Co-reporter:Kimberly M. Lovell, Kevin J. Frankowski, Edward L. Stahl, Stephen R. Slauson, Euna Yoo, Thomas E. Prisinzano, Jeffrey Aubé, and Laura M. Bohn
ACS Chemical Neuroscience 2015 Volume 6(Issue 8) pp:1411
Publication Date(Web):April 19, 2015
DOI:10.1021/acschemneuro.5b00092
Kappa opioid receptor (KOR) modulation is a promising target for drug discovery efforts due to KOR involvement in pain, depression, and addiction behaviors. We recently reported a new class of triazole KOR agonists that displays significant bias toward G protein signaling over βarrestin2 recruitment; interestingly, these compounds also induce less activation of ERK1/2 map kinases than the balanced agonist, U69,593. We have identified structure–activity relationships around the triazole scaffold that allows for decreasing the bias for G protein signaling over ERK1/2 activation while maintaining the bias for G protein signaling over βarrestin2 recruitment. The development of novel compounds, with different downstream signaling outcomes, independent of G protein/βarrestin2 bias, provides a more diverse pharmacological toolset for use in defining complex KOR signaling and elucidating the significance of KOR-mediated signaling.Keywords: arrestin; biased agonism; Functional selectivity; G protein coupling; GPCR; MAP kinase
Co-reporter:Charlie Fehl, Erin E. Hirt, Sze-Wan Li, Jeffrey Aubé
Tetrahedron Letters 2015 Volume 56(Issue 23) pp:3137-3140
Publication Date(Web):3 June 2015
DOI:10.1016/j.tetlet.2014.12.068
The intramolecular Schmidt reaction of ketones and tethered azides is an efficient method for the generation of amides and lactams. This reaction is catalyzed by Lewis acids, which tightly bind the strongly basic amide product and result in product inhibition. We report herein conditions to achieve a catalytic Schmidt reaction using substoichiometric amounts of the heat-stable Lewis acid Sc(OTf)3. This species was shown to effectively release products of the Schmidt reaction in a temperature-dependent fashion. Thus, heat was able to promote catalyst turnover. A brief substrate scope was conducted using these conditions.
Co-reporter:Stephen R. Slauson, Ryan Pemberton, Partha Ghosh, Dean J. Tantillo, and Jeffrey Aubé
The Journal of Organic Chemistry 2015 Volume 80(Issue 10) pp:5260-5271
Publication Date(Web):April 22, 2015
DOI:10.1021/acs.joc.5b00804
The development of the domino reaction between an aminoethyl-substituted diene and maleic anhydride to afford an N-substituted octahydroisoquinolin-1-one is described. A typical procedure involves the treatment of a 1-aminoethyl-substituted butadiene with maleic anhydride at 0 °C to room temperature for 20 min under low-solvent conditions, which affords a series of isoquinolinone carboxylic acids in moderate to excellent yields. NMR monitoring suggested that the reaction proceeded via an initial acylation step followed by an intramolecular Diels–Alder reaction. For the latter step, a significant rate difference was observed depending on whether the amino group was substituted by a phenyl or an alkyl (usually benzyl) substituent, with the former noted by NMR to be substantially slower. The Diels–Alder step was studied by density functional theory (DFT) methods, leading to the conclusion that the degree of preorganization in the starting acylated intermediate had the largest effect on the reaction barriers. In addition, the effect of electronics on the aromatic ring in N-phenyl substrates was studied computationally and experimentally. Overall, this protocol proved considerably more amenable to scale up compared to earlier methods by eliminating the requirement of microwave batch chemistry for this reaction as well as significantly reducing the quantity of solvent.
Co-reporter:Chan Woo Huh and Jeffrey Aubé
Chemical Science 2014 vol. 5(Issue 2) pp:699-706
Publication Date(Web):26 Nov 2013
DOI:10.1039/C3SC52805G
A means of generating an N-alkyl-N-arylaminodiazonium ion, which then loses nitrogen to form a reactive aryl nitrenium intermediate, is described. In this sequence, a set of triazenyl acetonitriles was synthesized by nucleophilic addition of a nitrile anion to an aryl azide followed by temperature-dependent alkylation at either of two nucleophilic triazenyl nitrogen atoms. An α,α-disubstituted acetonitrile and N-arylaminodiazonium moiety (or aryl nitrenium ion upon loss of N2) are embedded in the resulting 1,3,3-trisubsituted triazene, which undergoes liberation and recombination of these two components under acidic conditions to yield an α-arylated acetonitrile containing an all-carbon-quaternary center. We propose that the N-alkyl-N-arylaminodiazonium ion loses nitrogen to generate an aryl nitrenium species, which then reacts with an α,α-disubstituted acetonitrile either at the para- or meta-position of the aromatic ring to afford p-alkylaminoaryl acetonitrile derivatives or 3,3-substituted 1-methylindolin-2-imines, depending on the substrates and conditions.
Co-reporter:Rakesh H. Vekariya, Ruzhang Liu, and Jeffrey Aubé
Organic Letters 2014 Volume 16(Issue 7) pp:1844-1847
Publication Date(Web):March 17, 2014
DOI:10.1021/ol500011f
An intramolecular Huisgen cycloaddition of an interconverting set of isomeric allylic azides with alkynes affords substituted triazoles in high yield. The stereoisomeric vinyl-substituted triazoloxazines formed depend on the rate of cycloaddition of the different allylic azide precursors when the reaction is carried out under thermal conditions. In contrast, dimerized macrocyclic products were obtained when the reaction was done using copper(I)-catalyzed conditions, demonstrating the ability to control the reaction products through changing conditions.
Co-reporter:Gurpreet Singh, Angelica Meyer, and Jeffrey Aubé
The Journal of Organic Chemistry 2014 Volume 79(Issue 1) pp:452-458
Publication Date(Web):December 10, 2013
DOI:10.1021/jo402539p
Protected 4-hydroxycyclopentenones (4-HCPs) constitute an important class of intermediates in chemical synthesis. A route to this class of compound has been developed. Key steps include Noyori reduction (which establishes the stereochemistry of the product), ring-closing metathesis, and simple functional group conversions to provide a set of substituted 4-HCPs in either enantiomeric form.
Co-reporter:Michal Szostak and Jeffrey Aubé
Chemical Reviews 2013 Volume 113(Issue 8) pp:5701
Publication Date(Web):June 17, 2013
DOI:10.1021/cr4000144
Co-reporter:Hashim F. Motiwala ; Charlie Fehl ; Sze-Wan Li ; Erin Hirt ; Patrick Porubsky
Journal of the American Chemical Society 2013 Volume 135(Issue 24) pp:9000-9009
Publication Date(Web):May 20, 2013
DOI:10.1021/ja402848c
A method for carrying out the intramolecular Schmidt reaction of alkyl azides and ketones using a substoichiometric amount of catalyst is reported. Following extensive screening, the use of the strong hydrogen-bond-donating solvent hexafluoro-2-propanol was found to be consistent with low catalyst loadings, which ranged from 2.5 mol % for favorable substrates to 25 mol % for more difficult cases. Reaction optimization, broad substrate scope, and preliminary mechanistic studies of this improved version of the reaction are described.
Co-reporter:Hashim F. Motiwala, Joseph Bazzill, Abbas Samadi, Huaping Zhang, Barbara N. Timmermann, Mark S. Cohen, and Jeffrey Aubé
ACS Medicinal Chemistry Letters 2013 Volume 4(Issue 11) pp:1069-1073
Publication Date(Web):September 5, 2013
DOI:10.1021/ml400267q
The natural product withaferin A exhibits potent antitumor activity and other diverse pharmacological activities. The recently discovered withalongolide A, a C-19 hydroxylated congener of withaferin A, was recently reported to possess cytotoxic activity against head and neck squamous cell carcinomas. Semisynthetic acetylated analogues of withalongolide A were shown to be considerably more cytotoxic than the parent compound. To further explore the structure–activity relationships, 20 new semisynthetic analogues of withalongolide A were synthesized and evaluated for cytotoxic activity against four different cancer cell lines. A number of derivatives were found to be more potent than the parent compound and withaferin A.Keywords: DRO81-1; jaborosalactone; macrocycle; triacetate; withaferin A; Withalongolide A;
Co-reporter:Thomas C. Coombs, Cordelle Tanega, Min Shen, Jenna L. Wang, Douglas S. Auld, Samuel W. Gerritz, Frank J. Schoenen, Craig J. Thomas, Jeffrey Aubé
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 12) pp:3654-3661
Publication Date(Web):15 June 2013
DOI:10.1016/j.bmcl.2013.02.096
Substituted pyrimidine inhibitors of the Clk and Dyrk kinases have been developed, exploring structure–activity relationships around four different chemotypes. The most potent compounds have low-nanomolar inhibitory activity against Clk1, Clk2, Clk4, Dyrk1A and Dyrk1B. Kinome scans with 442 kinases using agents representing three of the chemotypes show these inhibitors to be highly selective for the Clk and Dyrk families. Further off-target pharmacological evaluation with ML315, the most selective agent, supports this conclusion.
Co-reporter:Ruzhang Liu ; Osvaldo Gutierrez ; Dean J. Tantillo
Journal of the American Chemical Society 2012 Volume 134(Issue 15) pp:6528-6531
Publication Date(Web):April 10, 2012
DOI:10.1021/ja300369c
Pre-equilibration of an interconverting set of isomeric allylic azides is coupled with an intramolecular Schmidt reaction to afford substituted lactams stereoselectively. The effect of substitution and a preliminary mechanistic study are reported. The synthetic potential of this method is demonstrated in the context of an enantioselective synthesis of an advanced intermediate leading toward pinnaic acid.
Co-reporter:Kevin J. Frankowski, Michael P. Hedrick, Palak Gosalia, Kelin Li, Shenghua Shi, David Whipple, Partha Ghosh, Thomas E. Prisinzano, Frank J. Schoenen, Ying Su, S. Vasile, Eduard Sergienko, Wilson Gray, Santosh Hariharan, Loribelle Milan, Susanne Heynen-Genel, Arianna Mangravita-Novo, Michael Vicchiarelli, Layton H. Smith, John M. Streicher, Marc G. Caron, Lawrence S. Barak, Laura M. Bohn, Thomas D. Y. Chung, and Jeffrey Aubé
ACS Chemical Neuroscience 2012 Volume 3(Issue 3) pp:221
Publication Date(Web):January 20, 2012
DOI:10.1021/cn200128x
Herein we present the outcome of a high throughput screening (HTS) campaign-based strategy for the rapid identification and optimization of selective and general chemotypes for both kappa (κ) opioid receptor (KOR) activation and inhibition. In this program, we have developed potent antagonists (IC50 < 120 nM) or agonists of high binding affinity (Ki < 3 nM). In contrast to many important KOR ligands, the compounds presented here are highly modular, readily synthesized, and, in most cases, achiral. The four new chemotypes hold promise for further development into chemical tools for studying the KOR or as potential therapeutic lead candidates.Keywords: high-throughput screening; Kappa opioid receptor agonist; kappa opioid receptor antagonist
Co-reporter:Jeffrey Aubé
ACS Medicinal Chemistry Letters 2012 Volume 3(Issue 6) pp:442
Publication Date(Web):May 24, 2012
DOI:10.1021/ml300114c
Drug repurposing is an approach to finding new uses for older drugs and has been gaining popularity in recent years. The role of traditional medicinal chemistry in the context of these efforts is considered.
Co-reporter:Hashim F. Motiwala, Belgin Gülgeze, and Jeffrey Aubé
The Journal of Organic Chemistry 2012 Volume 77(Issue 16) pp:7005-7022
Publication Date(Web):July 25, 2012
DOI:10.1021/jo3012336
The highly regio- and chemoselective oxidation of activated C–H bonds has been observed via copper-catalyzed reactions of oxaziridines. The oxidation proceeded with a variety of substrates, primarily comprising allylic and benzylic examples, as well as one example of an otherwise unactivated tertiary C–H bond. The mechanism of the reaction is proposed to involve single-electron transfer to the oxaziridines to generate a copper-bound radical anion, followed by hydrogen atom abstraction and collapse to products, with regeneration of the catalyst by a final single-electron transfer event. The involvement of allylic radical intermediates was supported by a radical-trapping experiment with TEMPO.
Co-reporter: Jeffrey Aubé
Angewandte Chemie 2012 Volume 124( Issue 13) pp:3117-3119
Publication Date(Web):
DOI:10.1002/ange.201108173
Co-reporter: Jeffrey Aubé
Angewandte Chemie International Edition 2012 Volume 51( Issue 13) pp:3063-3065
Publication Date(Web):
DOI:10.1002/anie.201108173
Co-reporter:Erik Fenster, Charlie Fehl, and Jeffrey Aubé
Organic Letters 2011 Volume 13(Issue 10) pp:2614-2617
Publication Date(Web):April 25, 2011
DOI:10.1021/ol200725m
A tandem Prins/Friedel–Crafts reaction useful for the construction of the indeno-tetrahydropyridine core of the haouamine alkaloids and a formal synthesis of (−)-haouamine A are described.
Co-reporter:Michal Szostak and Jeffrey Aubé
Organic & Biomolecular Chemistry 2011 vol. 9(Issue 1) pp:27-35
Publication Date(Web):11 Nov 2010
DOI:10.1039/C0OB00215A
Medium-bridged twisted lactams, in which a non-planar amide bond is achieved by incorporating the nitrogen atom at the bridgehead position in a medium-sized heterocycle, offer an attractive setting in which to study the properties of distorted amide linkages. This Emerging Area article will describe progress in the preparation and study of these compounds. This work shows that compounds containing an even moderately distorted amide bond display useful and unusual chemical properties while retaining a measure of stability that enables their study.
Co-reporter:Dr. Thomas O. Painter;Dr. Paul D. Thornton;Mario Orestano;Dr. Conrad Santini; Michael G. Organ; Jeffrey Aubé
Chemistry - A European Journal 2011 Volume 17( Issue 35) pp:9595-9598
Publication Date(Web):
DOI:10.1002/chem.201100768
Co-reporter:Chan Woo Huh, Chad Schroeder, Gurpreet Singh, and Jeffrey Aubé
The Journal of Organic Chemistry 2011 Volume 76(Issue 9) pp:3160-3165
Publication Date(Web):March 21, 2011
DOI:10.1021/jo102597d
The stereoselectivity of nucleophilic additions to 3-azidoalkanals was investigated. Non-chelating, BF3·OEt2-mediated Sakurai addition to 3-azidoalkanals afforded 1,3-anti products, whereas use of a chelating Lewis acid, TiCl4, resulted in 1,3-syn products with moderate selectivity. A boat-like chelation structure of the 3-azidoalkanal with the Lewis acid is proposed to be consistent with the 1,3-syn selectivity of the reactions. Mukaiyama aldol addition to 3-azidohexanal generated 1,3-anti products regardless of the chelating ability of the Lewis acid.
Co-reporter:Partha Ghosh and Jeffrey Aubé
The Journal of Organic Chemistry 2011 Volume 76(Issue 10) pp:4168-4172
Publication Date(Web):April 14, 2011
DOI:10.1021/jo200433w
A method for the optical resolution of carboxylic acids is described. Condensation of racemic carboxylic acids with chiral terminal propargyl alcohols gave separable diastereomeric esters. Chromatographic separation followed by heating the individual diastereomers in methanol with catalytic copper(I) halide regenerated the carboxylic acids in good yields and in enantiomeric ratios of ≥94%. This method is particularly useful for the resolution of carboxylic acids that are incompatible with conventional ester hydrolysis.
Co-reporter:Kevin J. Frankowski;Vincent Setola;Jon M. Evans;Ben Neuenswander;Bryan L. Roth
PNAS 2011 108 (17 ) pp:6727-6732
Publication Date(Web):2011-04-26
DOI:10.1073/pnas.1016558108
Reported biological activities of Stemona natural products, such as antitussive activity, inspired the development of synthetic methods to access several alkaloids
within this family and in so doing develop a general route to the core skeleta shared by the class of natural products. The
chemistry was subsequently adapted to afford a series of analogue sets bearing simplified, diverse Stemona-inspired skeleta. Over 100 of these analogues were subjected to general G protein-coupled receptor profiling along with the
known antitussive compound, neostenine; this led to the identification of hit compounds targeting several receptor types.
The particularly rich hit subset for sigma receptors was expanded with two focused library sets, which resulted in the discovery
of a fully synthetic, potent chemotype of sigma ligands. This collaborative effort combined the development of synthetic methods
with extensive, flexible screening resources and exemplifies the role of natural products in bioactivity mining.
Co-reporter:Kevin J. Frankowski;Vincent Setola;Jon M. Evans;Ben Neuenswander;Bryan L. Roth
PNAS 2011 108 (17 ) pp:6727-6732
Publication Date(Web):2011-04-26
DOI:10.1073/pnas.1016558108
Reported biological activities of Stemona natural products, such as antitussive activity, inspired the development of synthetic methods to access several alkaloids
within this family and in so doing develop a general route to the core skeleta shared by the class of natural products. The
