Co-reporter:Petra E. Jans, Adelphe M. Mfuh, Hadi D. Arman, Corena V. Shaffer, Oleg V. Larionov, and Susan L. Mooberry
Journal of Natural Products March 24, 2017 Volume 80(Issue 3) pp:676-676
Publication Date(Web):January 4, 2017
DOI:10.1021/acs.jnatprod.6b00963
The trichodermamides are modified dipeptides isolated from a wide variety of fungi, including Trichoderma virens. Previous studies reported that trichodermamide B (2) initiated cytotoxicity in HCT-116 colorectal cancer cells, while trichodermamide A (1) was devoid of activity. We recently developed an efficient total synthesis for the trichodermamides A–C (1–3). Multiple intermediates and analogues were produced, and they were evaluated for biological effects to identify additional structure–activity relationships and the possibility that a simplified analogue would retain the biological effects of 2. The antiproliferative effects of 18 compounds were evaluated, and the results show that 2 and four other compounds are active in HeLa cells, with IC50 values in the range of 1.4–21 μM. Mechanism of action studies of 2 and the other active analogues revealed different spectra of activity. At the IC85 concentration, 2 caused S-phase accumulation and cell death in HeLa cells, suggesting response to DNA double-strand breaks. The analogues did not cause S-phase accumulation or induction of DNA damage repair pathways, consistent with an alternate mode of action. The mechanistic differences are hypothesized to be due to the chlorohydrin moiety in 2, which is lacking in the analogues, which could form a DNA-reactive epoxide.
Co-reporter:April L. RisingerJing Li, Lin Du, Raymond Benavides, Andrew J. Robles, Robert H. Cichewicz, John G. KuhnSusan L. Mooberry
Journal of Natural Products 2017 Volume 80(Issue 2) pp:
Publication Date(Web):January 23, 2017
DOI:10.1021/acs.jnatprod.6b00944
The taccalonolides are microtubule stabilizers that covalently bind tubulin and circumvent clinically relevant forms of resistance to other drugs of this class. Efforts are under way to identify a taccalonolide with optimal properties for clinical development. The structurally similar taccalonolides AF and AJ have comparable microtubule-stabilizing activities in vitro, but taccalonolide AF has excellent in vivo antitumor efficacy when administered systemically, while taccalonolide AJ does not elicit this activity even at maximum tolerated dose. The hypothesis that pharmacokinetic differences underlie the differential efficacies of taccalonolides AF and AJ was tested. The effects of serum on their in vivo potency, metabolism by human liver microsomes and in vivo pharmacokinetic properties were evaluated. Taccalonolides AF and AJ were found to have elimination half-lives of 44 and 8.1 min, respectively. Furthermore, taccalonolide AJ was found to have excellent and highly persistent antitumor efficacy when administered directly to the tumor, suggesting that the lack of antitumor efficacy seen with systemic administration of AJ is likely due to its short half-life in vivo. These results help define why some, but not all, taccalonolides inhibit the growth of tumors at systemically tolerable doses and prompt studies to further improve their pharmacokinetic profile and antitumor efficacy.
Co-reporter:Ravi Kumar Vyas Devambatla, Wei Li, Nilesh Zaware, Shruti Choudhary, Ernest Hamel, Susan L. Mooberry, Aleem Gangjee
Bioorganic & Medicinal Chemistry Letters 2017 Volume 27, Issue 15(Issue 15) pp:
Publication Date(Web):1 August 2017
DOI:10.1016/j.bmcl.2017.05.085
To identify the structural features of 9H-pyrimido[4,5-b]indoles as microtubule depolymerizers, pyrimido[4,5-b]indoles 2–8 with varied substituents at the 2-, 4- and 5-positions were designed and synthesized. Nucleophilic displacement of 2,5-substituted-4-chloro-pyrimido[4,5-b]indoles with appropriate arylamines was the final step employed in the synthesis of target compounds 2–8. Compounds 2 and 6 had two-digit nanomolar potency (IC50) against MDA-MB-435, SK-OV-3 and HeLa cancer cells in vitro. Compounds 2 and 6 also depolymerized microtubules comparable to the lead compound 1. Compounds 2, 3, 6 and 8 were effective in cells expressing P-glycoprotein or the βIII isotype of tubulin, mechanisms that are associated with clinical drug resistance to microtubule targeting drugs. Proton NMR and molecular modeling studies were employed to identify the structural basis for the microtubule depolymerizing activity of pyrimido[4,5-b]indoles.Download high-res image (64KB)Download full-size image
Co-reporter:Ravi Kumar Vyas Devambatla; Ojas A. Namjoshi; Shruti Choudhary; Ernest Hamel; Corena V. Shaffer; Cristina C. Rohena; Susan L. Mooberry;Aleem Gangjee
Journal of Medicinal Chemistry 2016 Volume 59(Issue 12) pp:5752-5765
Publication Date(Web):May 23, 2016
DOI:10.1021/acs.jmedchem.6b00237
The design, synthesis, and biological evaluations of eight 4-substituted 5-methyl-furo[2,3-d]pyrimidines are reported. Synthesis involved N4-alkylation of N-aryl-5-methylfuro[2,3-d]pyrimidin-4-amines, obtained from Ullmann coupling of 4-amino-5-methylfuro[2,3-d]pyrimidine and appropriate aryl iodides. Compounds 3, 4, and 9 showed potent microtubule depolymerizing activities, while compounds 6–8 had slightly lower potency. Compounds 4, 6, 7, and 9 inhibited tubulin assembly with IC50 values comparable to that of combretastatin A-4 (CA-4). Compounds 3, 4, and 6–9 circumvented Pgp and βIII-tubulin mediated drug resistance, mechanisms that can limit the efficacy of paclitaxel, docetaxel, and the vinca alkaloids. In the NCI 60-cell line panel, compound 3 exhibited GI50 values less than 10 nM in 47 of the cell lines. In an MDA-MB-435 xenograft model, compound 3 had statistically significant antitumor effects. The biological effects of 3 identify it as a novel, potent microtubule depolymerizing agent with antitumor activity.
