Co-reporter:Cong Zhou; Joshua L. Avins; Paul C. Klauser; Benjamin M. Brandsen; Yujeong Lee
Journal of the American Chemical Society 2016 Volume 138(Issue 7) pp:2106-2109
Publication Date(Web):February 8, 2016
DOI:10.1021/jacs.5b12647
DNA catalysts (deoxyribozymes) for a variety of reactions have been identified by in vitro selection. However, for certain reactions this identification has not been achieved. One important example is DNA-catalyzed amide hydrolysis, for which a previous selection experiment instead led to DNA-catalyzed DNA phosphodiester hydrolysis. Subsequent efforts in which the selection strategy deliberately avoided phosphodiester hydrolysis led to DNA-catalyzed ester and aromatic amide hydrolysis, but aliphatic amide hydrolysis has been elusive. In the present study, we show that including modified nucleotides that bear protein-like functional groups (any one of primary amino, carboxyl, or primary hydroxyl) enables identification of amide-hydrolyzing deoxyribozymes. In one case, the same deoxyribozyme sequence without the modifications still retains substantial catalytic activity. Overall, these findings establish the utility of introducing protein-like functional groups into deoxyribozymes for identifying new catalytic function. The results also suggest the longer-term feasibility of deoxyribozymes as artificial proteases.
Co-reporter:Anthony R. Hesser, Benjamin M. Brandsen, Shannon M. Walsh, Puzhou Wang and Scott K. Silverman
Chemical Communications 2016 vol. 52(Issue 68) pp:10439-10439
Publication Date(Web):04 Aug 2016
DOI:10.1039/C6CC90354A
Correction for ‘DNA-catalyzed glycosylation using aryl glycoside donors’ by Anthony R. Hesser et al., Chem. Commun., 2016, 52, 9259–9262.
Co-reporter:Anthony R. Hesser, Benjamin M. Brandsen, Shannon M. Walsh, Puzhou Wang and Scott K. Silverman
Chemical Communications 2016 vol. 52(Issue 59) pp:9259-9262
Publication Date(Web):22 Jun 2016
DOI:10.1039/C6CC04329A
We report the identification by in vitro selection of Zn2+/Mn2+-dependent deoxyribozymes that glycosylate the 3′-OH of a DNA oligonucleotide. Both β and α anomers of aryl glycosides can be used as the glycosyl donors. Individual deoxyribozymes are each specific for a particular donor anomer.
Co-reporter:Scott K. Silverman
Accounts of Chemical Research 2015 Volume 48(Issue 5) pp:1369
Publication Date(Web):May 5, 2015
DOI:10.1021/acs.accounts.5b00090
Catalysis is a fundamental chemical concept, and many kinds of catalysts have considerable practical value. Developing entirely new catalysts is an exciting challenge. Rational design and screening have provided many new small-molecule catalysts, and directed evolution has been used to optimize or redefine the function of many protein enzymes. However, these approaches have inherent limitations that prompt the pursuit of different kinds of catalysts using other experimental methods.Nature evolved RNA enzymes, or ribozymes, for key catalytic roles that in modern biology are limited to phosphodiester cleavage/ligation and amide bond formation. Artificial DNA enzymes, or deoxyribozymes, have great promise for a broad range of catalytic activities. They can be identified from unbiased (random) sequence populations as long as the appropriate in vitro selection strategies can be implemented for their identification. Notably, in vitro selection is different in key conceptual and practical ways from rational design, screening, and directed evolution. This Account describes the development by in vitro selection of DNA catalysts for many different kinds of covalent modification reactions of peptide and protein substrates, inspired in part by our earlier work with DNA-catalyzed RNA ligation reactions.In one set of studies, we have sought DNA-catalyzed peptide backbone cleavage, with the long-term goal of artificial DNA-based proteases. We originally anticipated that amide hydrolysis should be readily achieved, but in vitro selection instead surprisingly led to deoxyribozymes for DNA phosphodiester hydrolysis; this was unexpected because uncatalyzed amide bond hydrolysis is 105-fold faster. After developing a suitable selection approach that actively avoids DNA hydrolysis, we were able to identify deoxyribozymes for hydrolysis of esters and aromatic amides (anilides). Aliphatic amide cleavage remains an ongoing focus, including via inclusion of chemically modified DNA nucleotides in the catalyst, which we have recently found to enable this cleavage reaction. In numerous other efforts, we have investigated DNA-catalyzed peptide side chain modification reactions. Key successes include nucleopeptide formation (attachment of oligonucleotides to peptide side chains) and phosphatase and kinase activities (removal and attachment of phosphoryl groups to side chains).Through all of these efforts, we have learned the importance of careful selection design, including the frequent need to develop specific “capture” reactions that enable the selection process to provide only those DNA sequences that have the desired catalytic functions. We have established strategies for identifying deoxyribozymes that accept discrete peptide and protein substrates, and we have obtained data to inform the key choice of random region length at the outset of selection experiments. Finally, we have demonstrated the viability of modular deoxyribozymes that include a small-molecule-binding aptamer domain, although the value of such modularity is found to be minimal, with implications for many selection endeavors.Advances such as those summarized in this Account reveal that DNA has considerable catalytic abilities for biochemically relevant reactions, specifically including covalent protein modifications. Moreover, DNA has substantially different, and in many ways better, characteristics than do small molecules or proteins for a catalyst that is obtained “from scratch” without demanding any existing information on catalyst structure or mechanism. Therefore, prospects are very strong for continued development and eventual practical applications of deoxyribozymes for peptide and protein modification.
