Co-reporter:Jiaolin Cui, Hua Peng, Zhiliang Song, Zhengliang Du, Yimin Chao, and Gang Chen
Chemistry of Materials September 12, 2017 Volume 29(Issue 17) pp:7467-7467
Publication Date(Web):August 9, 2017
DOI:10.1021/acs.chemmater.7b02467
γ-In2Se3 is selected as a thermoelectric candidate because it has a unique crystal structure and thermal stability at relatively high temperatures. In this work, we have prepared lithiated γ-In2Se3 through chemical diffusion and investigated its band structures and thermoelectric performance. After lithiation of γ-In2Se3 in a lithium acetate (CH3COOLi) solution at 50 °C for 30 h, we have observed a high Hall carrier concentration (nH) of ≤1.71 × 1018 cm–3 at room temperature, which is ∼4 orders of magnitude higher than that of pristine γ-In2Se3. The enhancement of nH is directly responsible for the remarkable improvement in electrical conductivity and can be elucidated as the Fermi level (Fr) unpinning and moving toward the conduction band through the dominant interstitial occupation of Li+ in the γ-In2Se3 lattice. Combined with the minimum lattice thermal conductivity (κL = 0.30–0.34 W K–1 m–1) at ∼923 K, the highest ZT value of 0.62–0.67 is attained, which is approximately 9–10 times that of pristine γ-In2Se3, proving that the lithiation in γ-In2Se3 is an effective approach to the improvement in thermoelectric performance.
•Effect of temperature on polaron recombination is studied.•After collision, an exciton or a mixed state of polaron pair and exciton is formed.•After including temperature effect, the yield of exciton is significantly improved.•Driven by the thermal random force, the exciton could perform a random walk.•With the increase of temperature, the exciton becomes unstable.The microcosmic mechanism of electroluminescence in polymer light emitting diodes (PLEDs) is the recombination of the oppositely charged polarons. In previous studies, it has been demonstrated that the temperature-induced irregular lattice vibration may have non-negligible influence on polaron dynamics. Nevertheless, there are few reports about thermal effect on recombination process between polaron pair, although it is very important for the performance of PLEDs. In this paper, we adopt the modified one-dimensional tight-binding model, including to which the thermal random force, and explore the temperature effect on polaron collision driven by electric field with different strengths. The dynamical simulation is performed by using the non-adiabatic evolution method. The results show that under the influence of electric field, the oppositely charged polarons could recombine into either an exciton with one lattice distortion, or the mixed state of polaron pair and exciton with two lattice distortions. It depends on both field strength and temperature. Anyway, after including temperature effect, a significant improvement of exciton yield is obtained. In addition, the new-formed exciton could perform a random walk along the polymer chain driven by the thermal random force when its strength is large enough. If we further increase the temperature, the stability of exciton would become worse.Download high-res image (729KB)Download full-size image
By using nonequilibrium Green’s function method in combination with density functional theory, the electron transport properties of single atomic chain connected silicene-silicane or germanene-germanane nanoribbon junction are theoretically investigated. Obvious rectifying behaviors with rectification ratios up to 104 can be obtained by asymmetrically hydrogenating electrodes of the nano junctions. The edge pattern of hydrogenated electrode is shown to play a significant role in determining the rectifying direction of these diodes. Besides, the germanium atoms constructed nano diodes show better rectifying performance than the respective devices constructed by silicon atoms. The rectifying mechanism for these devices is elucidated by analyzing the molecular projected self-consistent Hamiltonian, the transmission spectrum and the band structure of electrodes with applied bias.Download high-res image (143KB)Download full-size image
•Spin-polarized transport properties of Fe-oligoporphyrin dimer (Fe-P2TA) are studied.•High-efficiency magnetoresistance, spin filtering, and low-bias negative differential resistance effects are observed.•The system is a potential candidate for designing multifunctional spintronic devices.By applying the density functional theory and the nonequilibrium Green’s function formalism, we investigate the spin-polarized transport properties of a Fe-oligoporphyrin dimer (Fe-P2TA) sandwiched between two armchair single-walled carbon nanotube electrodes. The results show that the system can present high-efficiency magnetoresistance, spin-filtering, and low-bias negative differential resistance effects with the help of magnetic field modulation. The above results are explained by the evolution of the spin-polarized transmission spectra and the molecular projected self-consistent Hamiltonian eigenstates with applied bias. Therefore, the system provides the possibilities for a multifunctional molecular spintronic device design.We investigate the spin-polarized transport properties of a Fe-P2TA molecule sandwiched between two carbon nanotube electrodes. The results show that the system can present high-efficiency magnetoresistance, spin-filtering, and low-bias negative differential resistance effects with the help of magnetic field modulation.