Co-reporter:Shengxin Cai; April L. Risinger; Shalini Nair; Jiangnan Peng; Timothy J. C. Anderson; Lin Du; Douglas R. Powell; Susan L. Mooberry;Robert H. Cichewicz
Journal of Natural Products 2016 Volume 79(Issue 3) pp:490-498
Publication Date(Web):December 21, 2015
DOI:10.1021/acs.jnatprod.5b00874
Some of the most valuable antimalarial compounds, including quinine and artemisinin, originated from plants. While these drugs have served important roles over many years for the treatment of malaria, drug resistance has become a widespread problem. Therefore, a critical need exists to identify new compounds that have efficacy against drug-resistant malaria strains. In the current study, extracts prepared from plants readily obtained from local sources were screened for activity against Plasmodium falciparum. Bioassay-guided fractionation was used to identify 18 compounds from five plant species. These compounds included eight lupane triterpenes (1–8), four kaempferol 3-O-rhamnosides (10–13), four kaempferol 3-O-glucosides (14–17), and the known compounds amentoflavone and knipholone. These compounds were tested for their efficacy against multi-drug-resistant malaria parasites and counterscreened against HeLa cells to measure their antimalarial selectivity. Most notably, one of the new lupane triterpenes (3) isolated from the supercritical extract of Buxus sempervirens, the common boxwood, showed activity against both drug-sensitive and -resistant malaria strains at a concentration that was 75-fold more selective for the drug-resistant malaria parasites as compared to HeLa cells. This study demonstrates that new antimalarial compounds with efficacy against drug-resistant strains can be identified from native and introduced plant species in the United States, which traditionally have received scant investigation compared to more heavily explored tropical and semitropical botanical resources from around the world.
Co-reporter:Andrew J. Robles; Lin Du; Robert H. Cichewicz;Susan L. Mooberry
Journal of Natural Products 2016 Volume 79(Issue 7) pp:1822-1827
Publication Date(Web):June 16, 2016
DOI:10.1021/acs.jnatprod.6b00290
Triple-negative breast cancers are highly aggressive, and patients with these types of tumors have poor long-term survival. These breast cancers do not express estrogen or progesterone receptors and do not have gene amplification of human epidermal growth factor receptor 2; therefore, they do not respond to available targeted therapies. The lack of targeted therapies for triple-negative breast cancers stems from their heterogeneous nature and lack of a clear definition of driver defects. Studies have recently identified triple-negative breast cancer molecular subtypes based on gene expression profiling and representative cell lines, allowing for the identification of subtype-specific drug leads and molecular targets. We previously reported the identification of a new fungal metabolite named maximiscin (1) identified through a crowdsourcing program. New results show that 1 has selective cytotoxic efficacy against basal-like 1 MDA-MB-468 cells compared to cell lines modeling other triple-negative breast cancer molecular subtypes. This compound also exhibited antitumor efficacy in a xenograft mouse model. The mechanisms of action of 1 in MDA-MB-468 cells were investigated to identify potential molecular targets and affected pathways. Compound 1 caused accumulation of cells in the G1 phase of the cell cycle, suggesting induction of DNA damage. Indeed, treatment with 1 caused DNA double-strand breaks with concomitant activation of the DNA damage response pathways, indicated by phosphorylation of p53, Chk1, and Chk2. Collectively, these results suggest basal-like triple-negative breast cancer may be inherently sensitive to DNA-damaging agents relative to other triple-negative breast cancer subtypes. These results also demonstrate the potential of our citizen crowdsourcing program to identify new lead molecules for treating the subtypes of triple-negative breast cancer.