Co-reporter:Jagadeeswaran Chandrasekar; Adam C. Wylder
Journal of the American Chemical Society 2015 Volume 137(Issue 30) pp:9575-9578
Publication Date(Web):July 22, 2015
DOI:10.1021/jacs.5b06308
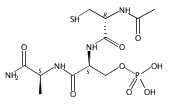
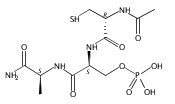
Dehydroalanine (Dha) is a nonproteinogenic electrophilic amino acid that is a synthetic intermediate or product in the biosynthesis of several bioactive cyclic peptides such as lantibiotics, thiopeptides, and microcystins. Dha also enables labeling of proteins and synthesis of post-translationally modified proteins and their analogues. However, current chemical approaches to introducing Dha into peptides have substantial limitations. Using in vitro selection, here we show that DNA can catalyze Zn2+ or Zn2+/Mn2+-dependent formation of Dha from phosphoserine (pSer), i.e., exhibit pSer lyase activity, a fundamentally new DNA-catalyzed reaction. Two new pSer lyase deoxyribozymes, named Dha-forming deoxyribozymes 1 and 2 (DhaDz1 and DhaDz2), each function with multiple turnover on the model hexapeptide substrate that was used during selection. Using DhaDz1, we generated Dha from pSer within an unrelated linear 13-mer peptide. Subsequent base-promoted intramolecular cyclization of homocysteine into Dha formed a stable cystathionine (thioether) analogue of the complement inhibitor compstatin. These findings establish the fundamental catalytic ability of DNA to eliminate phosphate from pSer to form Dha and suggest that with further development, pSer lyase deoxyribozymes will have broad practical utility for site-specific enzymatic synthesis of Dha from pSer in peptide substrates.
Co-reporter:Spurti U. Akki, Charles J. Werth, and Scott K. Silverman
Environmental Science & Technology 2015 Volume 49(Issue 16) pp:9905-9913
Publication Date(Web):July 16, 2015
DOI:10.1021/acs.est.5b02401
We used in vitro selection to identify new DNA aptamers for two endocrine-disrupting compounds often found in treated and natural waters, 17β-estradiol (E2) and 17α-ethynylestradiol (EE). We used equilibrium filtration to determine aptamer sensitivity/selectivity and dimethyl sulfate (DMS) probing to explore aptamer binding sites. The new E2 aptamers are at least 74-fold more sensitive for E2 than is a previously reported DNA aptamer, with dissociation constants (Kd values) of 0.6 μM. Similarly, the EE aptamers are highly sensitive for EE, with Kd of 0.5–1.0 μM. Selectivity values indicate that the E2 aptamers bind E2 and a structural analogue, estrone (E1), equally well and are up to 74-fold selective over EE. One EE aptamer is 53-fold more selective for EE over E2 or E1, but the other binds EE, E2, and E1 with similar affinity. The new aptamers do not lose sensitivity or selectivity in natural water from a local lake, despite the presence of natural organic matter (∼4 mg/L TOC). DMS probing suggests that E2 binding occurs in relatively flexible single-stranded DNA regions, an important finding for rational redesign of aptamers and their incorporation into sensing platforms. This is the first report of aptamers with strong selectivity for E2 and E1 over EE, or with strong selectivity for EE over E2 and E1. Such selectivity is important for achieving the goal of creating practically useful DNA-based sensors that can distinguish structurally similar estrogenic compounds in natural waters.
Co-reporter:Shannon M. Walsh;Stephanie N. Konecki
Journal of Molecular Evolution 2015 Volume 81( Issue 5-6) pp:218-224
Publication Date(Web):2015 December
DOI:10.1007/s00239-015-9699-3
Deoxyribozymes (DNA enzymes) have been developed for a growing variety of chemical reactions, including with peptide substrates. We recently described the first tyrosine kinase deoxyribozymes, which lacked the ability to discriminate among peptide substrates on the basis of the amino acids surrounding the tyrosine residue. Those deoxyribozymes were identified by in vitro selection using a DNA-anchored peptide substrate in which the residues neighboring tyrosine were all alanine. Here, we performed in vitro selection for tyrosine kinase activity using three peptide substrates in which the neighboring residues included a variety of side chains. For one of these three peptides, we found numerous deoxyribozymes that discriminate strongly in favor of phosphorylating tyrosine when the surrounding residues are specifically those used in the selection process. Three different short peptide sequence motifs of 2–4 amino acids were required for catalysis by three unique deoxyribozymes. For a second peptide substrate, the selection process led to one deoxyribozyme which exhibits partial discrimination among peptide sequences. These findings establish the feasibility of identifying DNA enzymes that catalyze sequence-selective tyrosine phosphorylation, which suggests the downstream practical utility of such deoxyribozymes. More broadly, this outcome reinforces the conclusion that nucleic acid catalysts can discriminate among peptide substrates in the context of biochemically relevant reactions.
Co-reporter:Victor Dokukin and Scott K. Silverman
Chemical Communications 2014 vol. 50(Issue 66) pp:9317-9320
Publication Date(Web):30 Jun 2014
DOI:10.1039/C4CC04253K
We assess the utility of integrating a predetermined aptamer DNA module adjacent to a random catalytic DNA region for identifying new deoxyribozymes by in vitro selection. By placing a known ATP aptamer next to an N40 random region, an explicitly modular DNA catalyst for tyrosine side chain phosphorylation is identified. The results have implications for broader identification of deoxyribozymes that function with small-molecule substrates.
