Co-reporter:Haoqi Chen, Bin Li, and Jinlong Yang
ACS Applied Materials & Interfaces November 8, 2017 Volume 9(Issue 44) pp:38999-38999
Publication Date(Web):October 16, 2017
DOI:10.1021/acsami.7b11454
Black phosphorus is a promising candidate for future nanoelectronics with a moderate electronic band gap and a high carrier mobility. Introducing the magnetism into black phosphorus will widely expand its application scope and may present a bright prospect in spintronic nanodevices. Here, we report our first-principles calculations of spin-polarized electronic structure of monolayer black phosphorus (phosphorene) adsorbed on a magnetic europium oxide (EuO) substrate. Effective spin injection into the phosphorene is realized by means of interaction with the nearby EuO(111) surface, i.e., proximity effect, which results in spin-polarized electrons in the 3p orbitals of phosphorene, with the spin polarization at Fermi level beyond 30%, together with an exchange-splitting energy of ∼0.184 eV for conduction-band minimum of the adsorbed phosphorene corresponding to an energy region where only one spin channel is conductive. The energy region of these exchange-splitting and spin-polarized band gaps of the adsorbed phosphorene can be effectively modulated by in-plane strain. Intrinsically high and anisotropic carrier mobilities at the conduction-band minimum of the phosphorene also become spin-polarized mainly due to spin polarization of deformation potentials and are not depressed significantly after the adsorption. These extraordinary properties would endow black phosphorus with great potentials in the future spintronic nanodevices.Keywords: black phosphorus; carrier mobility; magnetic substrate; proximity effect; spin injection;
Co-reporter:Haidi Wang, Bin LiJinlong Yang
The Journal of Physical Chemistry C 2017 Volume 121(Issue 6) pp:
Publication Date(Web):January 18, 2017
DOI:10.1021/acs.jpcc.6b12864
In recent years, carbon-based complex nanostructures have been explored due to many of their unique properties and related applications. Here we employ theoretical simulation based on density functional theory to investigate electronic, optical, and mechanical properties of a new type of the carbon-based complex nanostructure, i.e., experimentally fabricated one-dimensional complex material of hydrogenated diamond nanowires encapsulated in carbon nanotubes (CNW@CNT). The complex structure CNW@CNT is found to possess metallicity for the outer CNT and wide band gap nature for the inner CNW simultaneously. Under uniaxial strain a specific insulator-to-metal transition occurs for the inner CNW in the complex structure, with threshold value much smaller than that for the individual insulator. This effect is interpreted as that the strain induces relative shifting of bands of CNW and CNT and even charge transfer between them, making the valence band of CNW become not fully occupied. The inner CNW in the complex structure has optical absorption only in the ultraviolet waveband. The further examinations on the conductive bands reveal existences of nearly free-electron states which entirely dominate the conductive bands of the inner CNW and suggest that the electron–hole separation will happen in the CNW@CNT upon the ultraviolet illumination. The simulation results also reveal higher Young’s modulus of the CNW@CNT and the individual CNW even larger than those of CNT and graphene. We propose a simple parallel spring model to establish the relationship between the Young’s modulus of the complex structure and the one of its component which should be helpful to future predictions for other complex structures. The potential applications of this new type of carbon-based complex structure as a multifunctional integrated nanomaterial in future nanoelectronics, nano-optoelectronics, and nanoelectromechanics are discussed.
Co-reporter:Xiaohui Li
The Journal of Physical Chemistry C 2016 Volume 120(Issue 11) pp:6039-6049
Publication Date(Web):March 3, 2016
DOI:10.1021/acs.jpcc.5b12163