Co-reporter:Corena V. Shaffer; Shengxin Cai; Jiangnan Peng; Andrew J. Robles; Rachel M. Hartley; Douglas R. Powell; Lin Du; Robert H. Cichewicz;Susan L. Mooberry
Journal of Natural Products 2016 Volume 79(Issue 3) pp:531-540
Publication Date(Web):January 19, 2016
DOI:10.1021/acs.jnatprod.5b00908
There remains a critical need for more effective therapies for the treatment of late-stage and metastatic prostate cancers. Three Texas native plants yielded three new and three known compounds with antiproliferative and cytotoxic activities against prostate cancer cells with IC50 values in the range of 1.7–35.0 μM. A new sesquiterpene named espadalide (1), isolated from Gochnatia hypoleuca, had low micromolar potency and was highly effective in clonogenic assays. Two known bioactive germacranolides (2 and 3) were additionally isolated from G. hypoleuca. Dalea frutescens yielded two new isoprenylated chalcones, named sanjuanolide (4) and sanjoseolide (5), and the known sesquiterpenediol verbesindiol (6) was isolated from Verbesina virginica. Mechanistic studies showed that 1–4 caused G2/M accumulation and the formation of abnormal mitotic spindles. Tubulin polymerization assays revealed that 4 increased the initial rate of tubulin polymerization, but did not change total tubulin polymer levels, and 1–3 had no effects on tubulin polymerization. Despite its cytotoxic activity, compound 6 did not initiate changes in cell cycle distribution and has a mechanism of action different from the other compounds. This study demonstrates that new compounds with significant biological activities germane to unmet oncological needs can be isolated from Texas native plants.
Co-reporter:Andrew J. Robles;Shengxin Cai;Robert H. Cichewicz;Susan L. Mooberry
Breast Cancer Research and Treatment 2016 Volume 157( Issue 3) pp:475-488
Publication Date(Web):2016/06/01
DOI:10.1007/s10549-016-3841-9
Triple-negative breast cancers (TNBC) are aggressive malignancies with no effective targeted therapies. Recent gene expression profiling of these heterogeneous cancers and the classification of cell line models now allows for the identification of compounds with selective activities against molecular subtypes of TNBC. The natural product deguelin was found to have selective activity against MDA-MB-453 and SUM-185PE cell lines, which both model the luminal androgen receptor (LAR) subtype of TNBC. Deguelin potently inhibited proliferation of these cells with GI50 values of 30 and 61 nM, in MDA-MB-453 and SUM-185PE cells, respectively. Deguelin had exceptionally high selectivity, 197 to 566-fold, for these cell lines compared to cell lines representing other TNBC subtypes. Deguelin’s mechanisms of action were investigated to determine how it produced these potent and selective effects. Our results show that deguelin has dual activities, inhibiting PI3K/Akt/mTOR signaling, and decreasing androgen receptor levels and nuclear localization. Based on these data, we hypothesized that the combination of the mTOR inhibitor rapamycin and the antiandrogen enzalutamide would have efficacy in LAR models. Rapamycin and enzalutamide showed additive effects in MDA-MB-453 cells, and both drugs had potent antitumor efficacy in a LAR xenograft model. These results suggest that the combination of antiandrogens and mTOR inhibitors might be an effective strategy for the treatment of androgen receptor-expressing TNBC.
Co-reporter:Andrew J. Robles, Jiangnan Peng, Rachel M. Hartley, Brigette Lee, and Susan L. Mooberry
Journal of Natural Products 2015 Volume 78(Issue 3) pp:388-395
Publication Date(Web):February 16, 2015
DOI:10.1021/np500768s
A new tricyclic sesquiterpene, named meleucanthin (1), was isolated from an extract of the leaves and branches of Melampodium leucanthum, along with four known germacranolide sesquiterpene lactones, leucanthin-A (2), leucanthin-B (3), melampodin-A acetate (4), and 3α-hydroxyenhydrin (5). The chemical structure of 1 was elucidated by analysis of 1D and 2D NMR and mass spectrometric data. All compounds exhibited antiproliferative and cytotoxic efficacy against PC-3 and DU 145 prostate cancer cells, as well as HeLa cervical cancer cells, with IC50 values ranging from 0.18 to 9 μM. These compounds were effective in clonogenic assays and displayed high cellular persistence. They were also found to be capable of circumventing P-glycoprotein-mediated drug resistance. Mechanism of action studies showed that 4 caused an accumulation of cells in the G2/M phase of the cell cycle, and 2–5 caused the formation of abnormal mitotic spindles. These results suggest the cytotoxic effects of these germacranolides involve inhibition of mitotic spindle function, and it is likely that other mechanisms additionally contribute to cell death. These studies also demonstrate the possibility of isolating new, biologically active compounds from indigenous Texas plants.
Co-reporter:Xin Zhang, Sudhir Raghavan, Michael Ihnat, Ernest Hamel, Cynthia Zammiello, Anja Bastian, Susan L. Mooberry, Aleem Gangjee
Bioorganic & Medicinal Chemistry 2015 Volume 23(Issue 10) pp:2408-2423
Publication Date(Web):15 May 2015
DOI:10.1016/j.bmc.2015.03.061
A series of eleven conformationally restricted, 4-substituted 2,6-dimethylfuro[2,3-d]pyrimidines was designed to explore the bioactive conformation required for dual inhibition of microtubule assembly and receptor tyrosine kinases (RTKs), and their biological activities are reported. All three rotatable single bonds in the lead compound 1 were sequentially restricted to address the role of each in SAR for microtubule and RTK inhibitory effects. Compounds 2, 3, 7 and 10 showed microtubule depolymerizing activity comparable to or better than the lead 1, some with nanomolar EC50 values. While compound 8 had no effect on microtubules, 8 and 10 both showed potent RTK inhibition with nanomolar IC50s. These compounds confirm that the bioactive conformation for RTK inhibition is different from that for tubulin inhibition. The tetrahydroquinoline analog 10 showed the most potent dual tubulin and RTK inhibitory activities (low nanomolar inhibition of EGFR, VEGFR2 and PDGFR-β). Compound 10 has highly potent activity against many NCI cancer cell lines, including several chemo-resistant cell lines, and could serve as a lead for further preclinical studies.