Co-reporter:Benjamin M. Brsen;Tania E. Velez;Dr. Amit Sachdeva;Nora A. Ibrahim ; Scott K. Silverman
Angewandte Chemie International Edition 2014 Volume 53( Issue 34) pp:9045-9050
Publication Date(Web):
DOI:10.1002/anie.201404622
Abstract
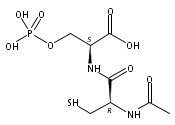
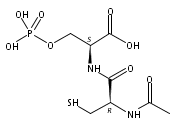
Catalyzing the covalent modification of aliphatic amino groups, such as the lysine (Lys) side chain, by nucleic acids has been challenging to achieve. Such catalysis will be valuable, for example, for the practical preparation of Lys-modified proteins. We previously reported the DNA-catalyzed modification of the tyrosine and serine hydroxy side chains, but Lys modification has been elusive. Herein, we show that increasing the reactivity of the electrophilic reaction partner by using 5′-phosphorimidazolide (5′-Imp) rather than 5′-triphosphate (5′-ppp) enables the DNA-catalyzed modification of Lys in a DNA-anchored peptide substrate. The DNA-catalyzed reaction of Lys with 5′-Imp is observed in an architecture in which the nucleophile and electrophile are not preorganized. In contrast, previous efforts showed that catalysis was not observed when Lys and 5′-ppp were used in a preorganized arrangement. Therefore, substrate reactivity is more important than preorganization in this context. These findings will assist ongoing efforts to identify DNA catalysts for reactions of protein substrates at lysine side chains.
Co-reporter:Chih-Chi Chu;On Yi Wong ; Scott K. Silverman
ChemBioChem 2014 Volume 15( Issue 13) pp:1905-1910
Publication Date(Web):
DOI:10.1002/cbic.201402255
Abstract
We report DNA catalysts (deoxyribozymes) that join tyrosine-containing peptides to RNA and DNA in one step and without requiring protecting groups on either the peptide or the nucleic acid. Our previous efforts towards this goal required tethering the peptide to a DNA anchor oligonucleotide. Here, we established direct in vitro selection for deoxyribozymes that use untethered, free peptide substrates. This approach enables imposition of selection pressure via reduced peptide concentration and leads to preparatively useful lower apparent Km values of ∼100 μM peptide. Use of phosphorimidazolide (Imp) rather than triphosphate as the electrophile enables reactivity of either terminus (5′ or 3′) of both RNA and DNA. Our findings establish a generalizable means of joining unprotected peptide to nucleic acid in one step by using DNA catalysts identified by in vitro selection.
Co-reporter:Benjamin M. Brsen;Tania E. Velez;Dr. Amit Sachdeva;Nora A. Ibrahim ; Scott K. Silverman
Angewandte Chemie 2014 Volume 126( Issue 34) pp:9191-9196
Publication Date(Web):
DOI:10.1002/ange.201404622
Abstract
Catalyzing the covalent modification of aliphatic amino groups, such as the lysine (Lys) side chain, by nucleic acids has been challenging to achieve. Such catalysis will be valuable, for example, for the practical preparation of Lys-modified proteins. We previously reported the DNA-catalyzed modification of the tyrosine and serine hydroxy side chains, but Lys modification has been elusive. Herein, we show that increasing the reactivity of the electrophilic reaction partner by using 5′-phosphorimidazolide (5′-Imp) rather than 5′-triphosphate (5′-ppp) enables the DNA-catalyzed modification of Lys in a DNA-anchored peptide substrate. The DNA-catalyzed reaction of Lys with 5′-Imp is observed in an architecture in which the nucleophile and electrophile are not preorganized. In contrast, previous efforts showed that catalysis was not observed when Lys and 5′-ppp were used in a preorganized arrangement. Therefore, substrate reactivity is more important than preorganization in this context. These findings will assist ongoing efforts to identify DNA catalysts for reactions of protein substrates at lysine side chains.
Co-reporter:Darren J. Parker ; Ying Xiao ; John M. Aguilar
Journal of the American Chemical Society 2013 Volume 135(Issue 23) pp:8472-8475
Publication Date(Web):May 22, 2013
DOI:10.1021/ja4032488
We recently used in vitro selection to identify many deoxyribozymes that catalyze DNA phosphodiester bond hydrolysis and create 5′-phosphate and 3′-hydroxyl termini. Alternatively, numerous deoxyribozymes have been identified for catalysis of RNA cleavage by 2′-hydroxyl transesterification, forming 2′,3′-cyclic phosphate and 5′-hydroxyl termini. In this study, we investigated the ability of DNA to catalyze RNA cleavage by hydrolysis rather than transesterification, although normally the hydrolysis reaction is substantially disfavored relative to transesterification. Via a series of in vitro selection experiments, we found that reselection of a DNA-hydrolyzing deoxyribozyme leads either to transesterification or hydrolysis, depending on exclusion or inclusion of a stringent selection pressure for hydrolysis. An entirely new selection starting from a random DNA pool, using an all-RNA substrate and imposing the same selection pressure, also leads to RNA hydrolysis. Collectively, these results establish experimentally that small DNA sequences have the catalytic ability to direct a chemical reaction down a disfavored pathway, even when a more favorable mechanism is readily available. Our view of DNA catalysis is therefore expanded beyond merely increasing the rates of reactions that would have occurred more slowly without the catalyst.
