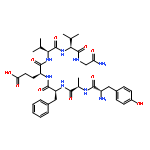
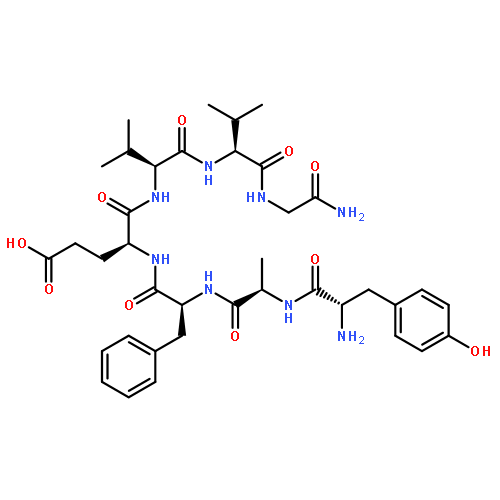
Co-reporter:Noriki Kutsumura, Ryuichiro Ohshita, Jumpei Horiuchi, Kotaro Tateno, Naoshi Yamamoto, Tsuyoshi Saitoh, Yasuyuki Nagumo, Hidetoshi Kawai, Hiroshi Nagase
Tetrahedron 2017 Volume 73, Issue 34(Issue 34) pp:
Publication Date(Web):24 August 2017
DOI:10.1016/j.tet.2017.07.016
The synthesis of novel adamantane-like cage compounds consisting of phosphorus, sulfur, and carbon atoms was developed. We examined the reaction of a variety of acetophenone derivatives with P4S10 in refluxing benzene. A novel noradamantane-like cage compound was also synthesized, when the reaction of 2’-methoxyacetophenone with P4S10 was performed in refluxing toluene. In addition, by using the adamantane-like cage compound, 4,4’-dimethoxybenzophenone and N,N-dimethylbenzamide were successfully transformed into the corresponding thioketone (98%) and benzothioamide (89%), respectively.Download high-res image (222KB)Download full-size image
Co-reporter:Naoshi Yamamoto, Takahiro Okada, Yukimasa Harada, Noriki Kutsumura, Satomi Imaide, Tsuyoshi Saitoh, Hideaki Fujii, Hiroshi Nagase
Tetrahedron 2017 Volume 73, Issue 39(Issue 39) pp:
Publication Date(Web):28 September 2017
DOI:10.1016/j.tet.2017.08.014
(–)-Galanthamine (4) was synthesized from naltrexone (1) in 18 steps with 3% total yield by overcoming many specific side reactions derived from the 4,5-epoxymorphinan skeleton. The key features are cleavage of the D-ring by the Hofmann elimination and the following the one-pot C9–C10 and C9–14 bond cleavages concomitant with the C9 removal by the OsO4–NaIO4 combination reaction. Then, the treatment with zinc powder in acetic acid led to not only removal of the 2,2,2-trichloroethoxycarbonyl (Troc) group, but also reductive amination of the resulting imine to give the desired 7-membered ring.Download high-res image (207KB)Download full-size image
Co-reporter:Yoshikazu Watanabe, Kohei Hayashida, Daisuke Saito, Toshihiro Takahashi, Junichi Sakai, Eriko Nakata, Takashi Kanda, Takashi Iwai, Shigeto Hirayama, Hideaki Fujii, Tomio Yamakawa, Hiroshi Nagase
Bioorganic & Medicinal Chemistry Letters 2017 Volume 27, Issue 15(Issue 15) pp:
Publication Date(Web):1 August 2017
DOI:10.1016/j.bmcl.2017.05.072
We designed and synthesized novel δ opioid receptor (DOR) agonists 3a–i with an azatricyclodecane skeleton, which was a novel structural class of DOR agonists. Among them, 3b exhibited high values of binding affinity and potent agonistic activity for the DOR that were approximately equivalent to those of 2 which bore an oxazatricyclodecane skeleton. In vitro assays using the blood-brain barrier (BBB) permeability test kit supported the idea that 3b achieved an excellent BBB permeability by converting an oxygen atom of 2 to a carbon atom (methylene group) in the core skeleton. As a result, 3b showed potent antinociceptive effects.Download high-res image (129KB)Download full-size image
Co-reporter:Naoshi Yamamoto, Sayaka Ohrui, Takahiro Okada, Masahiro Yata, Tsuyoshi Saitoh, Noriki Kutsumura, Yasuyuki Nagumo, Yoko Irukayama-Tomobe, Yasuhiro Ogawa, Yukiko Ishikawa, Yurie Watanabe, Daichi Hayakawa, Hiroaki Gouda, Masashi Yanagisawa, Hiroshi Nagase
Bioorganic & Medicinal Chemistry Letters 2017 Volume 27, Issue 17(Issue 17) pp:
Publication Date(Web):1 September 2017
DOI:10.1016/j.bmcl.2017.07.011
The essential structure of the orexin 1 receptor (OX1R) antagonist YNT-707 (2) was clarified, particularly the roles to OX1R antagonist activities of the 3-OMe, the 4,5-epoxy ring, the 14-hydroxy group, and the orientation of the 6-amide side chain.The 3-OMe and 17-sulfonamide group were shown to be essential for the OX1R antagonistic activity. The 4,5-epoxy ring plays an important role for the active orientation of the 6-amide group. The 14-hydroxy group could lower the activity of the 6β-amide isomer by the interaction of the 14-hydroxy group with the 6-amide group, which could orient the 6-amide group toward the upper side of the C-ring.Finally, we proposed the difference in the active conformation between OX1R and κ opioid receptor (KOR), especially in the orientation of the 6-amide group which is expected to be a useful guide for medicinal chemists to design OX1R ligands.Download high-res image (172KB)Download full-size image
Co-reporter:Noriki Kutsumura, Yasuaki Koyama, Yasuyuki Nagumo, Ryo Nakajima, Yoshiyuki Miyata, Naoshi Yamamoto, Tsuyoshi Saitoh, Naoko Yoshida, Satoshi Iwata, Hiroshi Nagase
Bioorganic & Medicinal Chemistry 2017 Volume 25, Issue 16(Issue 16) pp:
Publication Date(Web):15 August 2017
DOI:10.1016/j.bmc.2017.06.026
The 7-benzylidenenaltrexone (BNTX) derivatives 2a–v, 3a–c, 13a–c, and 14a were synthesized from naltrexone (1) and evaluated for their antitrichomonal activity. The structure-activity-relationship studies found that 4-iodo-BNTX (2g) showed the highest activity (IC50 = 10.5 µM) and the affinity for the opioid receptor was less important for antitrichomonal activity against Trichomonas vaginalis. The morphinan skeleton bearing both the double bond for a Michael acceptor and the phenolic hydroxy group would be a specific template for development of antitrichomonal agents. In addition, the mechanism of the antitrichomonal activity of the BNTX derivatives may differ from that of the standard drug, metronidazole.Download high-res image (75KB)Download full-size image
Co-reporter:Hiroshi NagaseNaoshi Yamamoto, Masahiro Yata, Sayaka Ohrui, Takahiro Okada, Tsuyoshi Saitoh, Noriki Kutsumura, Yasuyuki NagumoYoko Irukayama-Tomobe, Yukiko Ishikawa, Yasuhiro Ogawa, Shigeto Hirayama, Daisuke Kuroda, Yurie Watanabe, Hiroaki Gouda, Masashi Yanagisawa
Journal of Medicinal Chemistry 2017 Volume 60(Issue 3) pp:
Publication Date(Web):January 4, 2017
DOI:10.1021/acs.jmedchem.6b01418
Nalfurafine, a κ-selective opioid receptor agonist, unexpectedly showed a selective antagonist activity toward the orexin 1 receptor (OX1R) (Ki = 250 nM). Modification of the 17-amino side chain of the opioid ligand to an arylsulfonyl group and the 6-furan acrylamide chain to 2-pyridyl acrylamide led to compound 71 with improvement of the antagonist activity (OX1R, Ki = 1.36 nM; OX2R, not active) without any detectable affinity for the opioid receptor. The dihydrosulfate salt of 71, freely soluble in water, attenuated the physical dependence of morphine. Furthermore, all of the active nalfurafine derivatives in this study had almost no activity for OX2R, which led to high OX1R selectivity. These results suggest that nalfurafine derivatives could be a useful series of lead compounds to develop highly selective OX1R antagonists.