Co-reporter:Cristina C. Rohena and Susan L. Mooberry
Natural Product Reports 2014 vol. 31(Issue 3) pp:335-355
Publication Date(Web):30 Jan 2014
DOI:10.1039/C3NP70092E
Covering: late 2008 to August 2013
Nature has yielded numerous classes of chemically distinct microtubule stabilizers. Several of these, including paclitaxel (Taxol) and docetaxel (Taxotere), are important drugs used in the treatment of cancer. New microtubule stabilizers and novel formulations of these agents continue to provide advances in cancer therapy. In this review we cover recent progress in the chemistry and biology of these diverse microtubule stabilizers focusing on the wide range of organisms that produce these compounds, their mechanisms of inhibiting microtubule-dependent processes, mechanisms of drug resistance, and their interactions with tubulin including their distinct binding sites and modes. A new potential role for microtubule stabilizers in neurodegenerative diseases is reviewed.
Co-reporter:Jiangnan Peng ; April L. Risinger ; Jing Li ;Susan L. Mooberry
Journal of Medicinal Chemistry 2014 Volume 57(Issue 14) pp:6141-6149
Publication Date(Web):June 24, 2014
DOI:10.1021/jm500619j
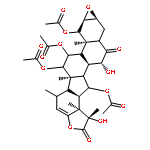
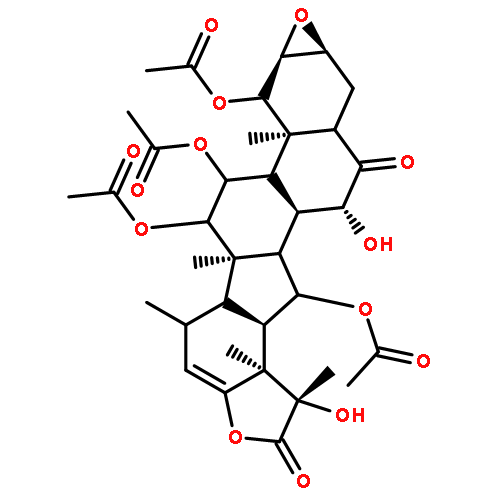
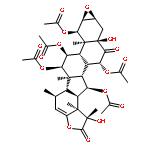
The taccalonolides are microtubule stabilizers isolated from plants of the genus Tacca. Taccalonolide AF is 231 times more potent than the major metabolite taccalonolide A and differs only by the oxidation of the C-22,23 double bond in A to an epoxy group in AF. In the current study, 10 other rare natural taccalonolides were epoxidized and in each case epoxidation improved potency. The epoxidation products of taccalonolide T and AI were the most potent, with IC50 values of 0.43 and 0.88 nM, respectively. These potent taccalonolides retained microtubule stabilizing effects, and T-epoxide demonstrated antitumor effects in a xenograft model of breast cancer. Additional reactions demonstrated that reduction of the C-6 ketone resulted in an inactive taccalonolide and that C-22,23 epoxidation restored its activity. These studies confirm the value of C-22,23 epoxidation as an effective strategy for increasing the potency of a wide range of structurally diverse taccalonolide microtubule stabilizers.
Co-reporter:Lin Du ; Andrew J. Robles ; Jarrod B. King ; Susan L. Mooberry ;Robert H. Cichewicz
Journal of Natural Products 2014 Volume 77(Issue 6) pp:1459-1466
Publication Date(Web):June 3, 2014
DOI:10.1021/np5002253
Two new dimeric epipolythiodiketopiperazines, preussiadins A (1) and B (2), together with two known diastereomers, leptosins C (6) and A (7), were obtained from the mycelia of a Preussia typharum isolate. The structures of the new compounds were established by spectroscopic methods, and the absolute configurations of 1 and 2 were assigned by chemical transformations and comparisons of quantum chemical ECD and VCD calculations to experimental data. Compound 1 exhibited potent cytotoxic activity in the NCI-60 cell line panel with an average LC50 value of 251 nM. Further studies demonstrated that 1 circumvents P-glycoprotein-mediated drug resistance, yet had no significant antitumor activity in a xenograft UACC-62 melanoma model.