Co-reporter:Shannon M. Walsh ; Amit Sachdeva
Journal of the American Chemical Society 2013 Volume 135(Issue 40) pp:14928-14931
Publication Date(Web):September 25, 2013
DOI:10.1021/ja407586u
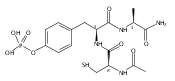
We show that DNA catalysts (deoxyribozymes, DNA enzymes) can phosphorylate tyrosine residues of peptides. Using in vitro selection, we identified deoxyribozymes that transfer the γ-phosphoryl group from a 5′-triphosphorylated donor (a pppRNA oligonucleotide or GTP) to the tyrosine hydroxyl acceptor of a tethered hexapeptide. Tyrosine kinase deoxyribozymes that use pppRNA were identified from each of N30, N40, and N50 random-sequence pools. Each deoxyribozyme requires Zn2+, and most additionally require Mn2+. The deoxyribozymes have little or no selectivity for the amino acid identities near the tyrosine, but they are highly selective for phosphorylating tyrosine rather than serine. Analogous GTP-dependent DNA catalysts were identified and found to have apparent Km(GTP) as low as ∼20 μM. These findings establish that DNA has the fundamental catalytic ability to phosphorylate the tyrosine side chain of a peptide substrate.
Co-reporter:Benjamin M. Brandsen ; Anthony R. Hesser ; Marissa A. Castner ; Madhavaiah Chandra
Journal of the American Chemical Society 2013 Volume 135(Issue 43) pp:16014-16017
Publication Date(Web):October 15, 2013
DOI:10.1021/ja4077233
We previously reported that DNA catalysts (deoxyribozymes) can hydrolyze DNA phosphodiester linkages, but DNA-catalyzed amide bond hydrolysis has been elusive. Here we used in vitro selection to identify DNA catalysts that hydrolyze ester linkages as well as DNA catalysts that hydrolyze aromatic amides, for which the leaving group is an aniline moiety. The aromatic amide-hydrolyzing deoxyribozymes were examined using linear free energy relationship analysis. The hydrolysis reaction is unaffected by substituents on the aromatic ring (ρ ≈ 0), suggesting general acid-catalyzed elimination as the likely rate-determining step of the addition–elimination hydrolysis mechanism. These findings establish that DNA has the catalytic ability to achieve hydrolysis of esters and aromatic amides as carbonyl-based substrates, and they suggest a mechanism-based approach to achieve DNA-catalyzed aliphatic amide hydrolysis.
Co-reporter:Jagadeeswaran Chandrasekar
PNAS 2013 Volume 110 (Issue 14 ) pp:5315-5320
Publication Date(Web):2013-04-02
DOI:10.1073/pnas.1221946110
Catalytic DNA sequences (deoxyribozymes, DNA enzymes, or DNAzymes) have been identified by in vitro selection for various
catalytic activities. Expanding the limits of DNA catalysis is an important fundamental objective and may facilitate practical
utility of catalysts that can be obtained from entirely unbiased (random) sequence populations. In this study, we show that
DNA can catalyze Zn2+-dependent phosphomonoester hydrolysis of tyrosine and serine side chains (i.e., exhibit phosphatase activity). The best deoxyribozyme
decreases the half-life for phosphoserine hydrolysis from as high as >1010 y to <1 h. The phosphatase activity also occurs with nonpeptidic substrates but with reduced efficiency, indicating a preference
for phosphopeptides. The newly identified deoxyribozymes can function with multiple turnover using free peptide substrates,
have activity in the presence of human cell lysate or BSA, and catalyze dephosphorylation of a larger protein substrate, suggesting
broader application of DNA catalysts as artificial phosphatases.
Co-reporter:Victor Dokukin and Scott K. Silverman
Chemical Science 2012 vol. 3(Issue 5) pp:1707-1714
Publication Date(Web):01 Mar 2012
DOI:10.1039/C2SC01067D
We report that micromolar concentrations of lanthanide ions can be required cofactors for DNA-hydrolyzing deoxyribozymes. Previous work identified deoxyribozymes that simultaneously require both Zn2+ and Mn2+ to achieve DNA-catalyzed DNA hydrolysis (1012 rate enhancement); a mutant of one such DNA catalyst requires only Zn2+. Here we show that in vitro selection in the presence of 10 μM lanthanide ion (Ce3+, Eu3+, or Yb3+) along with 1 mM Zn2+ leads to numerous DNA-hydrolyzing deoxyribozymes that strictly require the lanthanide ion as well as Zn2+ for catalytic activity. These DNA catalysts have a range of lanthanide dependences, including some deoxyribozymes that strongly favor one particular lanthanide ion (e.g., Ce3+ ≫ Eu3+ ≫ Yb3+) and others that function well with more than one lanthanide ion. Intriguingly, two of the Yb3+-dependent deoxyribozymes function well with Yb3+ alone (Kd,app ∼ 10 μM, in the absence of Zn2+) and have little or no activity with Eu3+ or Ce3+. In contrast to these selection outcomes when lanthanide ions were present, new selections with Zn2+ or Mn2+ alone, or Zn2+ with Mg2+/Ca2+, led primarily to deoxyribozymes that cleave DNA by deglycosylation and β-elimination rather than by hydrolysis, including several instances of depyrimidination. We conclude that lanthanide ions warrant closer attention as cofactors when identifying new nucleic acid catalysts, especially for applications in which high concentrations of polyvalent metal ion cofactors are undesirable.
Co-reporter:Amit Sachdeva and Scott K. Silverman
Organic & Biomolecular Chemistry 2012 vol. 10(Issue 1) pp:122-125
Publication Date(Web):14 Sep 2011
DOI:10.1039/C1OB06088K
During in vitro selection for DNA-catalyzed lysine reactivity, we identified a deoxyribozyme that instead catalyzes nucleophilic attack of a phosphoramidate functional group at a 5′-triphosphate-RNA, forming an unusual pyrophosphoramidate (N–PV–O–PV) linkage. This finding highlights the relatively poor nucleophilicity of nitrogen using nucleic acid catalysts, indicating a major challenge for future experimental investigation.