chemistry was subsequently adapted to afford a series of analogue sets bearing simplified, diverse Stemona-inspired skeleta. Over 100 of these analogues were subjected to general G protein-coupled receptor profiling along with the
known antitussive compound, neostenine; this led to the identification of hit compounds targeting several receptor types.
The particularly rich hit subset for sigma receptors was expanded with two focused library sets, which resulted in the discovery
of a fully synthetic, potent chemotype of sigma ligands. This collaborative effort combined the development of synthetic methods
with extensive, flexible screening resources and exemplifies the role of natural products in bioactivity mining.
Co-reporter:Dr. Thomas C. Coombs;Dr. Gerald H. Lushington;Dr. Justin Douglas;Dr. Jeffrey Aubé
Angewandte Chemie 2011 Volume 123( Issue 12) pp:2786-2789
Publication Date(Web):
DOI:10.1002/ange.201007133
Co-reporter:Dr. Thomas C. Coombs;Dr. Gerald H. Lushington;Dr. Justin Douglas;Dr. Jeffrey Aubé
Angewandte Chemie International Edition 2011 Volume 50( Issue 12) pp:2734-2737
Publication Date(Web):
DOI:10.1002/anie.201007133
Co-reporter:Michal Szostak ; Lei Yao
Journal of the American Chemical Society 2010 Volume 132(Issue 6) pp:2078-2084
Publication Date(Web):January 22, 2010
DOI:10.1021/ja909792h
The reactions of a series of strained bicyclic and tricyclic one-carbon bridged lactams with organometallic reagents have been investigated. These amides permit isolation of a number of remarkably stable hemiaminals upon nucleophilic addition to the twisted amide bonds present in the lactam precursors. The factors that affect the stability of the resulting bridged hemiaminals are presented. In some cases, the hemiaminals were found to collapse to the open-form amino ketones in a manner expected for traditional carboxylic acid derivatives. Transannular N···C═O interactions were also observed in some nine-membered amino ketones. Additionally, tricyclic bridged lactams were found to react with some nucleophiles that typically react with ketones but not with planar amides. The effect of geometry on the reactivity of amide bonds and the amide bond distortion range that marks the boundary of amide-like and ketone-like carbonyl reactivity of lactams are also discussed.
Co-reporter:Michal Szostak
Journal of the American Chemical Society 2010 Volume 132(Issue 8) pp:2530-2531
Publication Date(Web):February 3, 2010
DOI:10.1021/ja910654t
Intramolecular Schmidt reactions can be reliably steered toward bridged heterocycles containing orthoamides in high yields. The ketal tether enhances the control of regioselectivity in the migration of the bond distal to the reactive azide nucleophile, thus providing the first examples of the intramolecular Schmidt reaction proceeding with a complete regioselectivity en route to bridged products. The method is broad in scope and allows for systematic study of compounds that are analogous to elusive tetrahedral intermediates of amide addition reactions. Some initial reactivity and structural profiling of these compounds are also reported.
Co-reporter:Michal Szostak ; Lei Yao ; Victor W. Day ; Douglas R. Powell
Journal of the American Chemical Society 2010 Volume 132(Issue 26) pp:8836-8837
Publication Date(Web):June 10, 2010
DOI:10.1021/ja101690u
The straightforward protonation of lactams by treatment with acid and the full structural characterization of three resulting N-protonated lactams are disclosed. This work provides experimental evidence that N-protonation of amide bonds results in a dramatic increase in nonplanarity about the C−N amide bond. The resulting compounds are discussed in structural, spectroscopic, and reactivity terms. The data suggest that distortion of these amide bonds by ∼50° is sufficient for their effective N-activation.
Co-reporter:Angelica M. Meyer, Christopher E. Katz, Sze-Wan Li, David Vander Velde and Jeffrey Aubé
Organic Letters 2010 Volume 12(Issue 6) pp:1244-1247
Publication Date(Web):February 23, 2010
DOI:10.1021/ol100113r
The tricyclic core of the cylindricine or lepadiformine families of alkaloid natural products was assembled via a Prins addition/intramolecular Schmidt rearrangement under Lewis acid conditions. Both single-pot and two-stage variations of this process were examined, with particular attention to the stereochemical outcome of the processes. This technology has been applied to a formal total synthesis of lepadiformine A and a total synthesis of lepadiformine C.
Co-reporter:Kevin J. Frankowski, Partha Ghosh, Vincent Setola, Thuy B. Tran, Bryan L. Roth and Jeffrey Aubé
ACS Medicinal Chemistry Letters 2010 Volume 1(Issue 5) pp:189
Publication Date(Web):May 17, 2010
DOI:10.1021/ml100040t
Herein, we report that N-alkyl-octahydroisoquinolin-1-one-8-carboxamides are a novel class of readily synthesized, selective κ-opioid receptor (KOR) ligands. A striking feature of this class of compounds is the absence of any basic nitrogen atoms. Many of these compounds have demonstrated exclusive affinity for the KOR over not only the δ-opioid receptor and the μ-opioid receptor but also 38 other G protein-coupled receptor targets. The general binding affinity of this class of compounds for the KOR combined with a streamlined route for analogue synthesis provide strong motivation for pursuing this interesting new scaffold as a basis toward new probes targeting the KOR.Keywords (keywords): isoquinolones; κ-Opioid receptor
Co-reporter:Jennifer L. Treece, John R. Goodell, David Vander Velde, John A. Porco Jr. and Jeffrey Aubé
The Journal of Organic Chemistry 2010 Volume 75(Issue 6) pp:2028-2038
Publication Date(Web):February 17, 2010
DOI:10.1021/jo100087h
Polycyclic iminium ethers are ambident electrophilic intermediates that react with a range of nucleophiles in a highly condition-dependent manner to afford densely functionalized lactams. In an effort to expand the scope of reactivity and assist in the generation of new chemotypes from these intermediates, several iminium ethers were subjected to reaction screening using an automated microfluidics reaction platform. Application of this approach led to the discovery of several interesting reaction pathways involving the iminium ether intermediates that will be described.
Co-reporter:Michal Szostak
Journal of the American Chemical Society 2009 Volume 131(Issue 37) pp:13246-13247
Publication Date(Web):August 28, 2009
DOI:10.1021/ja906471q
The first example of a Corey−Chaykovsky epoxidation employing amides as substrates is described. Medium-bridged twisted amides serve as precursors to a family of isolable aminoepoxides. The reactivity of bridged spiro-epoxyamines is also investigated.
Co-reporter:Michal Szostak and Jeffrey Aubé
Organic Letters 2009 Volume 11(Issue 17) pp:3878-3881
Publication Date(Web):August 5, 2009
DOI:10.1021/ol901449y
The sequential RCM to construct a challenging medium-sized ring followed by a transannular cyclization across a medium-sized ring delivers previously unattainable twisted amides from simple acyclic precursors.
Co-reporter:Partha Ghosh, Weston R. Judd, Timothy Ribelin and Jeffrey Aubé
Organic Letters 2009 Volume 11(Issue 18) pp:4140-4142
Publication Date(Web):August 19, 2009
DOI:10.1021/ol901645j
Concise and asymmetric total synthesis of the title compounds are described. The key ring system was constructed using an intramolecular Schmidt reaction on a norbornenone derivative, which was subsequently subjected to ring-opening metathesis followed by reduction. An unusual isomerization of the C-6 ethyl group afforded the desired stereochemistry of the natural product. The synthesis is readily adaptable to analogue production.
Co-reporter:Michal Szostak, Lei Yao and Jeffrey Aubé
Organic Letters 2009 Volume 11(Issue 19) pp:4386-4389
Publication Date(Web):September 1, 2009
DOI:10.1021/ol901771b
The regiochemistry of the intramolecular Schmidt reaction of 2-azidoalkylketones is controlled by placing a thioether substituent at the position adjacent to the ketone to provide access to a family of unsubstituted medium bridged twisted amides. This outcome is ascribed to the presence of stabilizing through-space interactions between the diazonium cation and the n electrons on heteroatom and does not require a locked conformation of the ketone.
Co-reporter:Michal Szostak and Jeffrey Aubé
Chemical Communications 2009 (Issue 46) pp:7122-7124
Publication Date(Web):30 Oct 2009
DOI:10.1039/B917508C
The synthesis of a bridged thiolactam is described. This thiolactam is shown to undergo an unusual C–N bond cleavage reaction under conditions of its synthesis. Spectroscopic properties of the thiolactam indicate a considerable degree of twist of the thioamide bond.
Co-reporter:Chan Woo Huh, Gagandeep K. Somal, Christopher E. Katz, Huaxing Pei, Yibin Zeng, Justin T. Douglas and Jeffrey Aubé
The Journal of Organic Chemistry 2009 Volume 74(Issue 20) pp:7618-7626
Publication Date(Web):September 18, 2009
DOI:10.1021/jo901843w
A series of domino reactions in which the intramolecular Schmidt reaction is combined with either a Sakurai reaction, an aldol reaction, or both is reported. The Sakurai reaction of an allylsilane with an azido-containing enone under Lewis acidic conditions followed by protonation of the resulting titanium enolate species allowed for a subsequent intramolecular Schmidt reaction. Alternatively, the intermediate titanium enolate could undergo an aldol reaction followed by the intramolecular Schmidt reaction to form lactam products with multiple stereogenic centers. The stereochemical features of the titanium enolate aldol reaction with several 3-azidoaldehyde substrates during this domino process is discussed.
Co-reporter:Erik Fenster, Dinesh K. Rayabarapu, Mianji Zhang, Shubhasish Mukherjee, David Hill, Ben Neuenswander, Frank Schoenen, Paul R. Hanson and Jeffrey Aubé
ACS Combinatorial Science 2008 Volume 10(Issue 2) pp:230
Publication Date(Web):February 7, 2008
DOI:10.1021/cc700174c
The parallel synthesis of γ-turn-inspired peptidomimetic libraries has been demonstrated through two approaches toward the preparation of 1,4-diazepin-5-ones. In the first approach, 1,4-diazepin-5-ones scaffolds were prepared on gram scale and subsequently diversified to produce libraries. In a second approach, libraries of 1,4-diazepin-5-ones were produced directly through a three-component strategy that maximizes the diversity obtained in a single step.
Co-reporter:Kevin J. Frankowski, Benjamin Neuenswander and Jeffrey Aubé
ACS Combinatorial Science 2008 Volume 10(Issue 5) pp:721
Publication Date(Web):August 13, 2008
DOI:10.1021/cc800078h
A tandem Diels−Alder/Schmidt reaction provided an efficient route for the exploration of unnatural Stemona alkaloid analogues. Thus, a series of tricyclic scaffolds were efficiently prepared and then elaborated into seven sets of functionalized analogues. These derivatives incorporated appended heterocycles, such as indoles and quinolines, or other diversity-incorporating moieties such as carbamates and amines. Both the scaffold-generation sequence and the diversification steps could be manipulated to provide regio- and diastereochemically pure products.
Co-reporter:Anne Mengel, Oliver Reiser, Jeffrey Aubé
Bioorganic & Medicinal Chemistry Letters 2008 Volume 18(Issue 22) pp:5975-5977
Publication Date(Web):15 November 2008
DOI:10.1016/j.bmcl.2008.07.082
5-Aminovaleric acid and ornithine were evaluated as linkers for the cyclization of β-dipeptides. Two linked examples of β-Ala-β-Ala were prepared by standard coupling methods and their conformations probed by NMR, CD, and computational means. The data suggest that these non- or monosubstituted versions of the target compounds are flexible in solution.
Co-reporter:Timothy Ribelin;ChristopherE. Katz Dr.;DonnaG. English;Sherriel Smith;AnnaK. Manukyan;VictorW. Day Dr.;Benjamin Neuenswer;JenniferL. Poutsma ;Jeffrey Aubé
Angewandte Chemie International Edition 2008 Volume 47( Issue 33) pp:6233-6235
Publication Date(Web):
DOI:10.1002/anie.200801591
Co-reporter:Kevin J. Frankowski, Erin E. Hirt, Yibin Zeng, Ben Neuenswander, Drew Fowler, Frank Schoenen and Jeffrey Aubé
ACS Combinatorial Science 2007 Volume 9(Issue 6) pp:1188
Publication Date(Web):September 27, 2007
DOI:10.1021/cc700127f
A synthetic sequence was developed in which a diene containing an attached secondary amine was reacted with maleic anhydride to afford the title structures in one step. The reaction involves a Diels–Alder reaction combined with a transacylation reaction of the imide group. A series of six scaffolds was constructed using this methodology. Each scaffold was subsequently reacted with 12 amines to afford a library containing 72 compounds.
Co-reporter:Yibin Zeng, Qingshan Li, Robert P. Hanzlik, Jeffrey Aubé
Bioorganic & Medicinal Chemistry Letters 2005 Volume 15(Issue 12) pp:3034-3038
Publication Date(Web):15 June 2005
DOI:10.1016/j.bmcl.2005.04.031
A small library of 2,5-diketopiperazines based on previously reported calpain inhibitors was synthesized. In addition, a concise total synthesis of the structurally related natural product phevalin (2) was accomplished. Despite literature reports that some of the compounds prepared were calpain inhibitors, none of the library members were found to have significant activity against recombinant human calpain I.
Co-reporter:Jennifer E. Golden Dr. Dr.
Angewandte Chemie 2002 Volume 114(Issue 22) pp:
Publication Date(Web):12 NOV 2002
DOI:10.1002/1521-3757(20021115)114:22<4492::AID-ANGE4492>3.0.CO;2-B
Vier Stereozentren und drei Ringe werden in einem Schritt bei einer neuen formalen Totalsynthese von (±)-Stenin aufgebaut (siehe Schema). Diese Kaskadenreaktion besteht aus einer intramolekularen Diels-Alder-Reaktion und anschließender intramolekularer Schmidt-Reaktion. Der so erhaltene Tricyclus ließ sich in wenigen Schritten in ein Intermediat einer bereits beschriebenen Synthese von Stenin überführen.
Co-reporter:Jennifer E. Golden Dr. Dr.
Angewandte Chemie International Edition 2002 Volume 41(Issue 22) pp:
Publication Date(Web):12 NOV 2002
DOI:10.1002/1521-3773(20021115)41:22<4316::AID-ANIE4316>3.0.CO;2-U
Four stereocenters and three rings are established in one step in a new formal synthesis of (±)-stenine (see scheme). This key reaction cascade consists of an intramolecular Diels–Alder reaction followed by an intramolecular Schmidt reaction. The resulting tricyclic intermediate was readily transformed into a known intermediate en route to stenine, thus completing the formal synthesis.
Co-reporter:Michal Szostak and Jeffrey Aubé
Chemical Communications 2009(Issue 46) pp:NaN7124-7124
Publication Date(Web):2009/10/30
DOI:10.1039/B917508C
The synthesis of a bridged thiolactam is described. This thiolactam is shown to undergo an unusual C–N bond cleavage reaction under conditions of its synthesis. Spectroscopic properties of the thiolactam indicate a considerable degree of twist of the thioamide bond.
Co-reporter:Michal Szostak and Jeffrey Aubé
Organic & Biomolecular Chemistry 2011 - vol. 9(Issue 1) pp:NaN35-35
Publication Date(Web):2010/11/11
DOI:10.1039/C0OB00215A
Medium-bridged twisted lactams, in which a non-planar amide bond is achieved by incorporating the nitrogen atom at the bridgehead position in a medium-sized heterocycle, offer an attractive setting in which to study the properties of distorted amide linkages. This Emerging Area article will describe progress in the preparation and study of these compounds. This work shows that compounds containing an even moderately distorted amide bond display useful and unusual chemical properties while retaining a measure of stability that enables their study.
Co-reporter:Chan Woo Huh and Jeffrey Aubé
Chemical Science (2010-Present) 2014 - vol. 5(Issue 2) pp:NaN706-706
Publication Date(Web):2013/11/26
DOI:10.1039/C3SC52805G
A means of generating an N-alkyl-N-arylaminodiazonium ion, which then loses nitrogen to form a reactive aryl nitrenium intermediate, is described. In this sequence, a set of triazenyl acetonitriles was synthesized by nucleophilic addition of a nitrile anion to an aryl azide followed by temperature-dependent alkylation at either of two nucleophilic triazenyl nitrogen atoms. An α,α-disubstituted acetonitrile and N-arylaminodiazonium moiety (or aryl nitrenium ion upon loss of N2) are embedded in the resulting 1,3,3-trisubsituted triazene, which undergoes liberation and recombination of these two components under acidic conditions to yield an α-arylated acetonitrile containing an all-carbon-quaternary center. We propose that the N-alkyl-N-arylaminodiazonium ion loses nitrogen to generate an aryl nitrenium species, which then reacts with an α,α-disubstituted acetonitrile either at the para- or meta-position of the aromatic ring to afford p-alkylaminoaryl acetonitrile derivatives or 3,3-substituted 1-methylindolin-2-imines, depending on the substrates and conditions.

