemical Communications 2006 (Issue 21) pp:2236-2238
Publication Date(Web):28 Apr 2006
DOI:10.1039/B600970K
A low pressure microflow system was developed for palladium-catalyzed multiphase carbonylation reactions in an ionic liquid. The microflow system resulted in superior selectivity and higher yields in carbonylative Sonogashira coupling and amidation reactions of aryl iodides compared to the conventional batch system.
Co-reporter:Takashi Doi, Takahide Fukuyama, Satoshi Minamino, Guillaume Husson and Ilhyong Ryu
Chemical Communications 2006 (Issue 17) pp:1875-1877
Publication Date(Web):21 Mar 2006
DOI:10.1039/B602103D
When primary unsaturated alcohols were treated with a catalytic amount of RuHCl(CO)(PPh3)3 in benzene under reflux, dimerization reactions took place to give α-hydroxymethyl ketones as major product.
Co-reporter:Sangmo Kim, Sunggak Kim, Noboru Otsuka,Ilhyong Ryu
Angewandte Chemie International Edition 2005 44(38) pp:6183-6186
Publication Date(Web):
DOI:10.1002/anie.200501606
Co-reporter:Sangmo Kim;Sunggak Kim Dr.;Noboru Otsuka Dr.
Angewandte Chemie 2005 Volume 117(Issue 38) pp:
Publication Date(Web):31 AUG 2005
DOI:10.1002/ange.200501606
Zinn, nein danke! Thiolester konnten mit der im Schema gezeigten zinnfreien Radikalcarbonylierung synthetisiert werden (V-40=Initiator). Dieser Ansatz lässt sich auf sequenzielle Radikalreaktionen ausdehnen, die Cyclisierung, Carbonylierung und das Abfangen von Acylradikalen durch Phenylbenzolthiosulfonat umfassen.
Co-reporter:Takuji Kawamoto and Ilhyong Ryu
Organic & Biomolecular Chemistry 2014 - vol. 12(Issue 48) pp:NaN9742-9742
Publication Date(Web):2014/10/01
DOI:10.1039/C4OB01784F
Borohydrides are an important class of reagents in both organic and inorganic chemistry. Though popular as hydride-transfer reagents for reduction, since earlier work from the 1970s, borohydride reagents have also been known to serve as hydrogen-transfer reagents. In pursuit of greener tin hydride substitutes, recent progress has been made to mediate radical C–C bond forming reactions, including Giese reactions, radical carbonylation and addition to HCHO reactions, with borohydride reagents. This review article focuses on state-of-the-art borohydride based radical reactions, also covering earlier work, kinetics and some DFT calculations with respect to the hydrogen transfer mechanism.
Co-reporter:Sohei Omura, Takahide Fukuyama, Yuji Murakami, Hiromi Okamoto and Ilhyong Ryu
Chemical Communications 2009(Issue 44) pp:NaN6743-6743
Publication Date(Web):2009/10/19
DOI:10.1039/B912850F
Lactonization of dialdehydes and keto aldehydes was effectively catalyzed by RuHCl(CO)(PPh3)3. A cascade lactonization sequence accompanied by cross-coupling with enones was also attained.
Co-reporter:Takashi Kippo and Ilhyong Ryu
Chemical Communications 2014 - vol. 50(Issue 45) pp:NaN5996-5996
Publication Date(Web):2014/04/16
DOI:10.1039/C4CC01597E
A bromine-radical mediated three-component coupling reaction was effectively achieved by the use of allenes, electron-deficient alkenes, and allyl bromides and led to the synthesis of 2-bromo-1,7-dienes in good to high yields. This protocol was extended to the three-component process using alkylidenecyclopropane, which gave 2-bromo-1,8-diene along with alkylidenecyclopentane.
Co-reporter:Takashi Kippo, Yuki Kimura, Ayami Maeda, Hiroshi Matsubara, Takahide Fukuyama and Ilhyong Ryu
Inorganic Chemistry Frontiers 2014 - vol. 1(Issue 7) pp:
Publication Date(Web):
DOI:10.1039/C4QO00138A
Co-reporter:Shinji Maetani, Takahide Fukuyama, Nobuyoshi Suzuki, Daisuke Ishihara and Ilhyong Ryu
Chemical Communications 2012 - vol. 48(Issue 19) pp:NaN2554-2554
Publication Date(Web):2012/01/10
DOI:10.1039/C2CC18093F