Co-reporter:Tania E. Velez, Jaydeep Singh, Ying Xiao, Emily C. Allen, On Yi Wong, Madhavaiah Chandra, Sarah C. Kwon, and Scott K. Silverman
ACS Combinatorial Science 2012 Volume 14(Issue 12) pp:680
Publication Date(Web):October 22, 2012
DOI:10.1021/co300111f
Functional nucleic acids are DNA and RNA aptamers that bind targets, or they are deoxyribozymes and ribozymes that have catalytic activity. These functional DNA and RNA sequences can be identified from random-sequence pools by in vitro selection, which requires choosing the length of the random region. Shorter random regions allow more complete coverage of sequence space but may not permit the structural complexity necessary for binding or catalysis. In contrast, longer random regions are sampled incompletely but may allow adoption of more complicated structures that enable function. In this study, we systematically examined random region length (N20 through N60) for two particular deoxyribozyme catalytic activities, DNA cleavage and tyrosine-RNA nucleopeptide linkage formation. For both activities, we previously identified deoxyribozymes using only N40 regions. In the case of DNA cleavage, here we found that shorter N20 and N30 regions allowed robust catalytic function, either by DNA hydrolysis or by DNA deglycosylation and strand scission via β-elimination, whereas longer N50 and N60 regions did not lead to catalytically active DNA sequences. Follow-up selections with N20, N30, and N40 regions revealed an interesting interplay of metal ion cofactors and random region length. Separately, for Tyr-RNA linkage formation, N30 and N60 regions provided catalytically active sequences, whereas N20 was unsuccessful, and the N40 deoxyribozymes were functionally superior (in terms of rate and yield) to N30 and N60. Collectively, the results indicate that with future in vitro selection experiments for DNA and RNA catalysts, and by extension for aptamers, random region length should be an important experimental variable.Keywords: deoxyribozyme catalysis; DNA and RNA aptamers; DNA catalysts; nucleic acids; random region length; RNA catalysts
Co-reporter:Dr. Amit Sachdeva;Dr. Madhavaiah Chra;Jagadeeswaran Chrasekar ; Scott K. Silverman
ChemBioChem 2012 Volume 13( Issue 5) pp:654-657
Publication Date(Web):
DOI:10.1002/cbic.201200048
Co-reporter:Ying Xiao, Emily C. Allen and Scott K. Silverman
Chemical Communications 2011 vol. 47(Issue 6) pp:1749-1751
Publication Date(Web):01 Dec 2010
DOI:10.1039/C0CC04575F
A deoxyribozyme that hydrolyzes DNA phosphodiester linkages with a requirement for both Zn2+ and Mn2+ is switched by only two nucleotide mutations to require Zn2+ alone, demonstrating that DNA-catalyzed DNA hydrolysis can be achieved using only one metal ion cofactor.
Co-reporter:On Yi Wong, P. I. Pradeepkumar, and Scott K. Silverman
Biochemistry 2011 Volume 50(Issue 21) pp:
Publication Date(Web):April 21, 2011
DOI:10.1021/bi200585n
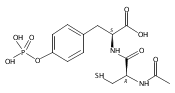
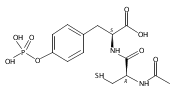
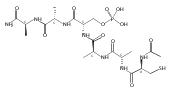
This study focuses on the development of DNA catalysts (deoxyribozymes) that modify side chains of peptide substrates, with the long-term goal of achieving DNA-catalyzed covalent protein modification. We recently described several deoxyribozymes that modify tyrosine (Tyr) or serine (Ser) side chains by catalyzing their reaction with 5′-triphosphorylated RNA, forming nucleopeptide linkages. In each previous case, the side chain was presented in a highly preorganized three-dimensional architecture such that the resulting deoxyribozymes inherently cannot function with free peptides or proteins, which do not maintain the preorganization. Here we describe in vitro selection of deoxyribozymes that catalyze Tyr side chain modification of tethered and free peptide substrates, where the approach can potentially be generalized for catalysis involving large proteins. Several new deoxyribozymes for Tyr modification (and several for Ser modification as well) were identified; progressively better catalytic activity was observed as the selection design was strategically changed. The best new deoxyribozyme, 15MZ36, catalyzes covalent Tyr modification of a free tripeptide substrate with a kobs of 0.50 h–1 (t1/2 of 83 min) and up to 65% yield. These findings represent an important advance by demonstrating, for the first time, DNA catalysis involving free peptide substrates. The new results suggest the feasibility of DNA-catalyzed covalent modification of side chains of large protein substrates and provide key insights into how to achieve this goal.
Co-reporter:On Yi Wong;Ama E. Mulcrone ; Scott K. Silverman
Angewandte Chemie International Edition 2011 Volume 50( Issue 49) pp:11679-11684
Publication Date(Web):
DOI:10.1002/anie.201104976
Co-reporter:On Yi Wong;Ama E. Mulcrone ; Scott K. Silverman
Angewandte Chemie 2011 Volume 123( Issue 49) pp:11883-11888
Publication Date(Web):
DOI:10.1002/ange.201104976
Co-reporter:Amit Sachdeva and Scott K. Silverman
Chemical Communications 2010 vol. 46(Issue 13) pp:2215-2217
Publication Date(Web):25 Feb 2010
DOI:10.1039/B927317D
New deoxyribozymes are shown to catalyze reactions of serine side chains, forming nucleopeptide linkages and discriminating between serine and tyrosine or between two competing serines.