Co-reporter:Takashi Nagahara; Tsuyoshi Saitoh; Noriki Kutsumura; Yoko Irukayama-Tomobe; Yasuhiro Ogawa; Daisuke Kuroda; Hiroaki Gouda; Hidetoshi Kumagai; Hideaki Fujii; Masashi Yanagisawa
Journal of Medicinal Chemistry 2015 Volume 58(Issue 20) pp:7931-7937
Publication Date(Web):August 12, 2015
DOI:10.1021/acs.jmedchem.5b00988
Orexins are a family of neuropeptides that regulate sleep/wakefulness, acting on two G-protein-coupled receptors, orexin receptors 1 (OX1R) and 2 (OX2R). Genetic and pharmacologic evidence suggests that orexin receptor agonists, especially OX2R agonist, will be useful for mechanistic therapy of the sleep disorder narcolepsy/cataplexy. We herein report the discovery of a potent (EC50 on OX2R is 0.023 μM) and OX2R-selective (OX1R/OX2R EC50 ratio is 70) agonist, 4′-methoxy-N,N-dimethyl-3′-[N-(3-{[2-(3-methylbenzamido)ethyl]amino}phenyl)sulfamoyl]-(1,1′-biphenyl)-3-carboxamide 26.
Co-reporter:Ryo Nakajima, Naoshi Yamamoto, Shigeto Hirayama, Takashi Iwai, Akiyoshi Saitoh, Yasuyuki Nagumo, Hideaki Fujii, Hiroshi Nagase
Bioorganic & Medicinal Chemistry 2015 Volume 23(Issue 19) pp:6271-6279
Publication Date(Web):1 October 2015
DOI:10.1016/j.bmc.2015.08.036
We designed and synthesized pentacyclic propellane derivatives with a 6-amide side chain to afford compounds with higher MOR/KOR ratio and lower sedative effects than nalfurafine. The obtained etheno-bridged derivative with a β-amide side chain, YNT-854, showed a higher MOR/KOR ratio than nalfurafine. YNT-854 also exhibited a higher dose ratio between the sedative effect and the analgesic effect than observed with nalfurafine, which may guide the future design of useful analgesics with a weaker sedative effect than nalfurafine.
Co-reporter:Noriki Kutsumura, Ryo Nakajima, Yasuaki Koyama, Yoshiyuki Miyata, Tsuyoshi Saitoh, Naoshi Yamamoto, Satoshi Iwata, Hideaki Fujii, Hiroshi Nagase
Bioorganic & Medicinal Chemistry Letters 2015 Volume 25(Issue 21) pp:4890-4892
Publication Date(Web):1 November 2015
DOI:10.1016/j.bmcl.2015.06.002
We evaluated antitrichomonal effects of δ opioid receptor (DOR) agonists and antagonists. Although all the agonists were inactive, the DOR antagonists BNTX (2a) and its derivatives 2b–d showed antitrichomonal activity with MIC of 20–40 μM. In addition, the development of a more effective synthetic method for the BNTX derivatives was achieved by using the Knoevenagel condensation.
Co-reporter:Hiroshi Nagase;Noriki Kutsumura
Archiv der Pharmazie 2015 Volume 348( Issue 6) pp:375-389
Publication Date(Web):
DOI:10.1002/ardp.201500031
We designed and synthesized novel triplet molecules with 1,3,5-trioxazatriquinane skeletons. One class comprises double-capped triplets with a morphinan skeleton; the other class comprises simple phenol derivatives with phenethylamine moieties. One compound with m-phenolic hydroxyl group, called SYK-146, is a highly selective, potent agonist for the κ receptor, with activity nearly equivalent to that of U-50488H. The o-phenolic isomer of SYK-146, called SYK-524, showed potent but non-selective agonistic activity for the opioid receptors. We also added several simple phenol derivatives to a library of compounds that target opioid receptors, and they showed high hit rates for the receptor. This library might also be expected to show high hit rates for other receptors.