We report our investigation of adsorption and self-assembly of a nonplanar molecule 2,3,5,6-tetra(2′-pyridyl)pyrazine (TPPZ) on a Au(111) surface using ultrahigh vacuum low-temperature scanning tunneling microscopy joint with density functional theory (DFT) calculations. We find that the nonplanar TPPZ molecules exhibit various adsorption configurations depending on the coverage of molecules. The molecules mainly adsorb at step edges with a flat-lying configuration at low coverages and gather into chiral trimers almost equidistantly separated from each other in the fcc domains accompanied by diffusive molecules in the hcp domains of the herringbone reconstructed Au(111) surface at a coverage of about 0.2 monolayer (ML) and then form two dominant types of ordered domains, i.e., stripe-like (S-phase) and honeycomb-like (H-phase) superstructures, which may reflect the chiral separation characteristics at a coverage of about 1 ML. In the trimers and ordered domains, the adsorption configurations of molecules become declining or almost erect, i.e., an “edge-on” configuration, quite different from the flat-lying configuration at low coverages. After annealing to 380 K the S-phase transfers to the H-phase, and the H-phase may persist after annealing up to 410 K, which can be attributed to the existence of C–H···N hydrogen bonds between the TPPZ molecules with the same chirality. Our observations can be energetically interpreted by considering the interplay of molecule–substrate interaction and intermolecular interaction including van der Waals interaction and hydrogen bonds on the basis of the DFT calculations, where the hydrogen bonds should be a key factor for the formation of the stable ordered H-phase with chiral separation.
Co-reporter:Ruiqi Zhang
The Journal of Physical Chemistry C 2015 Volume 119(Issue 5) pp:2871-2878
Publication Date(Web):January 14, 2015
DOI:10.1021/jp5116564
Density functional theory calculations have been carried out to investigate single-layer phosphorene functionalized with two kinds of organic molecules, i.e., an electrophilic molecule tetracyano-p-quinodimethane (TCNQ) as electron acceptor and a nucleophilic molecule tetrathiafulvalene (TTF) as electron donor. The TCNQ molecule introduces shallow acceptor states in the gap of phosphorene close to the valence band edge, which makes the doped system become a p-type semiconductor. However, when the TTF molecule is adsorbed on the phosphorene, the occupied molecular states introduced into the gap are of deep donor states so that effective n-doping for transport cannot be realized. This disadvantageous situation can be amended by applying an external out-of-plane electric field with direction from phosphorene to TTF, or an in-plane tensile strain, or their combination, under which the conduction band edge of the phosphorene moves closer to the TTF-derived donor states, and then the TTF-adsorbed phosphorene system becomes an n-type semiconductor. It is also noted that the out-of-plane electric field and in-plane strain can modulate the band gap of the TTF-adsorbed phosphorene markedly. The effective bipolar doping of single-layer phosphorene via molecular adsorption, especially n-doping against its native p-doping propensity, and the good response of band gap in the infrared waveband of the TTF-adsorbed phosphorene to the out-of-plane electric field and in-plane strain would broaden the way to the application of this new type of two-dimensional material in nanoelectronic and optoelectronic devices.
Co-reporter:Ruiqi Zhang, Zhenpeng Hu, Bin Li, and Jinlong Yang
The Journal of Physical Chemistry A 2014 Volume 118(Issue 39) pp:8953-8959
Publication Date(Web):April 7, 2014
DOI:10.1021/jp5018218
On the basis of Bardeen’s perturbation theory on electron tunneling and inspired by Paz et al.’s study, a new expression for the tunneling current between the scanning tunneling microscopy (STM) tip and sample has been obtained, and it provides us with an efficient method to simulate STM images. The method can be implemented in any code of first-principles computing software, which offers the wave functions of the tip and sample, calculated independently at the same footing, as input. By calculating the integral with fast Fourier transform (FFT), simulating the STM image of a given sample surface by a database of different tips on a PC turns out to be not a time-consuming work. Compared with Paz et al.’s method, our method abandons the application of the vacuum Green function and possesses better computing efficiency, fewer parameters, and more reasonable simulated results especially at lower computing cost. Simple tip–sample systems, such as H–H and Pd2–Ag2, are taken as benchmarks to test our method. The topographic images of a CO molecule adsorbed on a Cu(111) surface obtained by using a tungsten tip and a CO-terminated tip are also simulated, and the simulated results are in good agreement with the experimental ones.






























