The catalytic decarbonylation reaction of aliphatic carboxylic acids can be carried out in the presence of an iron complex, and it proceeds smoothly to give α-olefins with high selectivity.
Co-reporter:Megumi Okada, Takahide Fukuyama, Keiichi Yamada, Ilhyong Ryu, Davide Ravelli and Maurizio Fagnoni
Chemical Science (2010-Present) 2014 - vol. 5(Issue 7) pp:NaN2898-2898
Publication Date(Web):2014/05/08
DOI:10.1039/C4SC01072H
Sunlight-induced direct regioselective β-alkylation of cyclopentanones with electron-deficient alkenes was accomplished by using tetrabutylammonium decatungstate (TBADT) as the catalyst. The regiochemistry can be rationalized by a polar transition state for an SH2 reaction. In the presence of CO, the reaction gave the three-component β-acylation product in good yield.
Co-reporter:Ilhyong Ryu, Takahide Fukuyama, Mami Tojino, Yoshitaka Uenoyama, Yuka Yonamine, Nozomi Terasoma and Hiroshi Matsubara
Organic & Biomolecular Chemistry 2011 - vol. 9(Issue 10) pp:NaN3786-3786
Publication Date(Web):2011/03/23
DOI:10.1039/C1OB05145H
Tin hydride mediated radical carbonylation and cyclization reaction was investigated using a variety of ω-alkynyl amines as substrates. In this reaction α-methylene and α-stannylmethylene lactams having five to eight membered rings were obtained as principal products. In cases where the nitrogen has a substituent capable of giving stable radicals, such as an α-phenethyl group, the lactam ring formation again took place with extrusion of an α-phenethyl radical. Coupled with the subsequent protodestannylation procedure (TMSCl plus MeOH), these reactions provide a useful entry to α-methylene lactams with incorporation of CO as a lactam carbonyl group. In cases where the amines do not have a substituent acting as a radical leaving group, a reaction course involving a 1,4-H shift is chosen so as to liberate tin radicals ultimately. Thus the proposed mechanism involves (i) nucleophilic attack of amine nitrogen onto a carbonyl group of α,β-unsaturated acyl radicals/α-ketenyl radicals via lone pair–π* interaction, which leads to zwitterionic radical species, (ii) the subsequent proton shift from N to O to give hydroxyallyl radicals, (iii) 1,4-hydrogen shift from O to C, and (iv) β-scission to give lactams with liberation of tin radicals. DFT calculations reveal that the 1,4-hydrogen shifts, the key step of the reaction mechanism, can proceed under usual reaction conditions. On the other hand, an SHi type reaction to give lactams may be the result of the β-scission of the similar zwitterionic radical intermediates. DFT calculations also predict that an SHi type reaction would result when the intermediate has a good (radical) leaving group such as a phenethyl group.
Co-reporter:Matteo C. Quattrini, Saki Fujii, Keiichi Yamada, Takahide Fukuyama, Davide Ravelli, Maurizio Fagnoni and Ilhyong Ryu
Chemical Communications 2017 - vol. 53(Issue 15) pp:NaN2338-2338
Publication Date(Web):2017/01/25
DOI:10.1039/C6CC09725A
A facile sunlight-induced derivatization of heteroaromatics via photocatalyzed C–H functionalization in amides, ethers, alkanes and aldehydes is described. Tetrabutylammonium decatungstate (TBADT) was used as the photocatalyst and allowed to carry out the process under mild conditions.
Co-reporter:Shuhei Sumino, Takahito Ui and Ilhyong Ryu
Inorganic Chemistry Frontiers 2015 - vol. 2(Issue 9) pp:NaN1087-1087
Publication Date(Web):2015/07/14
DOI:10.1039/C5QO00185D
Aromatic β-keto esters were synthesized via a carbonylative cross-coupling reaction of alkyl iodides and arylboronic acids in the presence of a catalytic amount of Pd catalyst. A cooperative radical and Pd-catalyzed mechanism is proposed.