Co-reporter:Shigeto Hirayama, Naohisa Wada, Toru Nemoto, Takashi Iwai, Hideaki Fujii, and Hiroshi Nagase
ACS Medicinal Chemistry Letters 2014 Volume 5(Issue 8) pp:868
Publication Date(Web):June 26, 2014
DOI:10.1021/ml5000542
We designed and synthesized the 1,3,5-trioxazatriquinane derivatives with m-hydroxyphenyl groups. These compounds include the phenethylamine structure within them, which is a common structure observed in morphinan derivatives like morphine. Among the synthesized compounds, (−)-8c with two m-hydroxyphenyl groups selectively bound and exerted full agonist activity toward the κ opioid receptor (KOR). Subcutaneously administered (−)-8c exhibited significant antinociceptive effects via the KOR in a dose-dependent manner. These results suggest the emergence of a novel class of KOR agonist.Keywords: analgesics; Opioid; phenethylamine structure
Co-reporter:Shigeto Hirayama, Naohisa Wada, Naoya Kuroda, Takashi Iwai, Noriyuki Yamaotsu, Shuichi Hirono, Hideaki Fujii, Hiroshi Nagase
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 20) pp:4895-4898
Publication Date(Web):15 October 2014
DOI:10.1016/j.bmcl.2014.08.012
We designed and synthesized of 1,3,5-trioxazatriquinanes with o- or p-hydroxyphenyl rings as analogs of the κ opioid receptor agonist SYK-146 with m-hydroxyphenyl groups. Although almost all tested compounds did not bind to the opioid receptors, only 17b (SYK-524) with two o-hydroxyphenyl rings showed moderate or potent binding affinities and exhibited agonistic activities for the three opioid receptor types. Because the basicity of the nitrogen atom in the 1,3,5-trioxazatriquinane structure was predicted to be very low due to the electron withdrawing effect of the three oxygen atoms, SYK-524 was a novel non-morphinan and nonpeptidic opioid universal agonist lacking a basic nitrogen atom.
Co-reporter:Hiroshi Nagase, Ryo Nakajima, Naoshi Yamamoto, Shigeto Hirayama, Takashi Iwai, Toru Nemoto, Hiroaki Gouda, Shuichi Hirono, Hideaki Fujii
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 13) pp:2851-2854
Publication Date(Web):1 July 2014
DOI:10.1016/j.bmcl.2014.04.098
Indolopropellane 2 was reported to show almost no binding affinity to the δ opioid receptor (DOR) in spite of the fact that 2 has both the propellane fundamental skeleton (message part) with binding ability to the opioid receptors and a possible DOR address structure (indole moiety). We developed the working hypothesis that almost no binding affinity of 2 to the DOR would be derived from its possibly stable bent conformer. To enable the propellane skeleton to adopt an extended conformation which would reasonably interact with the DOR, quinolinopropellanes 3a–d were designed which had an additional pharmacophore, quinoline nitrogen. The calculated binding free energies of ligand–DOR complexes strongly supported our working hypothesis. The synthesized quinolinopropellane 3a was a selective DOR full agonist, confirming our working hypothesis and the results of in silico investigation.
Co-reporter:Toru Nemoto, Yoshihiro Ida, Yusuke Iihara, Ryo Nakajima, Shigeto Hirayama, Takashi Iwai, Hideaki Fujii, Hiroshi Nagase
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 24) pp:7628-7647
Publication Date(Web):15 December 2013
DOI:10.1016/j.bmc.2013.10.032
We investigated the structure–activity relationship of KNT-127 (opioid δ agonist) derivatives with various 17-substituents which are different in length and size. The 17-substituent in KNT-127 derivatives exerted a great influence on the affinity and agonistic activity for the δ receptor. While the compounds with electron-donating 17-substituents showed higher affinities for the δ receptor than those with electron-withdrawing groups, KNT-127 derivatives with 17-fluoroalkyl groups (the high electron-withdrawing groups) showed high selectivities for the δ receptor among evaluated compounds. In addition, the basicity of nitrogen as well as the structure of the 17-N substituent such as the length and configuration at an asymmetric carbon atom contributed to agonist properties for the δ receptor. Thus, the analog with a 17-(3-ethoxypropyl) group showed the best selectively and potent agonistic activity for the δ receptor among KNT-127 derivatives. These findings should be useful for designing novel δ selective agonists.The KNT-127 derivative (SYK-320) with 17-(3-ethoxypropyl) group showed potent and the best selective agonistic activity for the δ receptor.