![4-Piperidinol, 1-[(4-nitrophenyl)sulfonyl]-](http://img.cochemist.com/ccimg/873600/873537-45-2.png)
![4-Piperidinol, 1-[(4-nitrophenyl)sulfonyl]-](http://img.cochemist.com/ccimg/873600/873537-45-2_b.png)
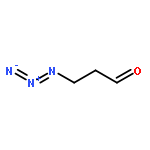
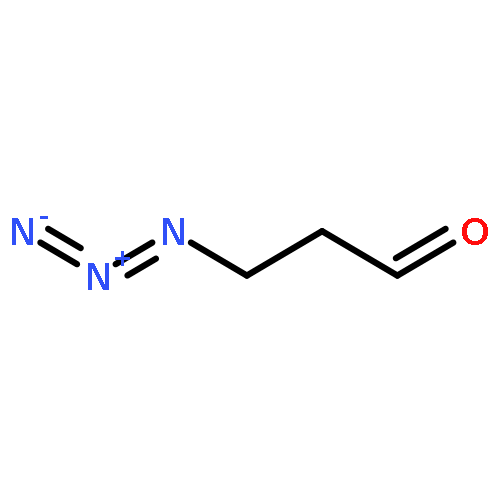
![SILANE, [[(1Z,2E)-5-AZIDO-1-PROPYLIDENE-2-PENTENYL]OXY]TRIMETHYL-](http://img.cochemist.com/ccimg/871400/871321-32-3.png)
![SILANE, [[(1Z,2E)-5-AZIDO-1-PROPYLIDENE-2-PENTENYL]OXY]TRIMETHYL-](http://img.cochemist.com/ccimg/871400/871321-32-3_b.png)