Co-reporter:Jing Li, April L. Risinger, Susan L. Mooberry
Bioorganic & Medicinal Chemistry 2014 Volume 22(Issue 18) pp:5091-5096
Publication Date(Web):15 September 2014
DOI:10.1016/j.bmc.2014.01.012
This review focuses on a relatively new class of microtubule stabilizers, the taccalonolides. The taccalonolides are highly oxygenated pentacyclic steroids isolated from plants of the genus Tacca. Originally identified in a cell-based phenotypic screen, the taccalonolides have many properties similar to other microtubule stabilizers. They increase the density of interphase microtubules, causing microtubule bundling, and form abnormal multi-polar mitotic spindles leading to mitotic arrest and, ultimately, apoptosis. However, the taccalonolides differ from other microtubule stabilizers in that they retain efficacy in taxane resistant cell lines and in vivo models. Binding studies with the newly identified, potent taccalonolide AJ demonstrated covalent binding to β-tubulin at or near the luminal and/or pore taxane binding site(s) which stabilizes microtubule protofilaments in a unique manner as compared to other microtubule stabilizers. The isolation and semi-synthesis of 21 taccalonolides helped to identify key structure activity relationships and the importance of multiple regions across the taccalonolide skeleton for optimal biological potency.
Co-reporter:Dr. Lin Du;Andrew J. Robles;Jarrod B. King;Dr. Douglas R. Powell;Dr. Andrew N. Miller;Dr. Susan L. Mooberry;Dr. Robert H. Cichewicz
Angewandte Chemie 2014 Volume 126( Issue 3) pp:823-828
Publication Date(Web):
DOI:10.1002/ange.201306549
Abstract
A fundamental component for success in drug discovery is the ability to assemble and screen compounds that encompass a broad swath of biologically relevant chemical-diversity space. Achieving this goal in a natural-products-based setting requires access to a wide range of biologically diverse specimens. For this reason, we introduced a crowdsourcing program in which citizen scientists furnish soil samples from which new microbial isolates are procured. Illustrating the strength of this approach, we obtained a unique fungal metabolite, maximiscin, from a crowdsourced Alaskan soil sample. Maximiscin, which exhibits a putative combination of polyketide synthase (PKS), non-ribosomal peptide synthetase (NRPS), and shikimate pathway components, was identified as an inhibitor of UACC-62 melanoma cells (LC50=0.93 μM). The metabolite also exhibited efficacy in a xenograft mouse model. These results underscore the value of building cooperative relationships between research teams and citizen scientists to enrich drug discovery efforts.
Co-reporter:Dr. Lin Du;Andrew J. Robles;Jarrod B. King;Dr. Douglas R. Powell;Dr. Andrew N. Miller;Dr. Susan L. Mooberry;Dr. Robert H. Cichewicz
Angewandte Chemie International Edition 2014 Volume 53( Issue 3) pp:804-809
Publication Date(Web):
DOI:10.1002/anie.201306549
Abstract
A fundamental component for success in drug discovery is the ability to assemble and screen compounds that encompass a broad swath of biologically relevant chemical-diversity space. Achieving this goal in a natural-products-based setting requires access to a wide range of biologically diverse specimens. For this reason, we introduced a crowdsourcing program in which citizen scientists furnish soil samples from which new microbial isolates are procured. Illustrating the strength of this approach, we obtained a unique fungal metabolite, maximiscin, from a crowdsourced Alaskan soil sample. Maximiscin, which exhibits a putative combination of polyketide synthase (PKS), non-ribosomal peptide synthetase (NRPS), and shikimate pathway components, was identified as an inhibitor of UACC-62 melanoma cells (LC50=0.93 μM). The metabolite also exhibited efficacy in a xenograft mouse model. These results underscore the value of building cooperative relationships between research teams and citizen scientists to enrich drug discovery efforts.
Co-reporter:Aleem Gangjee ; Ying Zhao ; Sudhir Raghavan ; Cristina C. Rohena ; Susan L. Mooberry ;Ernest Hamel
Journal of Medicinal Chemistry 2013 Volume 56(Issue 17) pp:6829-6844
Publication Date(Web):July 29, 2013
DOI:10.1021/jm400639z
A series of 21 substituted cyclopenta[d]pyrimidines were synthesized as an extension of our discovery of the parent compound (±)-1·HCl as an anti-microtubule agent. The structure–activity relationship indicates that the N-methyl and a 4N-methoxy groups appear important for potent activity. In addition, the 6-substituent in the parent analogue is not necessary for activity. The most potent compound 30·HCl was a one to two digit nanomolar inhibitor of most tumor cell proliferations and was up to 7-fold more potent than the parent compound (±)-1·HCl. In addition, 30·HCl inhibited cancer cell proliferation regardless of Pgp or βIII-tubulin status, both of which are known to cause clinical resistance to several anti-tubulin agents. In vivo efficacy of 30·HCl was demonstrated against a triple negative breast cancer xenograft mouse model. Compound 30·HCl is water-soluble and easily synthesized and serves as a lead compound for further preclinical evaluation as an antitumor agent.