methyl]-N,N-diethyl-](http://img.cochemist.com/ccimg/156800/156727-74-1.png)
methyl]-N,N-diethyl-](http://img.cochemist.com/ccimg/156800/156727-74-1_b.png)
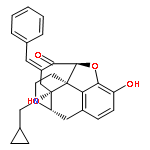
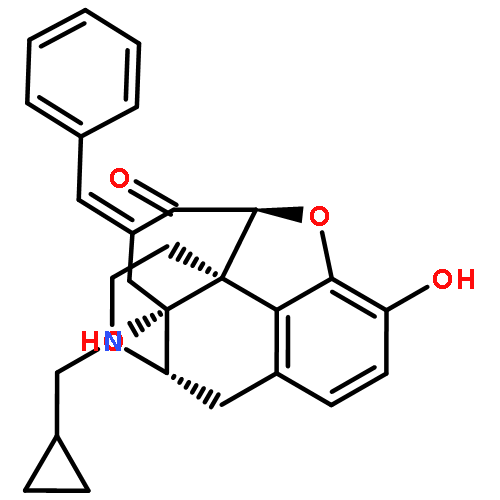
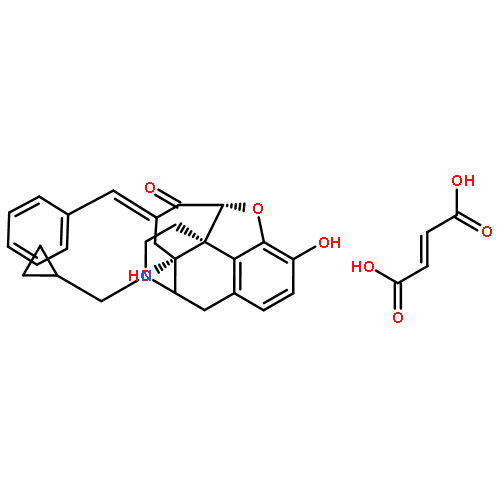
![2-Propenamide, N-[(5a,6b)-17-(cyclopropylmethyl)-4,5-epoxy-3,14-dihydroxymorphinan-6-yl]-3-(3-furanyl)-N-methyl-,hydrochloride (1:1), (2E)-](http://img.cochemist.com/ccimg/152700/152658-17-8.png)
![2-Propenamide, N-[(5a,6b)-17-(cyclopropylmethyl)-4,5-epoxy-3,14-dihydroxymorphinan-6-yl]-3-(3-furanyl)-N-methyl-,hydrochloride (1:1), (2E)-](http://img.cochemist.com/ccimg/152700/152658-17-8_b.png)
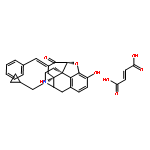
![4,8-Methano-8aH-bisbenzofuro[3,2-e:2',3'-g]isoquinoline-1,8a-diol,7-(cyclopropylmethyl)-5,6,7,8,9,14b-hexahydro-, (4bS,8R,8aS,14bR)-](http://img.cochemist.com/ccimg/111600/111555-58-9.png)
![4,8-Methano-8aH-bisbenzofuro[3,2-e:2',3'-g]isoquinoline-1,8a-diol,7-(cyclopropylmethyl)-5,6,7,8,9,14b-hexahydro-, (4bS,8R,8aS,14bR)-](http://img.cochemist.com/ccimg/111600/111555-58-9_b.png)