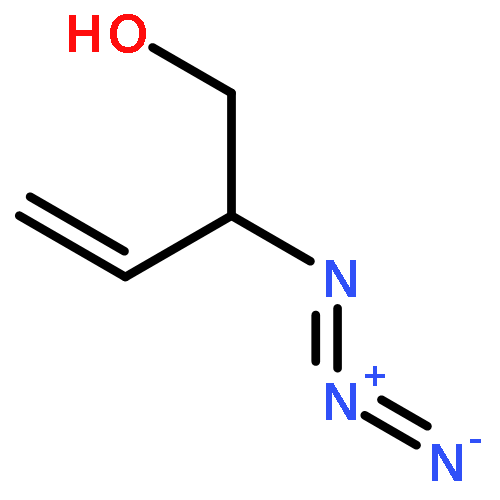



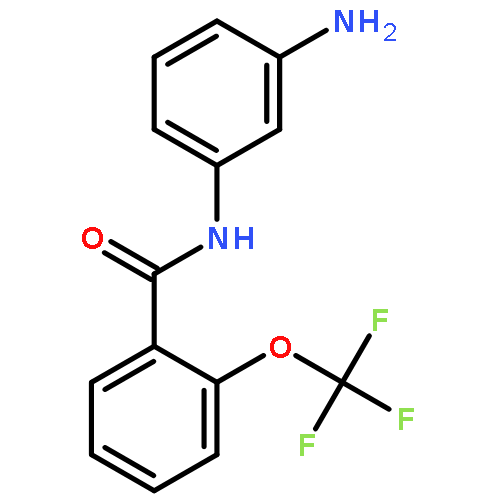
![Benzoic acid, 4-[(1,4-dihydro-2,4-dioxo-3(2H)-quinazolinyl)methyl]-](http://img.cochemist.com/ccimg/849300/849235-59-2.png)
![Benzoic acid, 4-[(1,4-dihydro-2,4-dioxo-3(2H)-quinazolinyl)methyl]-](http://img.cochemist.com/ccimg/849300/849235-59-2_b.png)
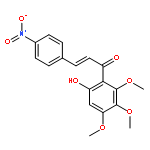