Co-reporter:Jiangnan Peng, April L. Risinger, Chenxiao Da, Gary A. Fest, Glen E. Kellogg, and Susan L. Mooberry
Journal of Natural Products 2013 Volume 76(Issue 12) pp:2189-2194
Publication Date(Web):December 4, 2013
DOI:10.1021/np4005085
Several biologically active compounds have been identified from Tacca species, including glycosides, diarylheptanoids, saponins, withanolides, and the taccalonolide class of microtubule stabilizers. More recently, two cytotoxic retro-dihydrochalcones named evelynin A (7) and taccabulin A (6) were isolated and their biological activities characterized, including the finding that taccabulin has microtubule destabilizing effects. Here we describe the identification and characterization of five new retro-chalcones, named taccabulins B–E (1–4) and evelynin B (5) from Tacca sp. extracts. Their structures were determined using 1D and 2D NMR as well as mass spectroscopic data and modeled into the colchicine binding site of tubulin. The antiproliferative and microtubule effects of each compound were determined experimentally and found to be well correlated with modeling studies. The isolation and biological characterization of several retro-dihydrochalcones facilitated preliminary structure–activity relationships for this compound class concerning its antiproliferative and microtubule depolymerizing activities.
Co-reporter:Jing Li, Jiangnan Peng, April L. Risinger, and Susan L. Mooberry
Journal of Natural Products 2013 Volume 76(Issue 7) pp:1369-1375
Publication Date(Web):July 15, 2013
DOI:10.1021/np400435t
The taccalonolides are microtubule stabilizers isolated from plants of the genus Tacca that show potent in vivo antitumor activity and the ability to overcome multiple mechanisms of drug resistance. The most potent taccalonolide identified to date, AJ, is a semisynthetic product generated from the major plant metabolite taccalonolide A in a two-step reaction. The first step involves hydrolysis of taccalonolide A to generate taccalonolide B, and then this product is oxidized to generate an epoxide group at C-22–C-23. To generate sufficient taccalonolide AJ for in vivo antitumor efficacy studies, the hydrolysis conditions for the conversion of taccalonolide A to B were optimized. During purification of the hydrolysis products, we identified the new taccalonolide AO (1) along with taccalonolide I. When the same hydrolysis reaction was performed on a taccalonolide E-enriched fraction, four new taccalonolides, assigned as AK, AL, AM, and AN (2–5), were obtained in addition to the expected product taccalonolide N. Biological assays were performed on each of the purified taccalonolides, which allowed for increased refinement of the structure–activity relationship of this class of compounds.
Co-reporter:Aleem Gangjee, Nilesh Zaware, Ravi Kumar Vyas Devambatla, Sudhir Raghavan, Cara D. Westbrook, Nicholas F. Dybdal-Hargreaves, Ernest Hamel, Susan L. Mooberry
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 4) pp:891-902
Publication Date(Web):15 February 2013
DOI:10.1016/j.bmc.2012.12.010
A series of fourteen N4-(substituted phenyl)-N4-alkyl/desalkyl-9H-pyrimido[4,5-b]indole-2,4-diamines was synthesized as potential microtubule targeting agents. The synthesis involved a Fisher indole cyclization of 2-amino-6-hydrazinylpyrimidin-4(3H)-one with cyclohexanone, followed by oxidation, chlorination and displacement with appropriate anilines. Compounds 6, 14 and 15 had low nanomolar potency against MDA-MB-435 tumor cells and depolymerized microtubules. Compound 6 additionally had nanomolar GI50 values against 57 of the NCI 60-tumor panel cell lines. Mechanistic studies showed that 6 inhibited tubulin polymerization and [3H]colchicine binding to tubulin. The most potent compounds were all effective in cells expressing P-glycoprotein or the βIII isotype of tubulin, which have been associated with clinical drug resistance. Modeling studies provided the potential interactions of 6, 14 and 15 within the colchicine site.
Co-reporter:Jiangnan Peng ; Rachel M. Hartley ; Gary A. Fest ;Susan L. Mooberry
Journal of Natural Products 2012 Volume 75(Issue 3) pp:494-496
Publication Date(Web):January 19, 2012
DOI:10.1021/np200796e
Three new O-prenylated flavonoids, amyrisins A–C (1–3), were isolated from the leaves and twigs of Amyris madrensis, along with the known compound polygamain (4). The structures of 1–3 were elucidated on the basis of the analysis of spectroscopic data interpretation. Amyrisins B (2) and C (3) showed moderate cytotoxicity against PC-3 and DU 145 prostate cancer cells with IC50 values of 17.5 and 23 μM, respectively, while amyrisin A (1) did not show any cytotoxicity at the highest concentration tested, 50 μM. Polygamain (4) exhibited potent antiproliferative and microtubule-depolymerizing activities.
