Modeling, Synthesis, and Biological Evaluation of Potential Retinoid X Receptor (RXR)-Selective Agonists: Analogues of 4-[1-(3,5,5,8,8-Pentamethyl-5,6,7,8-tetrahydro-2-naphthyl)ethynyl]benzoic Acid (Bexarotene) and 6-(Ethyl(5,5,8,8-tetrahydronaphthalen-2-yl)amino)nicotinic Acid (NEt-TMN)
Co-reporter:Michael C. Heck, Carl E. Wagner, Pritika H. Shahani, Mairi MacNeill, Aleksandra Grozic, Tamana Darwaiz, Micah Shimabuku, David G. Deans, Nathan M. Robinson, Samer H. Salama, Joseph W. Ziller, Ning Ma, Arjan van der Vaart, Pamela A. Marshall, and Peter W. Jurutka
Journal of Medicinal Chemistry 2016 Volume 59(Issue 19) pp:8924-8940
Publication Date(Web):September 3, 2016
DOI:10.1021/acs.jmedchem.6b00812
Sulfonic acid analogues of 4-[1-(3,5,5,8,8-pentamethyl-5,6,7,8-tetrahydro-2-naphthyl)ethynyl]benzoic acid (bexarotene, 1) as well as seven novel and two reported analogues of 6-(ethyl(5,5,8,8-tetramethyl-5,6,7,8-tetrahydronaphthalen-2-yl)amino)nicotinic acid (NEt-TMN) were synthesized and assessed for selective retinoid X receptor (RXR) agonism. Compound 1 is FDA-approved for treatment of cutaneous T-cell lymphoma (CTCL); however, 1 can provoke side effects by impacting RXR-dependent receptor pathways. All of the analogues in this study were evaluated for their potential to bind RXR through modeling and then assayed in an RXR–RXR mammalian-2-hybrid (M2H) system and in RXR-responsive element (RXRE)-mediated transcriptional experiments. The EC50 profiles for these unique analogues and their analogous effectiveness to inhibit proliferation in CTCL cells relative to 1 suggest that these compounds possess similar or even enhanced therapeutic potential. Several compounds also displayed more selective RXR activation with minimal cross-signaling of the retinoic acid receptor. These results suggest that modifications of potent RXR agonists such as NEt-TMN can lead to improved biological selectivity and potency compared with the known therapeutic.
Co-reporter:Shane Batie, Jamie H. Lee, Rabia A. Jama, Drew O. Browder, Luis A. Montano, Chanh C. Huynh, Lisa M. Marcus, Dorian G. Tsosie, Zeynab Mohammed, Vu Trang, Pamela A. Marshall, Peter W. Jurutka, Carl E. Wagner
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 3) pp:693-702
Publication Date(Web):1 February 2013
DOI:10.1016/j.bmc.2012.11.033
This report describes the synthesis of analogs of curcumin, and their analysis in acting as nuclear receptor specific agonists. Curcumin (CM), a turmeric-derived bioactive polyphenol found in curry, has recently been identified as a ligand for the vitamin D receptor (VDR), and it is possible that CM exerts some of its bioeffects via direct binding to VDR and/or other proteins in the nuclear receptor superfamily. Using mammalian-two-hybrid (M2H) and vitamin D responsive element (VDRE) biological assay systems, we tested CM and 11 CM synthetic analogs for their ability to activate VDR signaling. The M2H assay revealed that RXR and VDR association was induced by CM and several of its analogs. VDRE-based assays demonstrated that pure curcumin and eight CM analogs activated transcription of a luciferase plasmid at levels approaching that of the endocrine 1,25 dihydroxyvitamin D3 (1,25D) ligand in human colon cancer cells (HCT-116). Additional experiments were performed in HCT-116 utilizing various nuclear receptors and hormone responsive elements to determine the receptor specificity of curcumin binding. CM did not appear to activate transcription in a glucocorticoid responsive system. However, CM along with several analogs elicited transcriptional activation in retinoic acid and retinoid X receptor (RXR) responsive systems. M2H assays using RXR–RXR, VDR–SRC1 and VDR–DRIP revealed that CM and select analogs stimulate RXR homodimerization and VDR–coactivator interactions. These studies may lead to the discovery of novel curcumin analogs that activate nuclear receptors, including RXR, RAR and VDR, resulting in similar health benefits as those for vitamins A and D, such as lowering the risk of epithelial and colon cancers.
Co-reporter:Charles E. Deutch, Roy Krumbholz, Steve M. Schmid, Peter L. Bonate, Peter W. Jurutka
Enzyme and Microbial Technology (5 March 2010) Volume 46(Issues 3–4) pp:246-251
Publication Date(Web):5 March 2010
DOI:10.1016/j.enzmictec.2009.10.013
Tasidotin (ILX651) is a dolastatin analog active against several solid tumors. It is converted in vivo into two metabolites: M1, which lacks the carboxyl-terminal tert-butylamide group and is more active pharmacologically, and M2, which lacks this group and an adjacent proline residue. Both tasidotin and metabolite M1 were found to be competitive inhibitors of highly purified prolyl oligopeptidase (POP; EC 3.4.21.26) from Flavobacterium meningosepticum as measured by chromogenic and fluorogenic assays. HPLC analysis showed that POP converted tasidotin to M1 but not further to M2. Formation of M1 was linear for 120 min with a Vmax of 9.26 ng/mL min and an apparent Km of 0.238 mM (153 μg/mL). Several other enzymes known to cleave peptides at proline residues did not convert tasidotin to either M1 or M2. These results suggest that in addition to its known roles in the metabolism of physiologically active peptides and glutens, POP may function in drug metabolism and the level of POP activity in human tumor cells may determine their susceptibility to the pharmacologically active form of this drug.

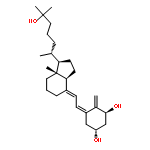
![(1r,3s,5z)-5-[(2e)-2-[(1r,3as,7ar)-1-[(2r)-6-hydroxy-6-methylheptan-2-yl]-7a-methyl-2,3,3a,5,6,7-hexahydro-1h-inden-4-ylidene]ethylidene]-4-methylidenecyclohexane-1,3-diol](http://img.cochemist.com/ccimg/32300/32222-06-3.png)
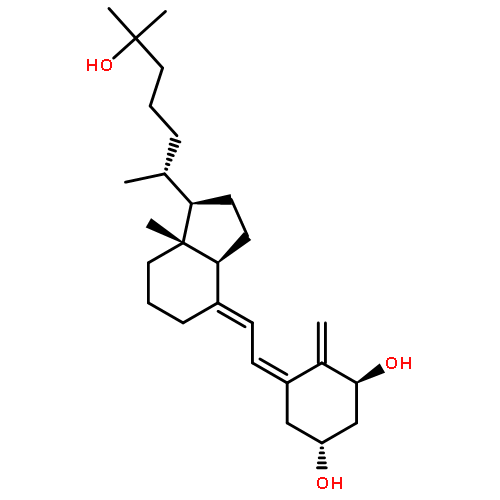
![(1r,3s,5z)-5-[(2e)-2-[(1r,3as,7ar)-1-[(2r)-6-hydroxy-6-methylheptan-2-yl]-7a-methyl-2,3,3a,5,6,7-hexahydro-1h-inden-4-ylidene]ethylidene]-4-methylidenecyclohexane-1,3-diol](http://img.cochemist.com/ccimg/32300/32222-06-3_b.png)