![Silane, [[(2E)-5-azido-1-methylene-2-pentenyl]oxy]trimethyl-](http://img.cochemist.com/ccimg/823900/823805-80-7.png)
![Silane, [[(2E)-5-azido-1-methylene-2-pentenyl]oxy]trimethyl-](http://img.cochemist.com/ccimg/823900/823805-80-7_b.png)
![1H-INDENE-1,3(2H)-DIONE, 2-HYDROXY-2-[(3-OXOESTR-4-EN-17-YL)OXY]-](http://img.cochemist.com/ccimg/813500/813460-00-3.png)
![1H-INDENE-1,3(2H)-DIONE, 2-HYDROXY-2-[(3-OXOESTR-4-EN-17-YL)OXY]-](http://img.cochemist.com/ccimg/813500/813460-00-3_b.png)
![2-[2-[methyl-[(2-methylpropan-2-yl)oxycarbonyl]amino]ethoxy]acetic Acid](http://img.cochemist.com/ccimg/756900/756874-17-6.png)
![2-[2-[methyl-[(2-methylpropan-2-yl)oxycarbonyl]amino]ethoxy]acetic Acid](http://img.cochemist.com/ccimg/756900/756874-17-6_b.png)

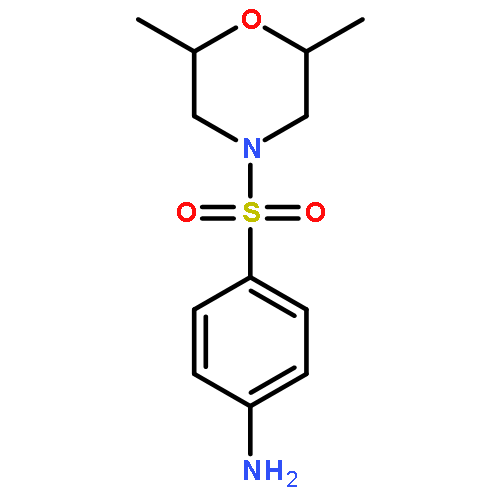





![2-Cyclopenten-1-ol, 4-[[tris(1-methylethyl)silyl]oxy]-, (1R,4S)-](http://img.cochemist.com/ccimg/645400/645390-09-6.png)
![2-Cyclopenten-1-ol, 4-[[tris(1-methylethyl)silyl]oxy]-, (1R,4S)-](http://img.cochemist.com/ccimg/645400/645390-09-6_b.png)
![2-Cyclopenten-1-one, 4-[[tris(1-methylethyl)silyl]oxy]-, (4S)-](http://img.cochemist.com/ccimg/645400/645389-89-5.png)
![2-Cyclopenten-1-one, 4-[[tris(1-methylethyl)silyl]oxy]-, (4S)-](http://img.cochemist.com/ccimg/645400/645389-89-5_b.png)
![Benzene, [1-[[(2E)-4-bromo-2-butenyl]oxy]-2-propynyl]-](http://img.cochemist.com/ccimg/516600/516517-93-4.png)
![Benzene, [1-[[(2E)-4-bromo-2-butenyl]oxy]-2-propynyl]-](http://img.cochemist.com/ccimg/516600/516517-93-4_b.png)
![7,10-METHANOPYRIDO[1,2-A]AZEPINE-6,9-DIONE,OCTAHYDRO-,(7S,10S,10AS)-(9CI)](http://img.cochemist.com/ccimg/450400/450392-91-3.png)
![7,10-METHANOPYRIDO[1,2-A]AZEPINE-6,9-DIONE,OCTAHYDRO-,(7S,10S,10AS)-(9CI)](http://img.cochemist.com/ccimg/450400/450392-91-3_b.png)










![2,6-dimethyl-4-[(4-nitrophenyl)sulfonyl]morpholine](http://img.cochemist.com/ccimg/346500/346464-60-6.png)
![2,6-dimethyl-4-[(4-nitrophenyl)sulfonyl]morpholine](http://img.cochemist.com/ccimg/346500/346464-60-6_b.png)
![1-Piperazinecarboxylic acid, 4-[2-(4-aminophenyl)ethyl]-,
1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/329100/329004-08-2.png)
![1-Piperazinecarboxylic acid, 4-[2-(4-aminophenyl)ethyl]-,
1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/329100/329004-08-2_b.png)








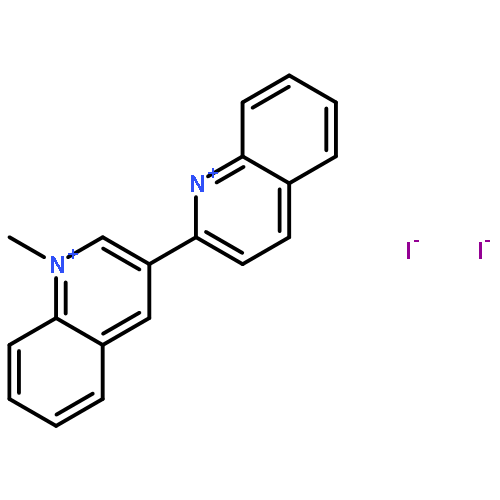
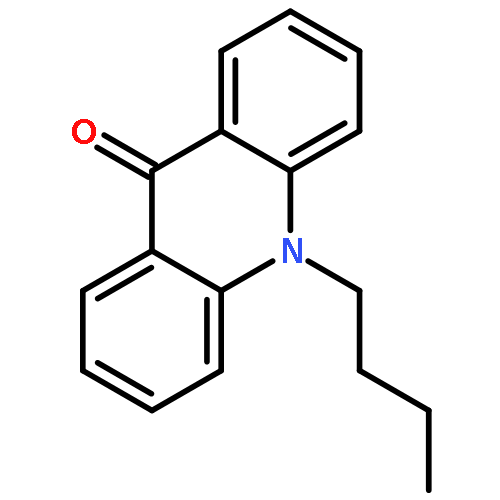
![[2,3'-Biquinolin]-4'(1'H)-one, 1'-butyl-](http://img.cochemist.com/ccimg/297800/297744-16-2.png)
![[2,3'-Biquinolin]-4'(1'H)-one, 1'-butyl-](http://img.cochemist.com/ccimg/297800/297744-16-2_b.png)






![Benzenamine, 4-[2-(4-methyl-1-piperazinyl)ethyl]-](/data/chemimg/462200/262368-48-9.png)
![Benzenamine, 4-[2-(4-methyl-1-piperazinyl)ethyl]-](/data/chemimg/462200/262368-48-9_b.png)











![Benzoic acid, 2-[(chloroacetyl)amino]-5-iodo-](http://img.cochemist.com/ccimg/175900/175850-45-0.png)
![Benzoic acid, 2-[(chloroacetyl)amino]-5-iodo-](http://img.cochemist.com/ccimg/175900/175850-45-0_b.png)
![1-Piperazinecarboxylicacid, 4-[(4-aminophenyl)sulfonyl]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/174000/173951-84-3.png)
![1-Piperazinecarboxylicacid, 4-[(4-aminophenyl)sulfonyl]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/174000/173951-84-3_b.png)
















![2-[(2-CHLOROACETYL)AMINO]-3-METHOXYBENZOIC ACID](http://img.cochemist.com/data/images/141023-39-4.png)
![2-[(2-CHLOROACETYL)AMINO]-3-METHOXYBENZOIC ACID](http://img.cochemist.com/data/images/141023-39-4_b.png)


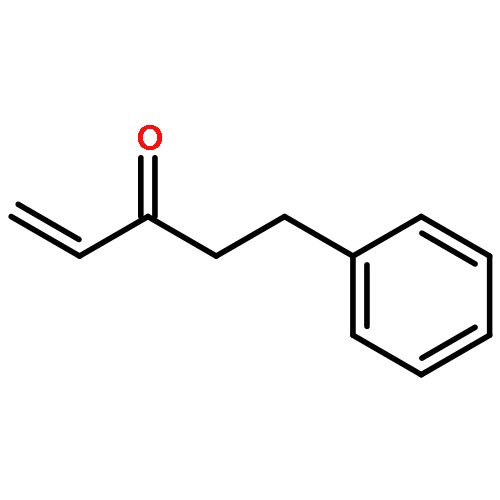
![Propanal, 3-[(4-methoxyphenyl)methoxy]-](http://img.cochemist.com/ccimg/128500/128461-65-4.png)
![Propanal, 3-[(4-methoxyphenyl)methoxy]-](http://img.cochemist.com/ccimg/128500/128461-65-4_b.png)
![Benzoic acid, 2-[(3,4-dimethoxyphenyl)thio]-](http://img.cochemist.com/ccimg/128000/127905-36-6.png)
![Benzoic acid, 2-[(3,4-dimethoxyphenyl)thio]-](http://img.cochemist.com/ccimg/128000/127905-36-6_b.png)

![5-[[2-[(2-METHYLPROPAN-2-YL)OXY]-2-OXOETHYL]AMINO]PENTANOIC ACID](http://img.cochemist.com/ccimg/124100/124073-08-1.png)
![5-[[2-[(2-METHYLPROPAN-2-YL)OXY]-2-OXOETHYL]AMINO]PENTANOIC ACID](http://img.cochemist.com/ccimg/124100/124073-08-1_b.png)