Co-reporter:Jing-hua Guo, Xin-Lu Cheng, Shu-Juan Li, and Gang Chen
The Journal of Physical Chemistry C 2016 Volume 120(Issue 31) pp:17153-17164
Publication Date(Web):July 19, 2016
DOI:10.1021/acs.jpcc.6b03389
It is recognized that “the bridge-building technique remains an art”, due to unclear microscopic nature of the bridging-spillover mechanism. In this paper, a theoretical model of a carbon bridge is designed to reveal the bridging-spillover mechanism in covalent organic frameworks (COFs) by the first-principles calculations and kinetic Monte Carlo (KMC) simulations. It is found that the activation barrier of H migration from a Pt cluster to carbon bridge ranges from 0.23 to 1.05 eV, which is much lower than the direct migration from the Pt6 cluster to COF-8 (activation barrier of 2.1 eV). The bare carbon atoms on carbon bridges are mainly responsible for facilitating hydrogen spillover. KMC simulations display that H would not directly migrate from the Pt6 cluster to COF-8 unless elevating the temperature to 700 K, in contrast to that of lower temperature (88–360 K) for H migration from the Pt6 cluster to carbon bridges. In addition, the introduction of oxygen impurities can reduce the H migration barrier from carbon bridges to COF-8. However, the migration can not carry out at room temperature (the reaction temperature >420 K). The results illustrate that the H atoms that overflowed from the catalyst tend to be adsorbed on carbon bridges rather than the COF-8 surface. The results may provide a useful guidance for understanding the bridging-spillover mechanism and offer a practical design of carbon bridges for hydrogen spillover.
Co-reporter:Q. H. Wu, P. Zhao, Y. Su, S. J. Li, J. H. Guo and G. Chen
RSC Advances 2015 vol. 5(Issue 65) pp:52938-52944
Publication Date(Web):10 Jun 2015
DOI:10.1039/C5RA07456H
Based on spin-polarized first-principles density functional theory in conjunction with the nonequilibrium Green's function method, the spin transport properties of transition metal (TM)–dibenzotetraaza[14]annulene (DBTAA) complexes (TM = Ti, V, Cr, Mn, Fe, Co, Ni, and Cu) sandwiched between two Au electrodes are investigated. The results show that Fe– and Co–DBTAA can display perfect spin filtering behavior in a wide bias voltage region. Moreover, it is found that the connected position of anchoring groups on the complexes affect significantly the spin filtering efficiency. The observed spin filtering behavior is explained by the spin-resolved transmission spectrum and molecular projected self-consistent Hamiltonian state analyses.
Co-reporter:Qiuhua Wu, Peng Zhao, Yan Su, Desheng Liu and Gang Chen
RSC Advances 2015 vol. 5(Issue 27) pp:20699-20703
Publication Date(Web):16 Feb 2015
DOI:10.1039/C4RA16845C
Based on spin-polarized first-principles density functional theory combined with nonequilibrium Green's function method, the thermal spin transport properties of a nitroxide radical-based molecule sandwiched between two Au electrodes are investigated. The results show that opposite spin currents can be induced by applying a temperature difference, rather than bias voltage, between two electrodes. Moreover, a pure spin current and a completely spin-polarized current can be realized by tuning the transverse gate voltage. These results indicate that the nitroxide radical-based molecule is a potential material for spin caloritronic and spintronic applications.
•Magnetic transport properties of DBTAA and transition metal-DBTAA with ZGNR electrodes are investigated.•DBTAA system can exhibit giant magnetoresistance, spin-filtering and spin-polarized current rectifying effects.•Introducing of TM atoms has obvious effects on these spin-related effects.Based on the density functional theory combined with the nonequilibrium Green’s function formalism, the magnetic transport properties of dibenzotetraaza[14]annulene (DBTAA) and transition metal (TM)-DBTAA (TM = Fe and Co) sandwiched between two ferromagnetic zigzag-edge graphene nanoribbon electrodes are investigated. The results show that giant magnetoresistance, spin-filtering and spin-polarized current rectifying effects can be realized simultaneously in the DBTAA system by modulating the external magnetic field. Introducing of TM atoms has obvious effects on these spin-related effects. The mechanisms of these intriguing phenomena are proposed and these phenomena would be instructive in the design of high-performance magnetic nanodevices.We investigate systematically the magnetic transport properties of DBTAA and transition metal (TM)-DBTAA (TM = Fe and Co) sandwiched between two ZGNR electrodes. The results show that giant magnetoresistance, spin-filtering and spin-polarized current rectifying effects can be realized simultaneously in the DBTAA system. And introducing of TM atoms has obvious effects on these effects.