![Benzene, pentafluoro[(4-methoxyphenyl)methyl]-](http://img.cochemist.com/ccimg/852200/852107-34-7.png)
![Benzene, pentafluoro[(4-methoxyphenyl)methyl]-](http://img.cochemist.com/ccimg/852200/852107-34-7_b.png)
![Furan,3-[2-(trimethylsilyl)ethynyl]-](http://img.cochemist.com/ccimg/465600/465521-19-1.png)
![Furan,3-[2-(trimethylsilyl)ethynyl]-](http://img.cochemist.com/ccimg/465600/465521-19-1_b.png)




![Benzene, 1-methyl-4-[(1E)-3-phenyl-1-propenyl]-](http://img.cochemist.com/ccimg/134600/134539-87-0.png)
![Benzene, 1-methyl-4-[(1E)-3-phenyl-1-propenyl]-](http://img.cochemist.com/ccimg/134600/134539-87-0_b.png)








![Benzene, 1,3-dimethoxy-5-[(4-methoxyphenyl)methyl]-](http://img.cochemist.com/ccimg/94600/94538-88-2.png)
![Benzene, 1,3-dimethoxy-5-[(4-methoxyphenyl)methyl]-](http://img.cochemist.com/ccimg/94600/94538-88-2_b.png)





![1-bromo-2-[(4-methoxyphenyl)methyl]-Benzene](http://img.cochemist.com/ccimg/68400/68355-78-2.png)
![1-bromo-2-[(4-methoxyphenyl)methyl]-Benzene](http://img.cochemist.com/ccimg/68400/68355-78-2_b.png)


![BENZENE, 1-BROMO-4-[(4-METHOXYPHENYL)METHYL]-](http://img.cochemist.com/ccimg/53100/53039-48-8.png)
![BENZENE, 1-BROMO-4-[(4-METHOXYPHENYL)METHYL]-](http://img.cochemist.com/ccimg/53100/53039-48-8_b.png)
![Benzonitrile, 4-[(4-methoxyphenyl)methyl]-](http://img.cochemist.com/ccimg/52200/52126-09-7.png)
![Benzonitrile, 4-[(4-methoxyphenyl)methyl]-](http://img.cochemist.com/ccimg/52200/52126-09-7_b.png)




![1-[(4-METHOXYPHENYL)METHYL]NAPHTHALENE](http://img.cochemist.com/ccimg/32900/32891-88-6.png)
![1-[(4-METHOXYPHENYL)METHYL]NAPHTHALENE](http://img.cochemist.com/ccimg/32900/32891-88-6_b.png)


![Benzene, 1-methoxy-4-[(4-methylphenyl)methyl]-](http://img.cochemist.com/ccimg/22900/22865-60-7.png)
![Benzene, 1-methoxy-4-[(4-methylphenyl)methyl]-](http://img.cochemist.com/ccimg/22900/22865-60-7_b.png)



















![3',5'-Dimethyl-[1,1'-biphenyl]-2-carboxylic acid](http://img.cochemist.com/ccimg/1183900/1183804-03-6.png)
![3',5'-Dimethyl-[1,1'-biphenyl]-2-carboxylic acid](http://img.cochemist.com/ccimg/1183900/1183804-03-6_b.png)







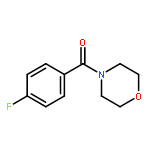
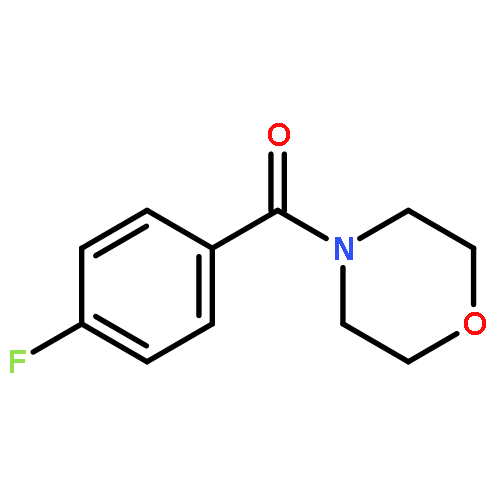
![Morpholine, 4-[4-(trifluoromethyl)benzoyl]-](http://img.cochemist.com/ccimg/447500/447455-27-8.png)
![Morpholine, 4-[4-(trifluoromethyl)benzoyl]-](http://img.cochemist.com/ccimg/447500/447455-27-8_b.png)


















![Decanenitrile, 2-[(trimethylsilyl)oxy]-](http://img.cochemist.com/ccimg/134600/134518-16-4.png)
![Decanenitrile, 2-[(trimethylsilyl)oxy]-](http://img.cochemist.com/ccimg/134600/134518-16-4_b.png)