Co-reporter:Michael D. Brenner, Mary S. Scanlan, Michelle K. Nahas, Taekjip Ha and Scott K. Silverman
Biochemistry 2010 Volume 49(Issue 8) pp:
Publication Date(Web):January 28, 2010
DOI:10.1021/bi9019912
Purine riboswitches are RNA regulatory elements that control purine metabolism in response to intracellular concentrations of the purine ligands. Conformational changes of the guanine riboswitch aptamer domain induced by guanine binding lead to transcriptional regulation of genes involved in guanine biosynthesis. The guanine riboswitch aptamer domain has three RNA helices designated P1, P2, and P3. An overall model for the Mg2+- and guanine-dependent relative orientations and dynamics of P1, P2, and P3 has not been reported, and the conformational role of guanine under physiologically relevant conditions has not been fully elucidated. In this study, an ensemble and single-molecule fluorescence resonance energy transfer (FRET) study was performed on three orthogonally labeled variants of the xpt guanine riboswitch aptamer domain. The combined FRET data support a model in which the unfolded state of the aptamer domain has a highly dynamic P2 helix that switches rapidly between two orientations relative to nondynamic P1 and P3. At ≪1 mM Mg2+ (in the presence of a saturating level of guanine) or ≥1 mM Mg2+ (in the absence of guanine), the riboswitch starts to adopt a folded conformation in which loop−loop interactions lock P2 and P3 into place. At >5 mM Mg2+, further compaction occurs in which P1 more closely approaches P3. Our data help to explain the biological role of guanine as stabilizing the globally folded aptamer domain conformation at physiologically relevant Mg2+ concentrations (≤1 mM), whereas in the absence of guanine, much higher Mg2+ concentrations are required to induce this folding event.
Co-reporter:Ying Xiao, Madhavaiah Chandra, and Scott K. Silverman
Biochemistry 2010 Volume 49(Issue 44) pp:
Publication Date(Web):October 6, 2010
DOI:10.1021/bi1013672
We recently reported the identification by in vitro selection of 10MD5, a deoxyribozyme that requires both Mn2+ and Zn2+ to hydrolyze a single-stranded DNA substrate with formation of 5′-phosphate and 3′-hydroxyl termini. DNA cleavage by 10MD5 proceeds with kobs = 2.7 h−1 and rate enhancement of 1012 over the uncatalyzed P−O hydrolysis reaction. 10MD5 has a very sharp pH optimum near 7.5, with greatly reduced DNA cleavage rate and yield when the pH is changed by only 0.1 unit in either direction. Here we have optimized 10MD5 by reselection (in vitro evolution), leading to variants with broader pH tolerance, which is important for practical DNA cleavage applications. Because of the extensive Watson−Crick complementarity between deoxyribozyme and substrate, the parent 10MD5 is inherently sequence-specific; i.e., it is able to cleave one DNA substrate sequence in preference to other sequences. 10MD5 is also site-specific because only one phosphodiester bond within the DNA substrate is cleaved, although here we show that intentionally creating Watson−Crick mismatches near the cleavage site relaxes the site specificity. Newly evolved 10MD5 variants such as 9NL27 are also sequence-specific. However, the 9NL27 site specificity is relaxed for some substrate sequences even when full Watson−Crick complementarity is maintained, corresponding to a functional compromise between pH tolerance and site specificity. The site specificity of 9NL27 may be restored by expanding its “recognition site” from ATG∧T (as for 10MD5) to ATG∧TT or larger, i.e., by considering 9NL27 to have reduced substrate sequence tolerance relative to 10MD5. These findings provide fundamental insights into the interplay among key deoxyribozyme characteristics of tolerance and selectivity, with implications for ongoing development of practical DNA-catalyzed DNA hydrolysis.
Co-reporter: Scott K. Silverman
Angewandte Chemie 2010 Volume 122( Issue 40) pp:7336-7359
Publication Date(Web):
DOI:10.1002/ange.200906345
Abstract
Die DNA (Desoxyribonukleinsäure) ist das genetische Material, das allen Organismen der Erde gemeinsam ist. Unser biologisches Verständnis der DNA ist umfangreich und wird weitreichend genutzt. In den letzten Jahren nun begannen Chemiker, die DNA auch für nicht-biologische Anwendungen in der Katalyse, der Kodierung und zur Stereokontrolle zu erschließen. Dieser Aufsatz fasst die wichtigsten Fortschritte auf diesen drei spannenden Forschungsfeldern zusammen, von denen sich jedes einen anderen bestimmten Aspekt der chemischen Eigenschaften der DNA zu Nutze macht.
Co-reporter: Scott K. Silverman
Angewandte Chemie International Edition 2010 Volume 49( Issue 40) pp:7180-7201
Publication Date(Web):
DOI:10.1002/anie.200906345
Abstract
DNA (deoxyribonucleic acid) is the genetic material common to all of Earth’s organisms. Our biological understanding of DNA is extensive and well-exploited. In recent years, chemists have begun to develop DNA for nonbiological applications in catalysis, encoding, and stereochemical control. This Review summarizes key advances in these three exciting research areas, each of which takes advantage of a different subset of DNA’s useful chemical properties.