Co-reporter:P. Zhao, Q. H. Wu, H. Y. Liu, D. S. Liu and G. Chen
Journal of Materials Chemistry A 2014 vol. 2(Issue 32) pp:6648-6654
Publication Date(Web):17 Jun 2014
DOI:10.1039/C4TC00895B
By using the nonequilibrium Green's function formalism in combination with the density functional theory, we have investigated the spin transport properties of a 4H-TAHDI-based multifunctional spintronic device constructed by contacting a 4H-TAHDI molecule with two ferromagnetic zigzag-edge graphene nanoribbon electrodes. The results show that perfect giant magnetoresistance, spin-filtering, bipolar spin-rectifying, and negative differential resistance effects can be realized simultaneously. The mechanisms were proposed for these interesting phenomena. Our results demonstrate that this system holds promise in the design of a high-performance multifunctional single-molecule spintronic device.
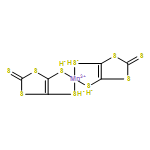
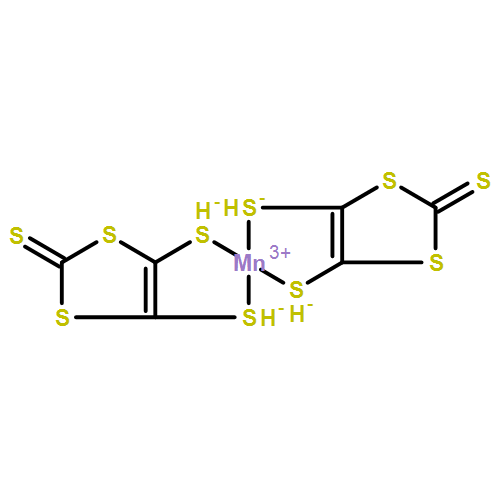
•Transport properties of a single-molecule magnet Mn(dmit)2 with two ferromagnetic ZGNR electrodes are studied.•The system presents large rectifying, giant magnetoresistance, spin-filtering and negative differential resistance effects.•An improved switching effect can also be realized by changing the orientation between planes of two dmit ligands.By applying the density functional theory and the nonequilibrium Green’s function formalism, we investigate the spin transport properties of a single-molecule magnet Mn(dmit)2 sandwiched between two ferromagnetic zigzag-edge graphene nanoribbon electrodes. The results show that the system can present large rectifying, giant magnetoresistance, spin-filtering and negative differential resistance effects with the help of magnetic field modulation. Moreover, an improved switching effect can also be realized by changing the orientation between planes of two dmit ligands. Therefore, the system will provide the possibilities for a multifunctional molecular device design.We investigate the spin transport properties of a single-molecule magnet Mn(dmit)2 sandwiched between two ferromagnetic zigzag-edge graphene nanoribbon electrodes. The results show that the system can present large rectifying, giant magnetoresistance, spin-filtering, negative differential resistance and switching effects.
Co-reporter:J.T. Jiang, S.L. Xiu, M.M. Zheng, T.T. Jia, H.Y. Liu, Y. Zhang, G. Chen
Chemical Physics Letters 2014 Volume 613() pp:74-79
Publication Date(Web):3 October 2014
DOI:10.1016/j.cplett.2014.08.060
•The indirect–direct bandgap transition can be realized in bilayer MoS2 superlattices.•Interlayer interaction affects the p orbital valence band of sulfur atom at Г point.•The bottom of conduction band could be folded from K to Г points.•The in-plane stretching can tune gap width by affecting the intralayer interaction.•Reducing layer interval can tune gap width by affecting the interlayer interaction.Using the band-folding analysis and the first-principles method, we have carefully studied the electronic properties of the bilayer MoS2 superlattices. In the (N,M) bilayer MoS2 superlattice, the bottom of the conduction band could be folded from K to Г points resulting in the direct bandgap semiconductor if both N and M are integer multiple of 3. Furthermore, the gap width could be tuned by the in-plane stretching and the perpendicular compressing. These studies could pave the path for designing the direct bandgap nanostructures and tuning their gap width toward the applications in the high-performance photoelectronic devices.