![Pyrazino[2,3-b]pyrazine, decahydro-1,4,5,8-tetranitro-](http://img.cochemist.com/ccimg/135900/135877-16-6.png)
![Pyrazino[2,3-b]pyrazine, decahydro-1,4,5,8-tetranitro-](http://img.cochemist.com/ccimg/135900/135877-16-6_b.png)
![5,2,6-(Iminomethenimino)-1H-imidazo[4,5-b]pyrazine,octahydro-1,3,4,7,8,10-hexanitro-](http://img.cochemist.com/ccimg/135300/135285-90-4.png)
![5,2,6-(Iminomethenimino)-1H-imidazo[4,5-b]pyrazine,octahydro-1,3,4,7,8,10-hexanitro-](http://img.cochemist.com/ccimg/135300/135285-90-4_b.png)






























![2-Phenylbenzo[d][1,2]selenazol-3(2H)-one](http://img.cochemist.com/ccimg/61000/60940-34-3.png)
![2-Phenylbenzo[d][1,2]selenazol-3(2H)-one](http://img.cochemist.com/ccimg/61000/60940-34-3_b.png)











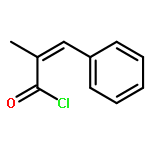
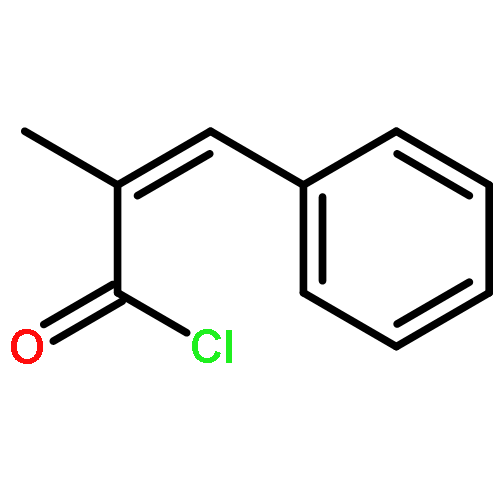


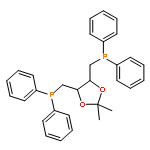
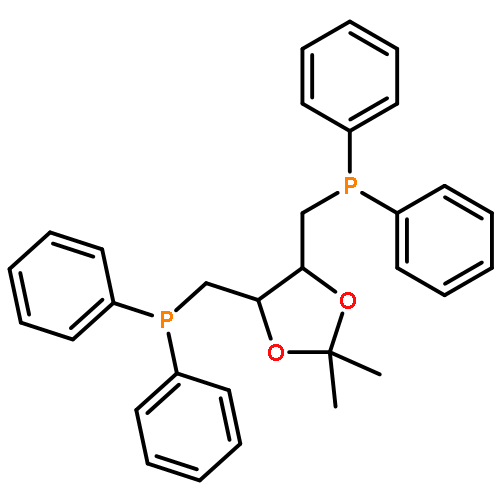







![Poly[oxy(1-oxo-1,6-hexanediyl)]](http://img.cochemist.com/ccimg/25300/25248-42-4.png)
![Poly[oxy(1-oxo-1,6-hexanediyl)]](http://img.cochemist.com/ccimg/25300/25248-42-4_b.png)












































![Decahydropyrazino[2,3-b]pyrazine](http://img.cochemist.com/ccimg/5500/5409-42-7.png)
![Decahydropyrazino[2,3-b]pyrazine](http://img.cochemist.com/ccimg/5500/5409-42-7_b.png)


![Spiro[2,3-dihydronaphthalene-4,1'-cyclopentane]-1-one](http://img.cochemist.com/ccimg/4900/4889-95-6.png)
![Spiro[2,3-dihydronaphthalene-4,1'-cyclopentane]-1-one](http://img.cochemist.com/ccimg/4900/4889-95-6_b.png)
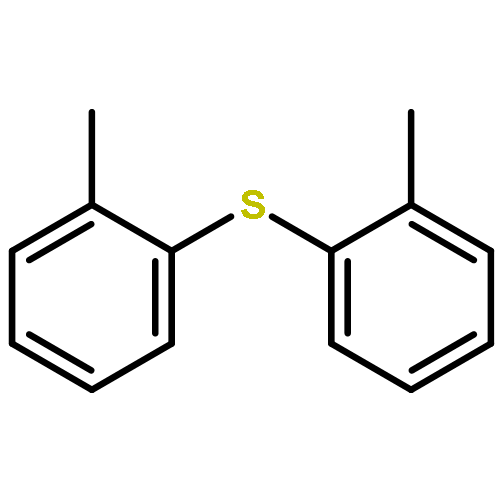
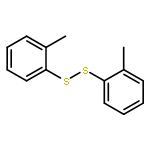
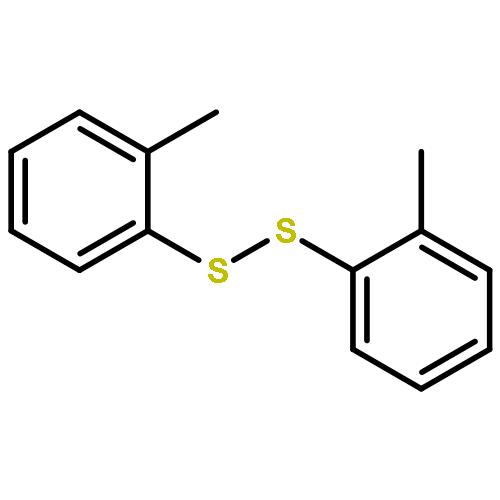
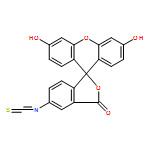





