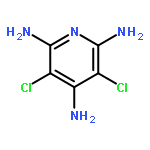
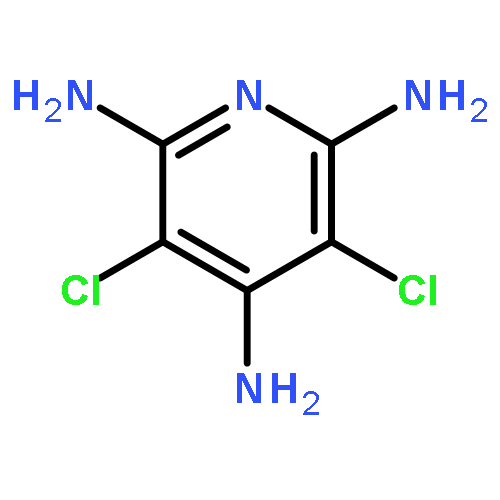
Co-reporter:Kenji Sugisaki;Kazuo Toyota;Kazunobu Sato;Daisuke Shiomi;Takeji Takui
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 44) pp:30128-30138
Publication Date(Web):2017/11/15
DOI:10.1039/C7CP05533A
Spin–orbit contributions to the zero-field splitting (ZFS) tensor (DSO tensor) of MIII(acac)3 complexes (M = V, Cr, Mn, Fe and Mo; acac = acetylacetonate anion) are evaluated by means of ab initio (a hybrid CASSCF/MRMP2) and DFT (Pederson–Khanna (PK) and natural orbital-based Pederson–Khanna (NOB-PK)) methods, focusing on the behaviour of DFT-based approaches to the DSO tensors against the valence d-electron configurations of the transition metal ions in octahedral coordination. Both the DFT-based approaches reproduce trends in the D tensors. Significantly, the differences between the theoretical and experimental D (D = DZZ − (DXX + DYY)/2) values are smaller in NOB-PK than in PK, emphasising the usefulness of the natural orbital-based approach to the D tensor calculations of transition metal ion complexes. In the case of d2 and d4 electronic configurations, the DSO(NOB-PK) values are considerably underestimated in the absolute magnitude, compared with the experimental ones. The DSO tensor analysis based on the orbital region partitioning technique (ORPT) revealed that the DSO contributions attributed to excitations from the singly occupied region (SOR) to the unoccupied region (UOR) are significantly underestimated in the DFT-based approaches to all the complexes under study. In the case of d3 and d5 configurations, the (SOR → UOR) excitations contribute in a nearly isotropic manner, which causes fortuitous error cancellations in the DFT-based DSO values. These results indicate that more efforts to develop DFT frameworks should be directed towards the reproduction of quantitative DSO tensors of transition metal complexes with various electronic configurations and local symmetries around metal ions.
Co-reporter:Kenji SugisakiKazuo Toyota, Kazunobu Sato, Daisuke Shiomi, Takeji Takui
The Journal of Physical Chemistry A 2016 Volume 120(Issue 49) pp:9857-9866
Publication Date(Web):November 15, 2016
DOI:10.1021/acs.jpca.6b10253
A quasi-restricted orbital (QRO) approach for the calculation of the spin–orbit term of zero-field splitting tensors (DSO tensors) by means of density functional theory (DFT) importantly features in the fact that it is free from spin contamination problems because it uses spin eigenfunctions for the zeroth order wave functions. In 2011, however, Schmitt and co-workers pointed out that in the originally proposed QRO working equation some possible excitations were not included in their sum-over-states procedure, which causes spurious DSO contributions from closed-shell subsystems located far from the magnetic molecule under study. We have revisited the derivation of the QRO working equation and modified it, making it include all possible types of excitations in the sum-over-states procedure. We have found that the spurious DSO contribution can be eliminated by taking into account contributions from all possible types of singly excited configuration state functions. We have also found that only the SOMO(α) → SOMO(β) excited configurations have nonzero contributions to the DSO tensors as long as α and β spin orbitals have the same spatial distributions and orbital energies. For the DSO tensor calculations, by using a ground state wave function free from spin contamination, we propose a natural orbital-based Pederson–Khanna (NOB-PK) method, which utilizes the single determinant wave function consisting of natural orbitals in conjunction with the Pederson–Khanna (PK) type perturbation treatment. Some relevant calculations revealed that the NOB-PK method can afford more accurate DSO tensors than the conventional PK method as well as the QRO approach in MnII complexes and ReIV-based single molecule magnets.
Co-reporter:Kenji Sugisaki, Satoru Yamamoto, Shigeaki Nakazawa, Kazuo Toyota, Kazunobu Sato, Daisuke Shiomi, and Takeji Takui
The Journal of Physical Chemistry A 2016 Volume 120(Issue 32) pp:6459-6466
Publication Date(Web):August 8, 2016
DOI:10.1021/acs.jpca.6b04932
Quantum computers are capable to efficiently perform full configuration interaction (FCI) calculations of atoms and molecules by using the quantum phase estimation (QPE) algorithm. Because the success probability of the QPE depends on the overlap between approximate and exact wave functions, efficient methods to prepare accurate initial guess wave functions enough to have sufficiently large overlap with the exact ones are highly desired. Here, we propose a quantum algorithm to construct the wave function consisting of one configuration state function, which is suitable for the initial guess wave function in QPE-based FCI calculations of open-shell molecules, based on the addition theorem of angular momentum. The proposed quantum algorithm enables us to prepare the wave function consisting of an exponential number of Slater determinants only by a polynomial number of quantum operations.
Co-reporter:Seigo Yamauchi, Mana Tanabe, Yasunori Ohba, Kenji Sugisaki, Kazuo Toyota, Kazunobu Sato, Takeji Takui, Irena Saltsman
Chemical Physics Letters 2012 Volume 521() pp:64-68
Publication Date(Web):10 January 2012
DOI:10.1016/j.cplett.2011.11.040
Abstract
The lowest excited triplet state of free-base 5,10,15-trispentafluorophenylcorrole (H3Cor) was studied in rigid glass by time-resolved electron paramagnetic resonance (TR-EPR) spectroscopy. Triplet sublevels were experimentally determined by using magnetophotoselection and liquid crystal. Quantum chemical calculations of zero-field splitting parameters D and E were used for the sublevel assignment. The out-of-plane sublevel Tz was found to be the lowest; namely, D is positive for H3Cor, in contrast to previous reports. The origin of the difference is discussed in detail. Preliminary TR-EPR experiments on rhodium corrole, accompanied by the quantum chemical calculations, emphasize the important contribution of spin-orbit couplings.