Co-reporter:Horrick Sharma, Tino W. Sanchez, Nouri Neamati, Mervi Detorio, Raymond F. Schinazi, Xiaolin Cheng, John K. Buolamwini
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 22) pp:6146-6151
Publication Date(Web):15 November 2013
DOI:10.1016/j.bmcl.2013.09.009
In the present study we report the synthesis of halogen-substituted phenanthrene β-diketo acids as new HIV-1 integrase inhibitors. The target phenanthrenes were obtained using both standard thermal- and microwave-assisted synthesis. 4-(6-Chlorophenanthren-2-yl)-2,4-dioxobutanoic acid (18) was the most active compound of the series, inhibiting both 3′-end processing (3′-P) and strand transfer (ST) with IC50 values of 5 and 1.3 μM, respectively. Docking studies revealed two predominant binding modes that were distinct from the binding modes of raltegravir and elvitegravir, and suggest a novel binding region in the IN active site. Moreover, these compounds are predicted not to interact significantly with some of the key amino acids (Q148 and N155) implicated in viral resistance. Therefore, this series of compounds can further be investigated for a possible chemotype to circumvent resistance to clinical HIV-1 IN inhibitors.
Co-reporter:Horrick Sharma, Xiaolin Cheng, and John K. Buolamwini
Journal of Chemical Information and Modeling 2012 Volume 52(Issue 2) pp:515-544
Publication Date(Web):January 18, 2012
DOI:10.1021/ci200485a
In the present study, we report the exploration of binding modes of potent HIV-1 integrase (IN) inhibitors MK-0518 (raltegravir) and GS-9137 (elvitegravir) as well as chalcone and related amide IN inhibitors we recently synthesized and the development of 3D-QSAR models for integrase inhibition. Homology models of DNA-bound HIV-1 IN were constructed on the basis of the X-ray crystal structure of the foamy virus IN-DNA complex (PDB ID: 3L2T) and used for docking. The binding modes of raltegravir and elvitegravir in our homology models are in accordance with those in the foamy virus structure revealing interactions important for inhibitor-IN binding. To gain further insights into the structural requirements for IN inhibition, three-dimensional quantitative structure activity relationship (3D-QSAR) studies were conducted using raltegravir, elvitegravir, and their analogs; our synthesized 3-keto salicylic acid IN inhibitor series; as well as other structurally related HIV-1 IN inhibitors. In the first part of the study with 103 compounds, atom-fit alignments, I and II, and docking-based alignment, III, were used to develop 3D-QSAR models 1, 2, and 3, respectively, each comprising comparative molecular field analysis (CoMFA) and comparative molecular similarity indices analysis (CoMSIA) 3D-QSARs. This initial analysis indicated that the docking-based (structure-based) model 3 performed better than the atom-fit (ligand-based) models 1 and 2, in terms of statistical significance and robustness. Thus, the docking-based alignment was then subsequently used with an expanded data set of 296 compounds for building a more comprehensive 3D-QSAR, model 4. Model 4 afforded good q2 values of 0.70 and 0.75 for CoMFA and CoMSIA 3D-QSARs, respectively, and showed good predictive performance on an external validation test set of 59 compounds with predictive r2 values up to 0.71. The HIV IN-DNA homology model of biological relevance and the comprehensive 3D-QSAR models developed in the present study provide insights and new predictive tools for structure-based design and optimization of IN inhibitors.
Co-reporter:Wenwei Lin and John K. Buolamwini
Bioconjugate Chemistry 2011 Volume 22(Issue 6) pp:1221
Publication Date(Web):May 3, 2011
DOI:10.1021/bc2000758
Nucleoside transporters are integral membrane glycoproteins that play critical roles in physiological nucleoside and nucleobase fluxes, and influence the efficacy of many nucleoside chemotherapy drugs. Fluorescent reporter ligands/substrates have been shown to be useful in the analysis of nucleoside transporter (NT) protein expression and discovery of new NT inhibitors. In this study, we have developed a novel dipyridamole (DP)-based equilibrative nucleoside transporter 1 (ENT1) fluorescent probe. The potent ENT1 and ENT2 inhibitor analogue of dipyridamole, 2,6-bis(diethanolamino)-4,8-diheptamethyleneiminopyrimido[5,4-d]pyrimidine (4, 8MDP), was modified to replace one β-hydroxyethyl group of the amino substituent at the 2-position with a β-aminoethyl group and then conjugated through the amino group to 6-(fluorescein-5-carboxamido)hexanoyl moiety to obtain a new fluorescent molecule, 2-diethanolamino-4,8-diheptamethyleneimino-2-(N-aminoethyl-N-ethanolamino)-6-(N,N-diethanolamino)pyrimido[5,4-d]pyrimidine-fluorescein conjugate, designated 8MDP-fluorescein (8MDP-fluor, 6). The binding affinities of 8MDP-fluor at ENT1 and ENT2 are reflected by the uridine uptake inhibitory Ki values of 52.1 nM and 285 nM, respectively. 8MDP-fluor was successfully demonstrated to be a flow cytometric probe for ENT1 comparable to the nitrobenzylmercaptopurine riboside (NBMPR) analogue ENT1 fluorescent probe SAENTA-X8-fluorescein (SAENTA-fluor, 1). This is the first reported dipyridamole-based ENT1 fluorescent probe, which adds a novel tool for probing ENT1, and possibly ENT2.