By using a combined method of density functional theory and nonequilibrium Green’s function formalism, we investigate the electronic transport properties of a gated C60 dimer molecule sandwiched between two gold electrodes. The results show that the gate voltage can strongly affect the electronic transport properties of the C60 dimer and change it from semiconducting to metallic. Negative differential resistance behaviors are obtained in such systems and can be modulated to occur at much lower bias by the gate voltage. The low bias negative differential resistance is analyzed from the calculated transmission spectra, projected density of states and the spatial distribution of molecular projected self-consistent Hamiltonian orbitals along with the voltage drop. These results provide a theoretical support to the design of low bias negative differential resistance molecular device by using the modulation of gate voltage.Graphical abstractWe present a systematic study of electronic transport properties of a gated C60 dimer molecule sandwiched between two gold electrodes.Highlights► Effect of gate voltage on electronic transport properties of the C60 dimer is studied. ► Gate voltage can change the system from semiconducting to metallic. ► Negative differential resistance behavior can be modulated to occur at much lower bias by the gate voltage.
Chemical Physics Letters 2013 Volume 565() pp:86-91
Publication Date(Web):5 April 2013
DOI:10.1016/j.cplett.2013.02.037
The Ti catalyst doped in the vicinity of surface step could split H2 with 0.37 eV activation energy and the resulting hydrogen atom could diffuse by overcoming 0.45 eV barrier. Our analysis on the energies of the Ti doping and the surface etching phenomenon suggest that Ti can remain as recycling active catalyst during the aluminum hydrogenation. The electronic properties of the intermediate state could account for the enhanced splitting properties. These studies on the role of surface step could contribute to the understanding of the mechanism of transition metal catalyzed hydrogen uptake in aluminum.Graphical abstractHighlights► The doping of Ti in surface step region can gain ∼0.36 eV higher doping energy. ► Activation energy is reduced to as low as 0.45 eV for aluminum hydrogenation. ► Ti doped in the lowest energy structure could remain as recycling active catalyst. ► H2 in intermediate state gains 0.3 e charge due to Kubas interaction mechanism.
Solid State Communications 2013 Volume 174() pp:5-9
Publication Date(Web):November 2013
DOI:10.1016/j.ssc.2013.09.020
•Spin-polarized transport properties of an endohedral Fe@C60 dimer are investigated.•Direction of the magnetic moments of two Fe atoms can be controlled by applying a magnetic field.•Magnetoresistance, spin filtering and negative differential resistance effects can be observed.Based on non-equilibrium Green's method and density functional theory, the spin-polarized transport properties of an endohedral Fe@C60 dimer-based spintronic device are investigated. The direction of the magnetic moments of the two Fe atoms can be controlled by applying a magnetic field. The interesting magnetoresistance, spin filtering and negative differential resistance effects can be observed in this device. These phenomena are explained by the evolution of spin-dependent transmission spectra and projected density of states with the increase in bias, as well as the voltage drop distributions.
Co-reporter:Peng Zhao, De-Sheng Liu, Shu-Juan Li, Gang Chen
Chemical Physics Letters 2012 Volume 554() pp:172-176
Publication Date(Web):3 December 2012
DOI:10.1016/j.cplett.2012.10.045
By applying nonequilibrium Green’s function formalism in combination with density functional theory, we have investigated the electronic transport properties of armchair graphene nanoribbon devices with periodic nitrogen-doping. Giant negative differential resistance behaviors with peak-to-valley ratio up to the order of 105 can be obtained in the mV bias regime by tuning the position and the concentration of the dopants. The negative differential resistance behavior is understood in terms of the evolution of the transmission spectrum and band structures with applied bias combined with the symmetry analyses of the Bloch wave functions of the corresponding subbands.Graphical abstractWe present a systematic study of the electronic transport properties of armchair graphene nanoribbon devices with periodic nitrogen-doping. Giant negative differential resistance behaviors with peak-to-valley ratio up to the order of 105 can be obtained in the mV bias regime by tuning the position and the concentration of the dopants.Highlights► Electronic transport properties of armchair graphene nanoribbon devices with periodic nitrogen-doping are studied. ► Transport properties are strongly dependent on the position and the concentration of the dopants. ► Giant NDR behaviors with peak-to-valley up to the order of 105 can be obtained in the mV bias regime.