![Benzoic acid,2-[2-(4,5-dihydro-3-methyl-5-oxo-1-phenyl-1H-pyrazol-4-yl)diazenyl]-](http://img.cochemist.com/ccimg/2900/2898-84-2.png)
![Benzoic acid,2-[2-(4,5-dihydro-3-methyl-5-oxo-1-phenyl-1H-pyrazol-4-yl)diazenyl]-](http://img.cochemist.com/ccimg/2900/2898-84-2_b.png)
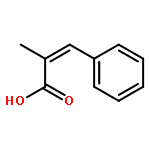
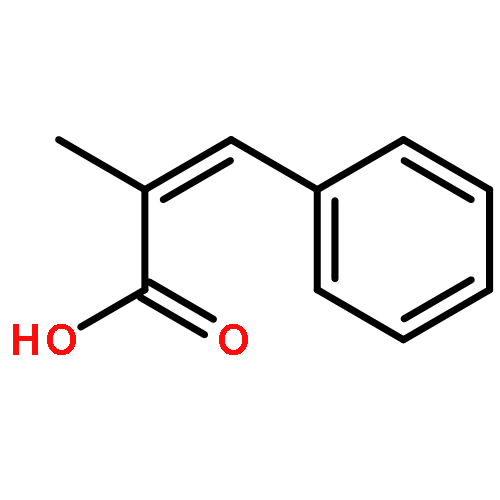




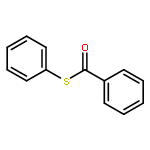
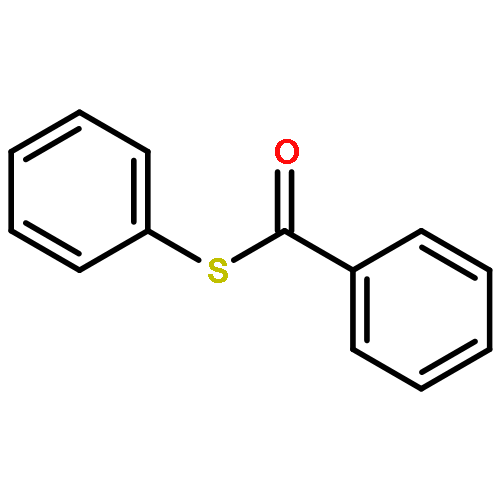




![2-Propanol, 1-[(1-methylethyl)amino]-3-(1-naphthalenyloxy)-](/data/chemimg/935900/525-66-6.png)
![2-Propanol, 1-[(1-methylethyl)amino]-3-(1-naphthalenyloxy)-](/data/chemimg/935900/525-66-6_b.png)
![Benzenesulfonic acid,4-[2-[4-(dimethylamino)phenyl]diazenyl]-](http://img.cochemist.com/ccimg/600/502-02-3.png)
![Benzenesulfonic acid,4-[2-[4-(dimethylamino)phenyl]diazenyl]-](http://img.cochemist.com/ccimg/600/502-02-3_b.png)
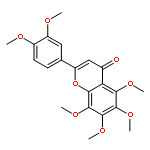

![3(2H)-Benzofuranone, 2-[(3,4-dihydroxyphenyl)methylene]-6-hydroxy-, (2Z)-](/data/chemimg/371600/120-05-8.png)
![3(2H)-Benzofuranone, 2-[(3,4-dihydroxyphenyl)methylene]-6-hydroxy-, (2Z)-](/data/chemimg/371600/120-05-8_b.png)



















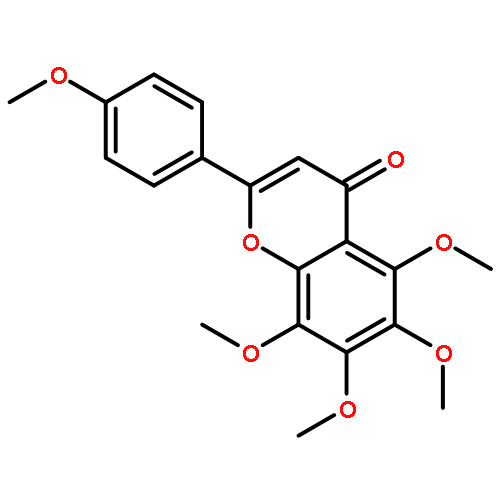
![1,4-Dithiino[2,3-b]-1,4-dithiin](http://img.cochemist.com/ccimg/300/255-55-0.png)
![1,4-Dithiino[2,3-b]-1,4-dithiin](http://img.cochemist.com/ccimg/300/255-55-0_b.png)