![Benzene, [(2-cyclohexylethyl)sulfonyl]-](http://img.cochemist.com/ccimg/126100/126002-58-2.png)
![Benzene, [(2-cyclohexylethyl)sulfonyl]-](http://img.cochemist.com/ccimg/126100/126002-58-2_b.png)




















![3-Methyl-[1,1'-biphenyl]-2-carboxylic acid](http://img.cochemist.com/ccimg/92300/92254-02-9.png)
![3-Methyl-[1,1'-biphenyl]-2-carboxylic acid](http://img.cochemist.com/ccimg/92300/92254-02-9_b.png)

![[1,1'-Biphenyl]-2-carboxylicacid, 4'-(trifluoromethyl)-, methyl ester](http://img.cochemist.com/ccimg/91800/91748-18-4.png)
![[1,1'-Biphenyl]-2-carboxylicacid, 4'-(trifluoromethyl)-, methyl ester](http://img.cochemist.com/ccimg/91800/91748-18-4_b.png)




![2‐(2‐iodethyl)‐[1,3]dioxane](http://img.cochemist.com/ccimg/79200/79148-55-3.png)
![2‐(2‐iodethyl)‐[1,3]dioxane](http://img.cochemist.com/ccimg/79200/79148-55-3_b.png)




![Benzene, [(iodomethyl)sulfonyl]-](http://img.cochemist.com/ccimg/65500/65492-21-9.png)
![Benzene, [(iodomethyl)sulfonyl]-](http://img.cochemist.com/ccimg/65500/65492-21-9_b.png)








































![TRISODIUM 5-[(Z)-(3-CARBOXYLATO-5-METHYL-4-OXO-2,5-CYCLOHEXADIEN-<WBR />1-YLIDENE)(2-SULFONATOPHENYL)METHYL]-2-HYDROXY-3-METHYLBENZOATE](http://img.cochemist.com/ccimg/35400/35329-42-1.png)
![TRISODIUM 5-[(Z)-(3-CARBOXYLATO-5-METHYL-4-OXO-2,5-CYCLOHEXADIEN-<WBR />1-YLIDENE)(2-SULFONATOPHENYL)METHYL]-2-HYDROXY-3-METHYLBENZOATE](http://img.cochemist.com/ccimg/35400/35329-42-1_b.png)





















![[1,1'-Biphenyl]-3-carboxylic acid, ethyl ester](http://img.cochemist.com/ccimg/20000/19926-50-2.png)
![[1,1'-Biphenyl]-3-carboxylic acid, ethyl ester](http://img.cochemist.com/ccimg/20000/19926-50-2_b.png)







![2-Butenoic acid, 4-oxo-4-tricyclo[3.3.1.13,7]dec-1-yl-, ethyl ester, (E)-](http://img.cochemist.com/ccimg/17900/17812-02-1.png)
![2-Butenoic acid, 4-oxo-4-tricyclo[3.3.1.13,7]dec-1-yl-, ethyl ester, (E)-](http://img.cochemist.com/ccimg/17900/17812-02-1_b.png)


![[1,1'-Biphenyl]-2-carboxylic acid, 4'-bromo-, methyl ester](http://img.cochemist.com/ccimg/17200/17103-26-3.png)
![[1,1'-Biphenyl]-2-carboxylic acid, 4'-bromo-, methyl ester](http://img.cochemist.com/ccimg/17200/17103-26-3_b.png)







![[1,1'-Bicyclohexyl]-3-one](http://img.cochemist.com/ccimg/7200/7122-93-2.png)
![[1,1'-Bicyclohexyl]-3-one](http://img.cochemist.com/ccimg/7200/7122-93-2_b.png)
![[1,1'-Biphenyl]-4-carboxylicacid, ethyl ester](http://img.cochemist.com/ccimg/6400/6301-56-0.png)
![[1,1'-Biphenyl]-4-carboxylicacid, ethyl ester](http://img.cochemist.com/ccimg/6400/6301-56-0_b.png)
![7H-benzo[c]fluoren-7-one](http://img.cochemist.com/ccimg/6100/6051-98-5.png)
![7H-benzo[c]fluoren-7-one](http://img.cochemist.com/ccimg/6100/6051-98-5_b.png)























![3-Fluoro-[1,1'-biphenyl]-2-carboxylic acid](http://img.cochemist.com/ccimg/1900/1841-56-1.png)
![3-Fluoro-[1,1'-biphenyl]-2-carboxylic acid](http://img.cochemist.com/ccimg/1900/1841-56-1_b.png)