Solid State Communications 2012 Volume 152(Issue 22) pp:2040-2044
Publication Date(Web):November 2012
DOI:10.1016/j.ssc.2012.08.013
Using first-principles density functional theory and non-equilibrium Green's function formalism for quantum transport calculation, we have investigated the electronic transport properties of (8,0), (9,0) and (13,0) zigzag single-walled carbon nanotube junctions with one undoped and one nitrogen-doped zigzag carbon nanotube electrode. Our results show that the transport properties are strongly dependent on the magnitude of energy gap of carbon nanotube. Large rectifying behavior can be obtained in the junction with large energy gap. The observed rectifying behavior are explained in terms of the evolution of the transmission spectra and energy band structures with applied bias voltage combined with molecular projected self-consistent Hamiltonian eigenstates analysis.Highlights► Transport properties of nitrogen-doped zigzag single-walled carbon nanotubes are studied. ► Transport properties strongly depend on the magnitude of energy gap of carbon nanotube. ► Large rectifying behavior occurs in the nitrogen-doped carbon nanotube with large energy gap.
Co-reporter:P. Zhao, D.S. Liu, Y. Zhang, Y. Su, H.Y. Liu, S.J. Li, G. Chen
Solid State Communications 2012 Volume 152(Issue 12) pp:1061-1066
Publication Date(Web):June 2012
DOI:10.1016/j.ssc.2012.03.018
Using first-principles density functional theory and non-equilibrium Green's function formalism for quantum transport calculation, we have investigated the electronic transport properties of heteronanotubes by joining a zigzag (6,0) carbon nanotube and a zigzag (6,0) boron nitride nanotube with different atomic compositions and joint configurations. Our results show that the atomic composition and joint configuration affect strongly the electronic transport properties. Obvious negative differential resistance behavior and large rectifying behavior are obtained in the heterostructure with certain composition and joint configuration. Moreover, tube length and tube radius can affect strongly the observed NDR and rectifying behaviors. The observed negative differential resistance and rectifying behaviors are explained in terms of the evolution of the transmission spectrum with applied bias combined with molecular projected self-consistent Hamiltonian states analysis.Highlights► Transport properties of heteronanotubes based on zigzag C- and BN-nanotubes are studied. ► Atomic composition and joint configuration affect strongly the transport properties. ► Obvious NDR and large rectifying behaviors are observed under certain conditions.
Co-reporter:P. Zhao, D. S. Liu, Y. Zhang, Y. Su, H. Y. Liu, S. J. Li, and G. Chen
The Journal of Physical Chemistry C 2012 Volume 116(Issue 14) pp:7968-7974
Publication Date(Web):March 5, 2012
DOI:10.1021/jp210880j
By applying the nonequilibrium Green function formalism combined with density functional theory, we have investigated the electronic transport properties of the C60 dimer and its endohedral complex Li@C60 dimer. Our results show that the doping of Li atoms significantly changes the transport properties of the C60 dimer. Negative differential resistance is found in such systems. Especially, the doping of Li atoms can lead to a much larger negative differential resistance at much lower bias, and it is quite evident from the plot of differential conductance versus bias. The negative differential resistance behavior is understood in terms of the evolution of the transmission spectrum and projected density of states spectrum with applied bias combined with molecular projected self-consistent Hamiltonian states analyses.
Chemical Physics Letters 2011 Volume 507(4–6) pp:260-264
Publication Date(Web):9 May 2011
DOI:10.1016/j.cplett.2011.04.011
Abstract
This Letter reports our detailed first-principles study on neutral and charged Ca8C12 clusters. Except a compact structure owning geometrical characters of the CaC2 bulk, all the other low-lying structures within 0.2 eV in relative total energy are hollow configurations. Most of the low-lying structures are calculated to be magnetic. The ground states are a D3d flat structure, a compact structure, and a D3d hollow cage-like structure for Ca8C12, , and clusters, respectively. The net charge on Ca ion, except the dangling one of the compact structure, is calculated to be around +1.4e.