![2,3-dihydro-2,2,9-trimethyl-1H-Pyrrolo[1,2-a]indol-1-one](http://img.cochemist.com/ccimg/119100/119043-71-9.png)
![2,3-dihydro-2,2,9-trimethyl-1H-Pyrrolo[1,2-a]indol-1-one](http://img.cochemist.com/ccimg/119100/119043-71-9_b.png)


![Octahydropyrrolo[1,2-a]azocin-5(1H)-one](http://img.cochemist.com/ccimg/111700/111633-59-1.png)
![Octahydropyrrolo[1,2-a]azocin-5(1H)-one](http://img.cochemist.com/ccimg/111700/111633-59-1_b.png)
![octahydro-5H-Pyrrolo[1,2-a]azepin-5-one](http://img.cochemist.com/ccimg/111700/111633-56-8.png)
![octahydro-5H-Pyrrolo[1,2-a]azepin-5-one](http://img.cochemist.com/ccimg/111700/111633-56-8_b.png)

![2-[(3-methyl-1h-indol-1-yl)methyl]benzoic Acid](http://img.cochemist.com/ccimg/109000/108973-42-8.png)
![2-[(3-methyl-1h-indol-1-yl)methyl]benzoic Acid](http://img.cochemist.com/ccimg/109000/108973-42-8_b.png)
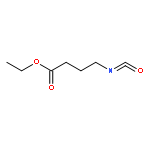


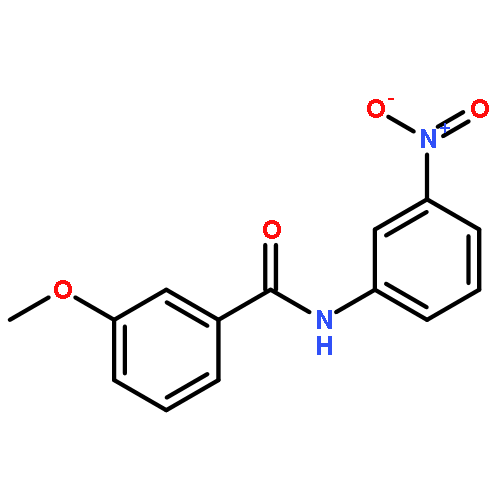



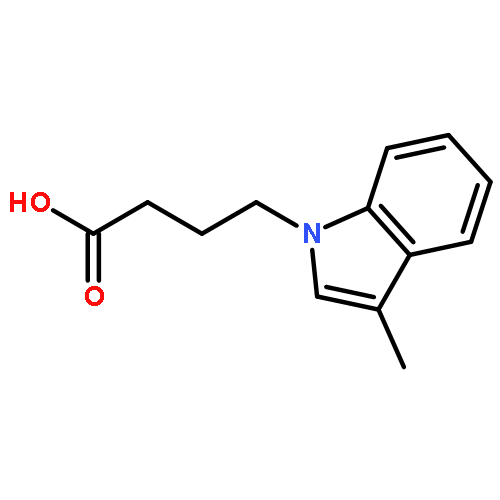
![5H-Cyclohepta[f]-1,3-benzodioxol-5-one, 6,7,8,9-tetrahydro-](http://img.cochemist.com/ccimg/93400/93399-51-0.png)
![5H-Cyclohepta[f]-1,3-benzodioxol-5-one, 6,7,8,9-tetrahydro-](http://img.cochemist.com/ccimg/93400/93399-51-0_b.png)



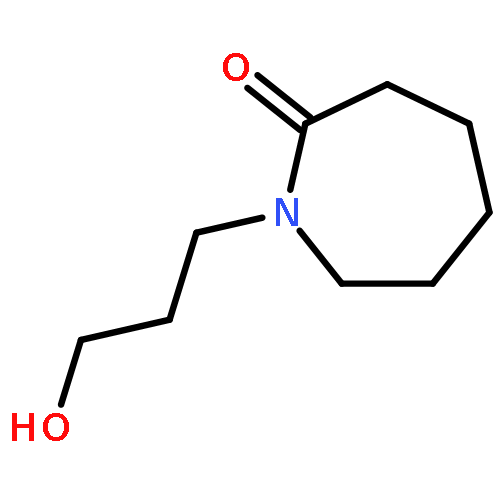





![[2,2'-Binaphthalene]-8,8'-dicarboxaldehyde,1,1',6,6',7,7'-hexahydroxy-3,3'-dimethyl-5,5'-bis(1-methylethyl)-, (2R)-](http://img.cochemist.com/ccimg/90200/90141-22-3.png)
![[2,2'-Binaphthalene]-8,8'-dicarboxaldehyde,1,1',6,6',7,7'-hexahydroxy-3,3'-dimethyl-5,5'-bis(1-methylethyl)-, (2R)-](http://img.cochemist.com/ccimg/90200/90141-22-3_b.png)







![Silane, (1,1-dimethylethyl)dimethyl[(1-methylene-2-propenyl)oxy]-](http://img.cochemist.com/ccimg/80800/80738-05-2.png)
![Silane, (1,1-dimethylethyl)dimethyl[(1-methylene-2-propenyl)oxy]-](http://img.cochemist.com/ccimg/80800/80738-05-2_b.png)



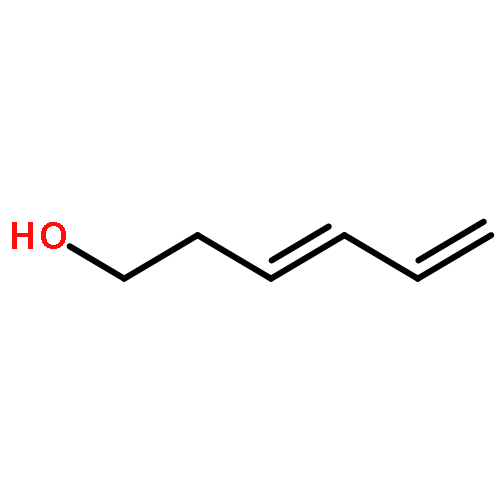
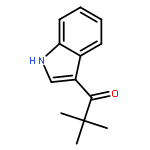

![2-(3,4-dichlorophenyl)-N-methyl-N-[2-(pyrrolidin-1-yl)cyclohexyl]acetamide](http://img.cochemist.com/ccimg/67200/67198-13-4.png)
![2-(3,4-dichlorophenyl)-N-methyl-N-[2-(pyrrolidin-1-yl)cyclohexyl]acetamide](http://img.cochemist.com/ccimg/67200/67198-13-4_b.png)

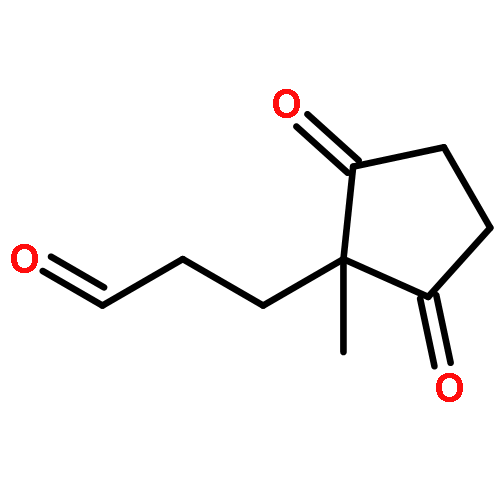
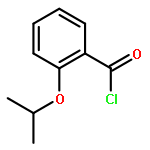





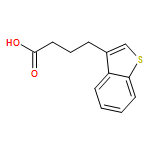
![Methanone, (2-methylbenzo[b]thien-3-yl)phenyl-](http://img.cochemist.com/ccimg/65700/65628-46-8.png)
![Methanone, (2-methylbenzo[b]thien-3-yl)phenyl-](http://img.cochemist.com/ccimg/65700/65628-46-8_b.png)
![2-Cyclopenten-1-ol, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, (1R,4R)-](http://img.cochemist.com/ccimg/61800/61740-30-5.png)
![2-Cyclopenten-1-ol, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, (1R,4R)-](http://img.cochemist.com/ccimg/61800/61740-30-5_b.png)
![1,2-Ethanediamine, N-[(4-chlorophenyl)methyl]-N-(1-methylethyl)-](http://img.cochemist.com/ccimg/61700/61694-91-5.png)
![1,2-Ethanediamine, N-[(4-chlorophenyl)methyl]-N-(1-methylethyl)-](http://img.cochemist.com/ccimg/61700/61694-91-5_b.png)



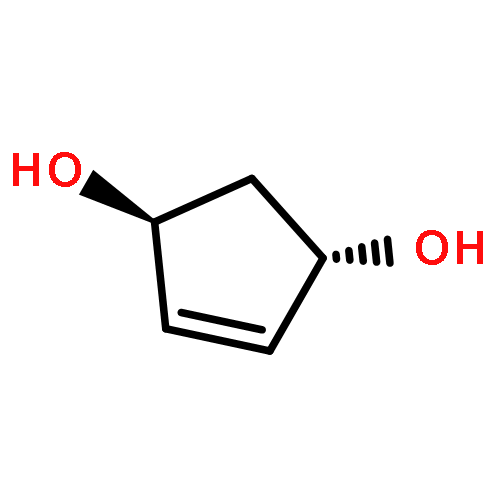
![2-Cyclopenten-1-one,4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, (4R)-](http://img.cochemist.com/ccimg/61400/61305-35-9.png)
![2-Cyclopenten-1-one,4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, (4R)-](http://img.cochemist.com/ccimg/61400/61305-35-9_b.png)