Co-reporter:Horrick Sharma, Shivaputra Patil, Tino W. Sanchez, Nouri Neamati, Raymond F. Schinazi, John K. Buolamwini
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 6) pp:2030-2045
Publication Date(Web):15 March 2011
DOI:10.1016/j.bmc.2011.01.047
HIV-1 integrase is one of the three most important enzymes required for viral replication and is therefore an attractive target for anti retroviral therapy. We herein report the design and synthesis of 3-keto salicylic acid chalcone derivatives as novel HIV-1 integrase inhibitors. The most active compound, 5-bromo-2-hydroxy-3-[3-(2,3,6-trichlorophenyl)acryloyl]benzoic acid (25) was selectively active against integrase strand transfer, with an IC50 of 3.7 μM. While most of the compounds exhibited strand transfer selectivity, a few were nonselective, such as 5-bromo-3-[3-(4-bromophenyl)acryloyl]-2-hydroxybenzoic acid (15), which was active against both 3′-processing and strand transfer with IC50 values of 11 ± 4 and 5 ± 2 μM, respectively. The compounds also inhibited HIV replication with potencies comparable with their integrase inhibitory potencies. Thus, 5-bromo-2-hydroxy-3-[3-(2,3,6-trichlorophenyl)acryloyl]benzoic acid (25) and 5-bromo-3-[3-(4-bromophenyl)acryloyl]-2-hydroxybenzoic acid (15) inhibited HIV-1 replication with EC50 values of 7.3 and 8.7 μM, respectively. A PHASE pharmacophore hypothesis was developed and validated by 3D-QSAR, which gave a predictive r2 of 0.57 for an external test set of ten compounds. Phamacophore derived molecular alignments were used for CoMFA and CoMSIA 3D-QSAR modeling. CoMSIA afforded the best model with q2 and r2 values of 0.54 and 0.94, respectively. This model predicted all the ten compounds of the test set within 0.56 log units of the actual pIC50 values; and can be used to guide the rational design of more potent novel 3-keto salicylic acid integrase inhibitors

![Benzoic acid,3,4,5-trimethoxy-,1,1'-[(tetrahydro-1H-1,4-diazepine-1,4(5H)-diyl)di-3,1-propanediyl] ester](http://img.cochemist.com/ccimg/35900/35898-87-4.png)
![Benzoic acid,3,4,5-trimethoxy-,1,1'-[(tetrahydro-1H-1,4-diazepine-1,4(5H)-diyl)di-3,1-propanediyl] ester](http://img.cochemist.com/ccimg/35900/35898-87-4_b.png)





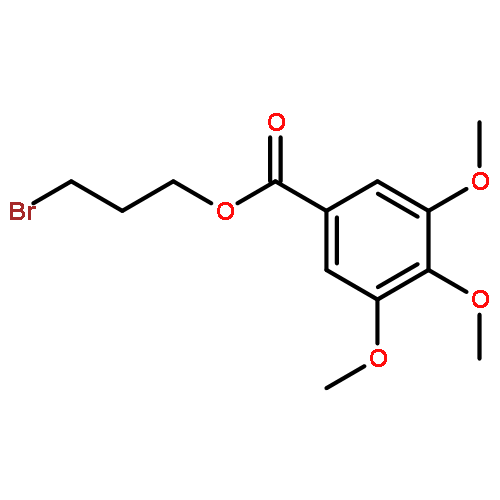
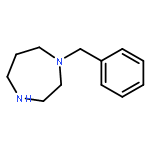
![2,5-Diazabicyclo[2.2.2]octane](http://img.cochemist.com/ccimg/700/658-24-2.png)
![2,5-Diazabicyclo[2.2.2]octane](http://img.cochemist.com/ccimg/700/658-24-2_b.png)