Co-reporter:Jing Li ; April L. Risinger ; Jiangnan Peng ; Zhongliang Chen ; Lihong Hu ;Susan L. Mooberry
Journal of the American Chemical Society 2011 Volume 133(Issue 47) pp:19064-19067
Publication Date(Web):November 1, 2011
DOI:10.1021/ja209045k
The taccalonolides are a class of microtubule stabilizing agents isolated from plants of the genus Tacca. In efforts to define their structure–activity relationships, we isolated five new taccalonolides, AC–AF and H2, from one fraction of an ethanol extract of Tacca plantaginea. The structures were elucidated using a combination of spectroscopic methods, including 1D and 2D NMR and HR-ESI-MS. Taccalonolide AJ, an epoxidation product of taccalonolide B, was generated by semisynthesis. Five of these taccalonolides demonstrated cellular microtubule-stabilizing activities and antiproliferative actions against cancer cells, with taccalonolide AJ exhibiting the highest potency with an IC50 value of 4.2 nM. The range of potencies of these compounds, from 4.2 nM to >50 μM, for the first time provides the opportunity to identify specific structural moieties crucial for potent biological activities as well as those that impede optimal cellular effects. In mechanistic assays, taccalonolides AF and AJ stimulated the polymerization of purified tubulin, an activity that had not previously been observed for taccalonolides A and B, providing the first evidence that this class of microtubule stabilizers can interact directly with tubulin/microtubules. Taccalonolides AF and AJ were able to enhance tubulin polymerization to the same extent as paclitaxel but exhibited a distinct kinetic profile, suggesting a distinct binding mode or the possibility of a new binding site. The potencies of taccalonolides AF and AJ and their direct interaction with tubulin, together with the previous excellent in vivo antitumor activity of this class, reveal the potential of the taccalonolides as new anticancer agents.
Co-reporter:Aleem Gangjee ; Ying Zhao ; Ernest Hamel ; Cara Westbrook ;Susan L. Mooberry
Journal of Medicinal Chemistry 2011 Volume 54(Issue 17) pp:6151-6155
Publication Date(Web):July 25, 2011
DOI:10.1021/jm2007722
(R,S)-1 is a potent antimitotic compound. (R)-1·HCl and (S)-1·HCl were synthesized from (R)- and (S)-3-methyladipic acid. Both enantiomers were potent inhibitors of cell proliferation and caused cellular microtubule loss and mitotic arrest. They inhibited purified tubulin assembly and the binding of [3H]colchicine to tubulin, with (S)-1 being about twice as potent. Cytotoxicity against 60 tumor cell lines, however, indicated that the (S)-isomer was 10- to 88-fold more potent than the (R)-isomer.
Co-reporter:Jiangnan Peng ; April L. Risinger ; Gary A. Fest ; Evelyn M. Jackson ; Gregory Helms ; Lisa A. Polin ;Susan L. Mooberry
Journal of Medicinal Chemistry 2011 Volume 54(Issue 17) pp:6117-6124
Publication Date(Web):July 29, 2011
DOI:10.1021/jm200757g
The taccalonolides are a unique class of microtubule stabilizers that do not bind directly to tubulin. Three new taccalonolides, Z, AA, and AB, along with two known compounds, taccalonolides R and T, were isolated from Tacca chantrieri and Tacca integrifolia. Taccalonolide structures were determined by 1D and 2D NMR methods. The biological activities of the new taccalonolides, as well as taccalonolides A, B, E, N, R, and T, were evaluated. All nine taccalonolides display microtubule stabilizing activity, but profound differences in antiproliferative potencies were noted, with IC50 values ranging from the low nanomolar range for taccalonolide AA (32 nM) to the low micromolar range for taccalonolide R (13 μM). These studies demonstrate that diverse taccalonolides possess microtubule stabilizing properties and that significant structure–activity relationships exist. In vivo antitumor evaluations of taccalonolides A, E, and N show that each of these molecules has in vivo antitumor activity.
Co-reporter:Lauren Lee ; Lyda M. Robb ; Megan Lee ; Ryan Davis ; Hilary Mackay ; Sameer Chavda ; Balaji Babu ; Erin L. O’Brien ; April L. Risinger ; Susan L. Mooberry ;Moses Lee
Journal of Medicinal Chemistry 2010 Volume 53(Issue 1) pp:325-334
Publication Date(Web):November 6, 2009
DOI:10.1021/jm901268n
A total of 24 novel 2,5-diaryl-1,3,4-oxadiazoline analogs of combretastatin A-4 (CA-4, 1) were designed, synthesized, and evaluated for biological activities. The compounds represent two structural classes; the Type I class has three methoxy groups on the A ring and the Type II class has a single methoxy group on the A ring. Biological evaluations demonstrate that multiple structural features control the biological potency. Four of the compounds, 2-(3′-bromophenyl)-5-(3′′,4′′,5′′-trimethoxyphenyl)-2-acetyl-2,3-dihydro-1,3,4-oxadiazoline (9l), 2-(2′,5′-dimethoxyphenyl)-5-(3′′-methoxyphenyl)-2-acetyl-2,3-dihydro-1,3,4-oxadiazoline (10h), 2-(3′,4′,5′-trimethoxyphenyl)-5-(3′′-methoxyphenyl)-2-acetyl-2,3-dihydro-1,3,4-oxadiazoline (10i), and 2-(3′,5′-dimethoxyphenyl)-5-(3′′-methoxyphenyl)-2-acetyl-2,3-dihydro-1,3,4-oxadiazoline (10j), have potent antiproliferative activities against multiple cancer cell lines. Mechanistic studies indicate that they retain the microtubule disrupting effects of compound 1, including microtubule loss, the formation of aberrant mitotic spindles, and mitotic arrest. Compound 10i inhibits purified tubulin polymerization and circumvents drug resistance mediated by P-glycoprotein and βIII tubulin expression. The oxadiazoline analog 10i is a promising lead candidate worthy of further investigation.