Co-reporter:Scott K. Silverman
Accounts of Chemical Research 2009 Volume 42(Issue 10) pp:1521
Publication Date(Web):July 2, 2009
DOI:10.1021/ar900052y
One of the chemist’s key motivations is to explore the forefront of catalysis. In this Account, we describe our laboratory’s efforts at one such forefront: the use of DNA as a catalyst. Natural biological catalysts include both protein enzymes and RNA enzymes (ribozymes), whereas nature apparently uses DNA solely for genetic information storage. Nevertheless, the chemical similarities between RNA and DNA naturally lead to laboratory examination of DNA as a catalyst, especially because DNA is more stable than RNA and is less costly and easier to synthesize. Many catalytically active DNA sequences (deoxyribozymes, also called DNAzymes) have been identified in the laboratory by in vitro selection, in which many random DNA sequences are evaluated in parallel to find those rare sequences that have a desired functional ability. Since 2001, our research group has pursued new deoxyribozymes for various chemical reactions. We consider DNA simply as a large biopolymer that can adopt intricate three-dimensional structure and, in the presence of appropriate metal ions, generate the chemical complexity required to achieve catalysis. Our initial efforts focused on deoxyribozymes that ligate two RNA substrates. In these studies, we used only substrates that are readily obtained biochemically. Highly active deoxyribozymes were identified, with emergent questions regarding chemical selectivity during RNA phosphodiester bond formation. Deoxyribozymes allow synthesis of interesting RNA products, such as branches and lariats, that are otherwise challenging to prepare. Our experiments have demonstrated that deoxyribozymes can have very high rate enhancements and chemical selectivities. We have also shown how the in vitro selection process itself can be directed toward desired goals, such as selective formation of native 3′−5′ RNA linkages. A final lesson is that unanticipated selection outcomes can be very interesting, highlighting the importance of allowing such opportunities in future experiments. More recently, we have begun using nonoligonucleotide substrates in our efforts with deoxyribozymes. We have especially focused on developing DNA catalysts for reactions of small molecules or amino acid side chains. For example, new deoxyribozymes have the catalytic power to create a nucleopeptide linkage between a tyrosine or serine side chain and the 5′-terminus of an RNA strand. Although considerable further work remains to establish DNA as a practical catalyst for small molecules and full-length proteins, the progress to date is very promising. The many lessons learned during the experiments described in this Account will help us and others to realize the full catalytic power of DNA.
Co-reporter:Elena Zelin and Scott K. Silverman
Chemical Communications 2009 (Issue 7) pp:767-769
Publication Date(Web):14 Jan 2009
DOI:10.1039/B820676G
Double-stranded DNA constraints enable efficient control of catalysis by a large multi-domain group I intron ribozyme.
Co-reporter:Anthony R. Hesser, Benjamin M. Brandsen, Shannon M. Walsh, Puzhou Wang and Scott K. Silverman
Chemical Communications 2016 - vol. 52(Issue 59) pp:NaN9262-9262
Publication Date(Web):2016/06/22
DOI:10.1039/C6CC04329A
We report the identification by in vitro selection of Zn2+/Mn2+-dependent deoxyribozymes that glycosylate the 3′-OH of a DNA oligonucleotide. Both β and α anomers of aryl glycosides can be used as the glycosyl donors. Individual deoxyribozymes are each specific for a particular donor anomer.
Co-reporter:Amit Sachdeva and Scott K. Silverman
Chemical Communications 2010 - vol. 46(Issue 13) pp:NaN2217-2217
Publication Date(Web):2010/02/25
DOI:10.1039/B927317D
New deoxyribozymes are shown to catalyze reactions of serine side chains, forming nucleopeptide linkages and discriminating between serine and tyrosine or between two competing serines.
Co-reporter:Ying Xiao, Emily C. Allen and Scott K. Silverman
Chemical Communications 2011 - vol. 47(Issue 6) pp:NaN1751-1751
Publication Date(Web):2010/12/01
DOI:10.1039/C0CC04575F
A deoxyribozyme that hydrolyzes DNA phosphodiester linkages with a requirement for both Zn2+ and Mn2+ is switched by only two nucleotide mutations to require Zn2+ alone, demonstrating that DNA-catalyzed DNA hydrolysis can be achieved using only one metal ion cofactor.
Co-reporter:Victor Dokukin and Scott K. Silverman
Chemical Science (2010-Present) 2012 - vol. 3(Issue 5) pp:NaN1714-1714
Publication Date(Web):2012/03/01
DOI:10.1039/C2SC01067D
We report that micromolar concentrations of lanthanide ions can be required cofactors for DNA-hydrolyzing deoxyribozymes. Previous work identified deoxyribozymes that simultaneously require both Zn2+ and Mn2+ to achieve DNA-catalyzed DNA hydrolysis (1012 rate enhancement); a mutant of one such DNA catalyst requires only Zn2+. Here we show that in vitro selection in the presence of 10 μM lanthanide ion (Ce3+, Eu3+, or Yb3+) along with 1 mM Zn2+ leads to numerous DNA-hydrolyzing deoxyribozymes that strictly require the lanthanide ion as well as Zn2+ for catalytic activity. These DNA catalysts have a range of lanthanide dependences, including some deoxyribozymes that strongly favor one particular lanthanide ion (e.g., Ce3+ ≫ Eu3+ ≫ Yb3+) and others that function well with more than one lanthanide ion. Intriguingly, two of the Yb3+-dependent deoxyribozymes function well with Yb3+ alone (Kd,app ∼ 10 μM, in the absence of Zn2+) and have little or no activity with Eu3+ or Ce3+. In contrast to these selection outcomes when lanthanide ions were present, new selections with Zn2+ or Mn2+ alone, or Zn2+ with Mg2+/Ca2+, led primarily to deoxyribozymes that cleave DNA by deglycosylation and β-elimination rather than by hydrolysis, including several instances of depyrimidination. We conclude that lanthanide ions warrant closer attention as cofactors when identifying new nucleic acid catalysts, especially for applications in which high concentrations of polyvalent metal ion cofactors are undesirable.