Co-reporter:Peng Zhao, Yan Su, Ying Zhang, Shu-Juan Li, Gang Chen
Chemical Physics Letters 2011 Volume 515(1–3) pp:159-162
Publication Date(Web):17 October 2011
DOI:10.1016/j.cplett.2011.09.034
Using first-principles density functional theory, we have investigated the catalytic oxidation of CO on Fe-embedded hexagonal boron nitride (h-BN) sheet. Fe atom can be constrained at a boron vacancy site of h-BN sheet with a high diffusion barrier (3.70 eV), and effectively activate the adsorbed O2 molecule. The reactions between the adsorbed O2 with CO via both Langmuir–Hinshelwood and Eley–Rideal mechanisms were comparably studied. The reaction proceeds via the more favorable Eley–Rideal mechanism with a two-step route (CO + O2 → CO2 + O and CO + O → CO2). The energy barriers are 0.56 and 0.61 eV, respectively.Graphical abstractWe have investigated theoretically the catalytic oxidation of CO on Fe-embedded h-BN sheet. The reaction proceeds rapidly via the Eley–Rideal mechanism due to the low energy barriers.Highlights► CO catalytic oxidation on Fe-embedded h-BN sheet is studied for the first time. ► Fe can be constrained at a B-vacancy, and effectively activates the O2 molecule. ► The oxidation could proceed rapidly because of the low energy barriers.
Solid State Communications (November 2012) Volume 152(Issue 22) pp:2040-2044
Publication Date(Web):1 November 2012
DOI:10.1016/j.ssc.2012.08.013
Using first-principles density functional theory and non-equilibrium Green's function formalism for quantum transport calculation, we have investigated the electronic transport properties of (8,0), (9,0) and (13,0) zigzag single-walled carbon nanotube junctions with one undoped and one nitrogen-doped zigzag carbon nanotube electrode. Our results show that the transport properties are strongly dependent on the magnitude of energy gap of carbon nanotube. Large rectifying behavior can be obtained in the junction with large energy gap. The observed rectifying behavior are explained in terms of the evolution of the transmission spectra and energy band structures with applied bias voltage combined with molecular projected self-consistent Hamiltonian eigenstates analysis.Highlights► Transport properties of nitrogen-doped zigzag single-walled carbon nanotubes are studied. ► Transport properties strongly depend on the magnitude of energy gap of carbon nanotube. ► Large rectifying behavior occurs in the nitrogen-doped carbon nanotube with large energy gap.
Physics Letters A (5 February 2017) Volume 381(Issue 5) pp:549-555
Publication Date(Web):5 February 2017
DOI:10.1016/j.physleta.2016.12.016
•Effect of interchain coupling on the single-excited state of polaron is studied.•When coupling strength exceeds critical value, the excited polaron is dissociated.•Soliton pair could be dissociated into polaron-like particle with strong coupling.•Reverse polarization of excited polaron is enhanced by weak interchain coupling.•Reverse polarization is obtained more easily in solid film of polymer molecules.Based on the one-dimensional extended Su–Schrieffer–Heeger model, we theoretically investigate the effect of interchain coupling on the formation and polarization of the single-excited state of polaron in conjugated polymers. It is found that there exists a turnover value of the coupling strength, over which the excited polaron could not be formed in either of the two coupled chains. Instead, a polaron-like particle is localized at the center of each chain. In addition, we also find that the reverse polarization of the excited polaron could be enhanced for some cases in polymer when the interchain coupling becomes strong until it exceeds the critical value.
Co-reporter:P. Zhao, Q. H. Wu, H. Y. Liu, D. S. Liu and G. Chen
Journal of Materials Chemistry A 2014 - vol. 2(Issue 32) pp:NaN6654-6654
Publication Date(Web):2014/06/17
DOI:10.1039/C4TC00895B
By using the nonequilibrium Green's function formalism in combination with the density functional theory, we have investigated the spin transport properties of a 4H-TAHDI-based multifunctional spintronic device constructed by contacting a 4H-TAHDI molecule with two ferromagnetic zigzag-edge graphene nanoribbon electrodes. The results show that perfect giant magnetoresistance, spin-filtering, bipolar spin-rectifying, and negative differential resistance effects can be realized simultaneously. The mechanisms were proposed for these interesting phenomena. Our results demonstrate that this system holds promise in the design of a high-performance multifunctional single-molecule spintronic device.