![SPIRO[3.5]NONAN-1-OL](http://img.cochemist.com/ccimg/60300/60211-19-0.png)
![SPIRO[3.5]NONAN-1-OL](http://img.cochemist.com/ccimg/60300/60211-19-0_b.png)






![SPIRO[CYCLOHEXANE-1,3'-[3H]INDOL]-2'(1'H)-IMINE, 1'-METHYL-](http://img.cochemist.com/ccimg/58500/58457-49-1.png)
![SPIRO[CYCLOHEXANE-1,3'-[3H]INDOL]-2'(1'H)-IMINE, 1'-METHYL-](http://img.cochemist.com/ccimg/58500/58457-49-1_b.png)















![Acetonitrile, [(1,1-dimethylethyl)(phenylmethyl)amino]-](http://img.cochemist.com/ccimg/50600/50587-77-4.png)
![Acetonitrile, [(1,1-dimethylethyl)(phenylmethyl)amino]-](http://img.cochemist.com/ccimg/50600/50587-77-4_b.png)










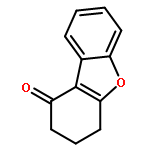
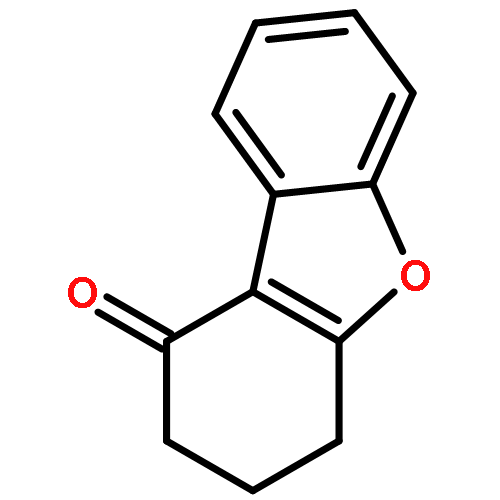





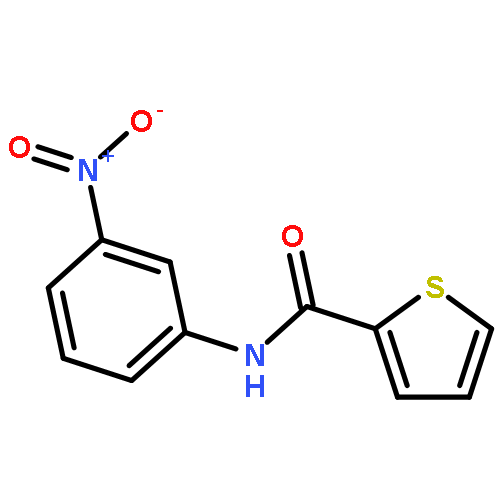




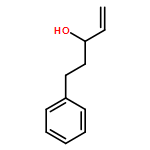









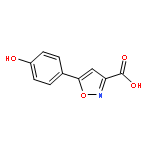



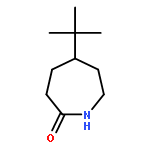







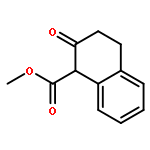
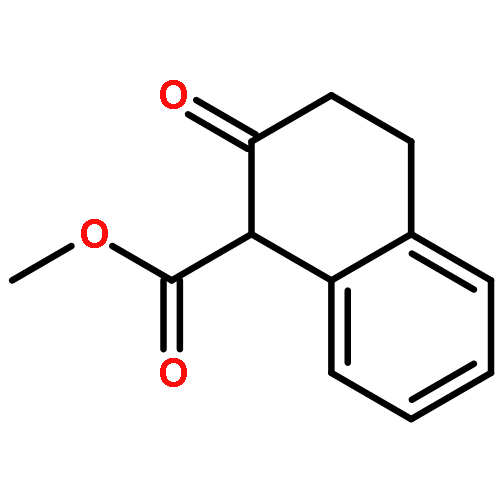
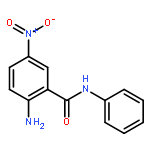
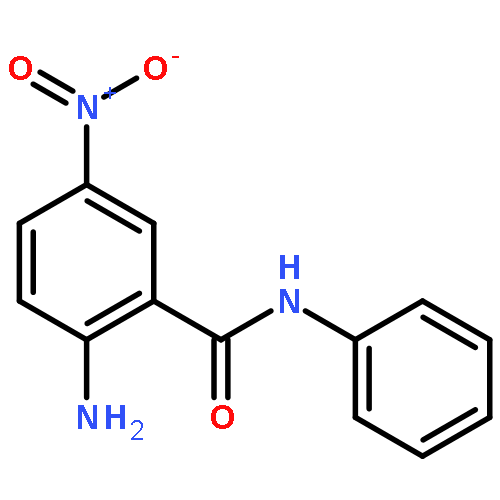
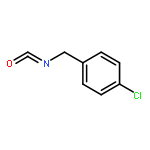


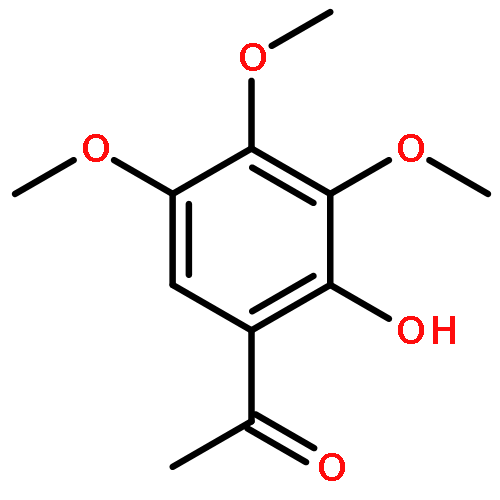
![Spiro[3.5]nonan-1-one](http://img.cochemist.com/ccimg/29900/29800-45-1.png)
![Spiro[3.5]nonan-1-one](http://img.cochemist.com/ccimg/29900/29800-45-1_b.png)


![Bicyclo[2.2.1]heptane-2,5-dione](http://img.cochemist.com/ccimg/28000/27943-47-1.png)
![Bicyclo[2.2.1]heptane-2,5-dione](http://img.cochemist.com/ccimg/28000/27943-47-1_b.png)












![Bicyclo[2.2.1]heptane-2,5-diol](http://img.cochemist.com/ccimg/21500/21462-09-9.png)
![Bicyclo[2.2.1]heptane-2,5-diol](http://img.cochemist.com/ccimg/21500/21462-09-9_b.png)


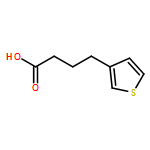
![8H-Cyclohepta[b]thiophen-8-one,4,5,6,7-tetrahydro-](http://img.cochemist.com/ccimg/21000/20905-96-8.png)
![8H-Cyclohepta[b]thiophen-8-one,4,5,6,7-tetrahydro-](http://img.cochemist.com/ccimg/21000/20905-96-8_b.png)
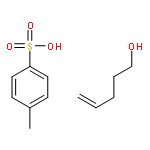
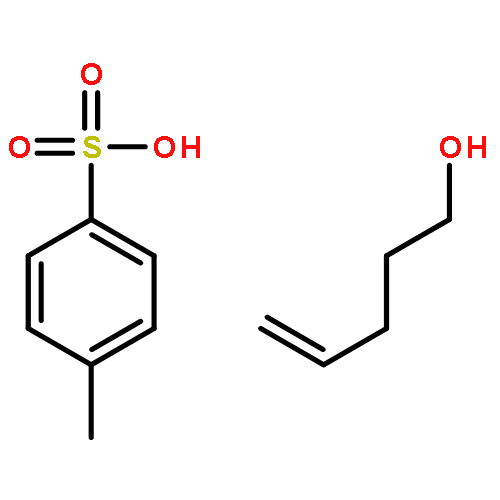


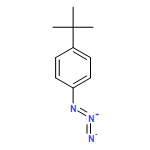




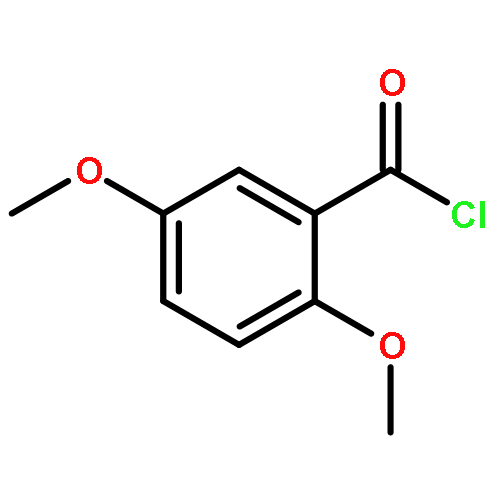






![1,4-dihydro-4-methyl-Cyclopent[b]indol-3(2H)-one](http://img.cochemist.com/ccimg/16300/16244-16-9.png)
![1,4-dihydro-4-methyl-Cyclopent[b]indol-3(2H)-one](http://img.cochemist.com/ccimg/16300/16244-16-9_b.png)
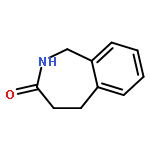
![4,5-Dihydro-1H-benzo[d]azepin-2(3H)-one](http://img.cochemist.com/ccimg/16000/15987-50-5.png)
![4,5-Dihydro-1H-benzo[d]azepin-2(3H)-one](http://img.cochemist.com/ccimg/16000/15987-50-5_b.png)