Co-reporter:Amit Sachdeva and Scott K. Silverman
Organic & Biomolecular Chemistry 2012 - vol. 10(Issue 1) pp:NaN125-125
Publication Date(Web):2011/09/14
DOI:10.1039/C1OB06088K
During in vitro selection for DNA-catalyzed lysine reactivity, we identified a deoxyribozyme that instead catalyzes nucleophilic attack of a phosphoramidate functional group at a 5′-triphosphate-RNA, forming an unusual pyrophosphoramidate (N–PV–O–PV) linkage. This finding highlights the relatively poor nucleophilicity of nitrogen using nucleic acid catalysts, indicating a major challenge for future experimental investigation.
Co-reporter:Elena Zelin and Scott K. Silverman
Chemical Communications 2009(Issue 7) pp:NaN769-769
Publication Date(Web):2009/01/14
DOI:10.1039/B820676G
Double-stranded DNA constraints enable efficient control of catalysis by a large multi-domain group I intron ribozyme.
Co-reporter:Anthony R. Hesser, Benjamin M. Brandsen, Shannon M. Walsh, Puzhou Wang and Scott K. Silverman
Chemical Communications 2016 - vol. 52(Issue 68) pp:NaN10439-10439
Publication Date(Web):2016/08/04
DOI:10.1039/C6CC90354A
Correction for ‘DNA-catalyzed glycosylation using aryl glycoside donors’ by Anthony R. Hesser et al., Chem. Commun., 2016, 52, 9259–9262.
Co-reporter:Victor Dokukin and Scott K. Silverman
Chemical Communications 2014 - vol. 50(Issue 66) pp:NaN9320-9320
Publication Date(Web):2014/06/30
DOI:10.1039/C4CC04253K
We assess the utility of integrating a predetermined aptamer DNA module adjacent to a random catalytic DNA region for identifying new deoxyribozymes by in vitro selection. By placing a known ATP aptamer next to an N40 random region, an explicitly modular DNA catalyst for tyrosine side chain phosphorylation is identified. The results have implications for broader identification of deoxyribozymes that function with small-molecule substrates.
.png)
![2-Propenoic acid,3-[1-(2-deoxy-b-D-erythro-pentofuranosyl)-1,2,3,4-tetrahydro-2,4-dioxo-5-pyrimidinyl]-,methyl ester, (2E)-](http://img.cochemist.com/ccimg/86200/86163-17-9.png)
![2-Propenoic acid,3-[1-(2-deoxy-b-D-erythro-pentofuranosyl)-1,2,3,4-tetrahydro-2,4-dioxo-5-pyrimidinyl]-,methyl ester, (2E)-](http://img.cochemist.com/ccimg/86200/86163-17-9_b.png)
![2-METHYL-2-PROPANYL 4-[4-CHLORO-2-(4,4-DIMETHYL-4,5-DIHYDRO-1,3-O<WBR />XAZOL-2-YL)PHENYL]-4-HYDROXY-1-PIPERIDINECARBOXYLATE](http://img.cochemist.com/ccimg/71500/71484-85-0.png)
![2-METHYL-2-PROPANYL 4-[4-CHLORO-2-(4,4-DIMETHYL-4,5-DIHYDRO-1,3-O<WBR />XAZOL-2-YL)PHENYL]-4-HYDROXY-1-PIPERIDINECARBOXYLATE](http://img.cochemist.com/ccimg/71500/71484-85-0_b.png)



![3',6'-Dihydroxy-3H-spiro[isobenzofuran-1,9'-xanthen]-3-one](http://img.cochemist.com/ccimg/2400/2321-07-5.png)
![3',6'-Dihydroxy-3H-spiro[isobenzofuran-1,9'-xanthen]-3-one](http://img.cochemist.com/ccimg/2400/2321-07-5_b.png)































































































































































































































![2-[(2s,3r,5r)-2-(hydroxymethyl)-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-3-yl]acetic Acid](http://img.cochemist.com/ccimg/195600/195519-89-2.png)
![2-[(2s,3r,5r)-2-(hydroxymethyl)-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-3-yl]acetic Acid](http://img.cochemist.com/ccimg/195600/195519-89-2_b.png)



![Methanaminium,N-[4-[[4-(dimethylamino)phenyl]phenylmethylene]-2,5-cyclohexadien-1-ylidene]-N-methyl-](http://img.cochemist.com/ccimg/10400/10309-95-2.png)
![Methanaminium,N-[4-[[4-(dimethylamino)phenyl]phenylmethylene]-2,5-cyclohexadien-1-ylidene]-N-methyl-](http://img.cochemist.com/ccimg/10400/10309-95-2_b.png)