Co-reporter:Aleem Gangjee ; Ying Zhao ; Lu Lin ; Sudhir Raghavan ; Elizabeth G. Roberts ; April L. Risinger ; Ernest Hamel ;Susan L. Mooberry
Journal of Medicinal Chemistry 2010 Volume 53(Issue 22) pp:8116-8128
Publication Date(Web):October 25, 2010
DOI:10.1021/jm101010n
Two classes of molecules were designed and synthesized based on a 6-CH3 cyclopenta[d]pyrimidine scaffold and a pyrrolo[2,3-d]pyrimidine scaffold. The pyrrolo[2,3-d]pyrimidines were synthesized by reacting ethyl 2-cyano-4,4-diethoxybutanoate and acetamidine, which in turn was chlorinated and reacted with the appropriate anilines to afford 1 and 2. The cyclopenta[d]pyrimidines were obtained from 3-methyladapic acid, followed by reaction with acetamidine to afford the cyclopenta[d]pyrimidine scaffold. Chlorination and reaction with appropriate anilines afforded (±)-3·HCl−(±)-7·HCl. Compounds 1 and (±)-3·HCl had potent antiproliferative activities in the nanomolar range. Compound (±)-3·HCl is significantly more potent than 1. Mechanistic studies showed that 1 and (±)-3·HCl cause loss of cellular microtubules, inhibit the polymerization of purified tubulin, and inhibit colchicine binding. Modeling studies show interactions of these compounds within the colchicine site. The identification of these new inhibitors that can also overcome clinically relevant mechanisms of drug resistance provides new scaffolds for colchicine site agents.
Co-reporter:Jiangnan Peng, Evelyn M. Jackson, David J. Babinski, April L. Risinger, Gregory Helms, Doug E. Frantz, and Susan L. Mooberry
Journal of Natural Products 2010 Volume 73(Issue 9) pp:1590-1592
Publication Date(Web):August 17, 2010
DOI:10.1021/np100350s
A new benzoquinone-type retro-dihydrochalcone, named evelynin, was isolated from the roots and rhizomes of Tacca chantrieri. The structure was elucidated on the basis of the analysis of spectroscopic data and confirmed by a simple one-step total synthesis. Evelynin exhibited cytotoxicity against four human cancer cell lines, MDA-MB-435 melanoma, MDA-MB-231 breast, PC-3 prostate, and HeLa cervical carcinoma cells, with IC50 values of 4.1, 3.9, 4.7, and 6.3 μM, respectively.
Co-reporter:April L. Risinger ; Jiangnan Peng ; Cristina C. Rohena ; Hector R. Aguilar ; Doug E. Frantz ;Susan L. Mooberry
Journal of Natural Products () pp:
Publication Date(Web):
DOI:10.1021/np4005079
The biosynthesis of secondary metabolites provides higher plants with mechanisms of defense against microbes, insects, and herbivores. One common cellular target of these molecules is the highly conserved microtubule cytoskeleton, and microtubule-targeting compounds with insecticidal, antifungal, nematicidal, and anticancer activities have been identified from plants. A new retro-dihydrochalcone, taccabulin A, with microtubule-destabilizing activity has been identified from the roots and rhizomes of Tacca species. This finding is notable because the microtubule-stabilizing taccalonolides are also isolated from these sources. This is the first report of an organism producing compounds with both microtubule-stabilizing and -destabilizing activities. A two-step chemical synthesis of taccabulin A was performed. Mechanistic studies showed that taccabulin A binds within the colchicine site on tubulin and has synergistic antiproliferative effects against cancer cells when combined with a taccalonolide, which binds to a different site on tubulin. Taccabulin A is effective in cells that are resistant to many other plant-derived compounds. The discovery of a natural source that contains both microtubule-stabilizing and -destabilizing small molecules is unprecedented and suggests that the synergistic action of these compounds was exploited by nature long before it was discovered in the laboratory.




















![OXIRENO[7,8]CYCLODECA[1,2-B]FURAN-3-CARBOXYLIC ACID, 4-(ACETYLOXY)-5-[[(2,3-DIMETHYLOXIRANYL)CARBONYL]OXY]-1A,4,5,5A,6,7,8A,10A-OCTAHYDRO-10-METHYL-6-METHYLENE-7-OXO-, METHYL ESTER, [1AR-[1AR*,2E,4S*,5S*(2R*,3R*),5AS*,8AR*,9E,10AS*]]-](http://img.cochemist.com/ccimg/35900/35878-52-5.png)
![OXIRENO[7,8]CYCLODECA[1,2-B]FURAN-3-CARBOXYLIC ACID, 4-(ACETYLOXY)-5-[[(2,3-DIMETHYLOXIRANYL)CARBONYL]OXY]-1A,4,5,5A,6,7,8A,10A-OCTAHYDRO-10-METHYL-6-METHYLENE-7-OXO-, METHYL ESTER, [1AR-[1AR*,2E,4S*,5S*(2R*,3R*),5AS*,8AR*,9E,10AS*]]-](http://img.cochemist.com/ccimg/35900/35878-52-5_b.png)