![Benzoic acid,2-[(2-chloroacetyl)amino]-](http://img.cochemist.com/ccimg/14500/14422-49-2.png)
![Benzoic acid,2-[(2-chloroacetyl)amino]-](http://img.cochemist.com/ccimg/14500/14422-49-2_b.png)



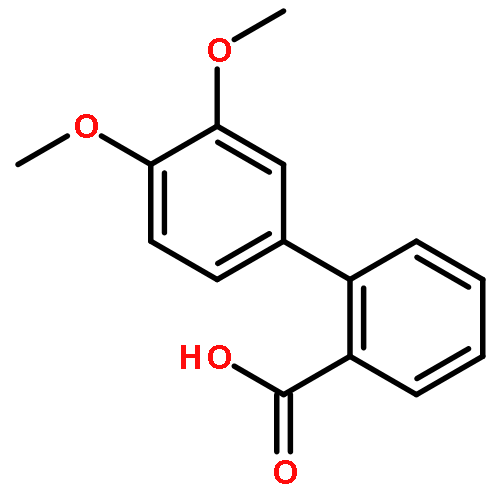
![[1,1'-BIPHENYL]-2-CARBONYL CHLORIDE-](http://img.cochemist.com/ccimg/14100/14002-52-9.png)
![[1,1'-BIPHENYL]-2-CARBONYL CHLORIDE-](http://img.cochemist.com/ccimg/14100/14002-52-9_b.png)





![Tetrazolo[1,5-a]azocine, 5,6,7,8,9,10-hexahydro-](http://img.cochemist.com/ccimg/7200/7198-75-6.png)
![Tetrazolo[1,5-a]azocine, 5,6,7,8,9,10-hexahydro-](http://img.cochemist.com/ccimg/7200/7198-75-6_b.png)
![5H-Tetrazolo[1,5-a]azonine,6,7,8,9,10,11-hexahydro-](http://img.cochemist.com/ccimg/7200/7140-70-7.png)
![5H-Tetrazolo[1,5-a]azonine,6,7,8,9,10,11-hexahydro-](http://img.cochemist.com/ccimg/7200/7140-70-7_b.png)
![METHYL 4-[2-HYDROXY-3-(ISOPROPYLAMINO)PROPOXY]BENZOATE HYDROCHLOR<WBR />IDE (1:1)](http://img.cochemist.com/ccimg/7100/7019-01-4.png)
![METHYL 4-[2-HYDROXY-3-(ISOPROPYLAMINO)PROPOXY]BENZOATE HYDROCHLOR<WBR />IDE (1:1)](http://img.cochemist.com/ccimg/7100/7019-01-4_b.png)






















![2,3-DIHYDROBENZO[F]THIOCHROMEN-1-ONE](http://img.cochemist.com/ccimg/3600/3528-20-9.png)
![2,3-DIHYDROBENZO[F]THIOCHROMEN-1-ONE](http://img.cochemist.com/ccimg/3600/3528-20-9_b.png)

















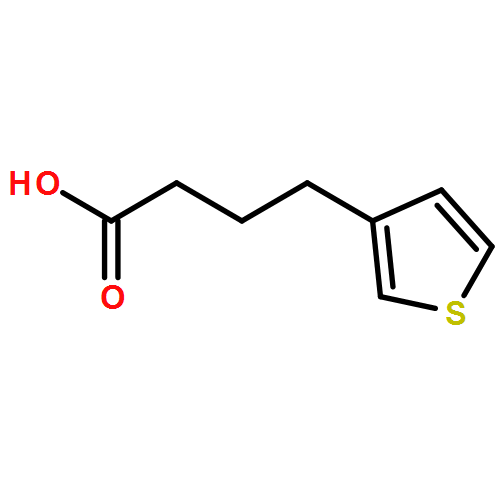
![5,6-Dihydrobenzo[b]thiophen-7(4H)-one](http://img.cochemist.com/ccimg/1500/1468-84-4.png)
![5,6-Dihydrobenzo[b]thiophen-7(4H)-one](http://img.cochemist.com/ccimg/1500/1468-84-4_b.png)
![3-Azaspiro[5.5]undecane hydrochloride](http://img.cochemist.com/ccimg/1200/1125-01-5.png)
![3-Azaspiro[5.5]undecane hydrochloride](http://img.cochemist.com/ccimg/1200/1125-01-5_b.png)
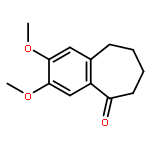
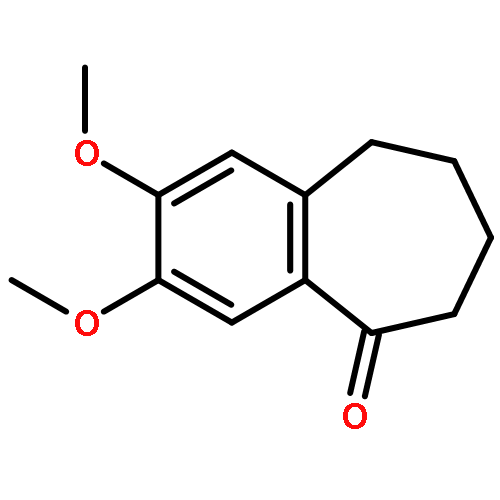





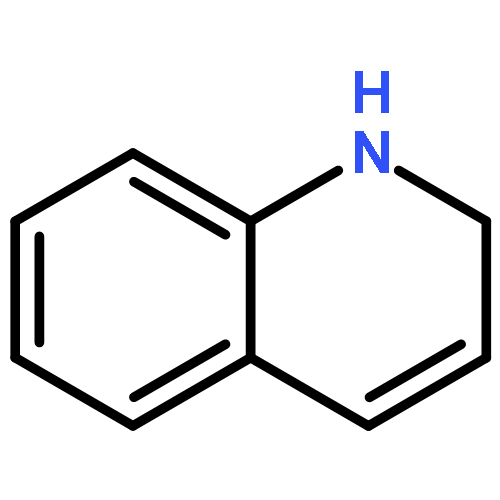


![TERT-BUTYL 4-[(6-CHLOROPYRIDAZIN-3-YL)AMINO]PIPERIDINE-1-CARBOXYLATE](http://img.cochemist.com/ccimg/200/133-05-1.png)
![TERT-BUTYL 4-[(6-CHLOROPYRIDAZIN-3-YL)AMINO]PIPERIDINE-1-CARBOXYLATE](http://img.cochemist.com/ccimg/200/133-05-1_b.png)



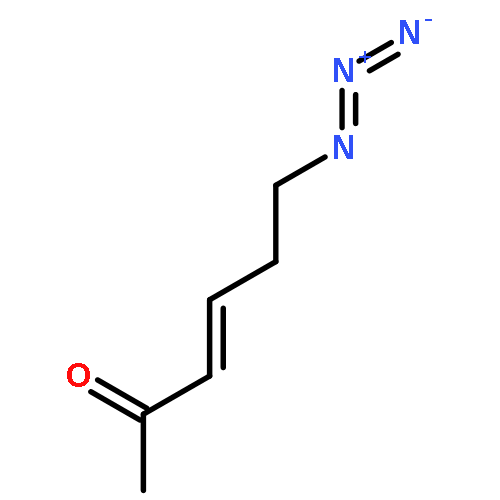
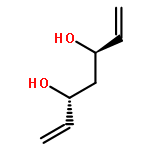
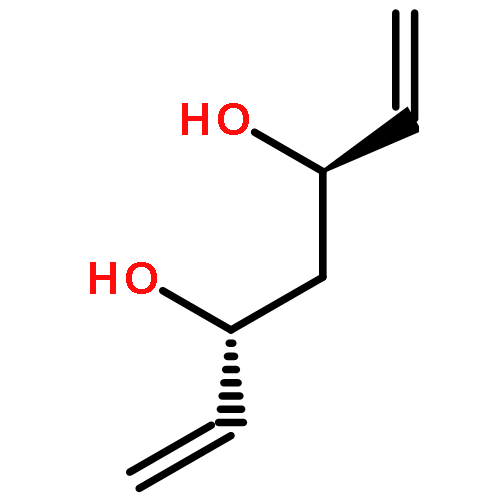
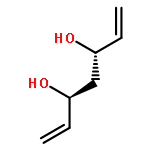



![Carbamic acid, methyl[3-(methylamino)propyl]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/123200/123183-72-2.png)
![Carbamic acid, methyl[3-(methylamino)propyl]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/123200/123183-72-2_b.png)
![4,8:11,15-Dimethano-20H-bisbenzofuro[2,3-a:3',2'-i]dipyrido[4,3-b:3',4'-h]carbazole-1,8a,10a,18-tetrol,7,12-bis(cyclopropylmethyl)-5,6,7,8,9,10,11,12,13,14,19a,20b-dodecahydro-,(4bS,8R,8aS,10aS,11R,14aS,19aR,20bR)-](http://img.cochemist.com/ccimg/105700/105618-26-6.png)
![4,8:11,15-Dimethano-20H-bisbenzofuro[2,3-a:3',2'-i]dipyrido[4,3-b:3',4'-h]carbazole-1,8a,10a,18-tetrol,7,12-bis(cyclopropylmethyl)-5,6,7,8,9,10,11,12,13,14,19a,20b-dodecahydro-,(4bS,8R,8aS,10aS,11R,14aS,19aR,20bR)-](http://img.cochemist.com/ccimg/105700/105618-26-6_b.png)





