Co-reporter:Qiao Zhang;Ilkeun Lee;Jianping Ge;Yadong Yin
Advanced Functional Materials 2010 Volume 20( Issue 14) pp:2201-2214
Publication Date(Web):
DOI:10.1002/adfm.201000428
Abstract
Nanoparticles of transition metals, particularly noble metals, are widely used in catalysis. However, enhancing their stability during catalytic reactions has been a challenge that has limited the full use of the benefits associated with their small size. In this Feature Article, a general “encapsulation and etching” strategy for the fabrication of nanocatalyst systems is introduced in which catalyst nanoparticles are protected within porous shells. The novelty of this approach lies in the use of chemical etching to assist the creation of mesopores in a protective oxide shell to promote efficient mass transfer to encapsulated metal nanoparticles. The etching process allows for the direct transformation of dense silica coatings into porous shells so that chemical species can reach the catalyst surface to participate in reactions while the shells act as physical barriers against aggregation of the catalyst particles. By using the surface-protected etching process, both yolk–shell and core–satellite type nanoreactors are synthesized and their utilization in liquid- and gas-phase catalysis is demonstrated. The thermal and chemical stability of the metallic cores during catalytic reactions is also investigated, and further work is carried out to enhance recyclability via the introduction of superparamagnetic components into the nanoreactor framework.
Co-reporter:Franklin Anariba ; Hugo Tiznado ; James R. Diers ; Izabela Schmidt ; Ana Z. Muresan ; Jonathan S. Lindsey ; Francisco Zaera ;David F. Bocian
The Journal of Physical Chemistry C 2008 Volume 112(Issue 25) pp:9474-9485
Publication Date(Web):June 4, 2008
DOI:10.1021/jp802428y
The promise of molecular electronics has stimulated a variety of approaches for making hybrid junctions wherein molecules are sandwiched between two metal contacts or a metal and a semiconductor contact. However, the fate of molecules subsequent to deposition of a top metal contact has generally not been well characterized. Toward this goal, the interaction of evaporated Cu, Ag, and Au films deposited in varying thicknesses (3, 5, and 8 nm) on a series of monolayer-coverage porphyrins covalently attached to Si(100) substrates was investigated. Each porphyrin contains a triallyl tripod attached to the porphyrin via a p-phenylene unit, which anchors the porphyrin to the Si(100) substrate via hydrosilylation of the allyl groups. All of the porphyrins are Zn chelates bearing meso p-tolyl substituents orthogonal (lateral) to the tripodal surface anchor, but contain different substituents in the meso position opposite (distal) to the surface anchor, p-tolyl, α,α,α-trifluoro-p-tolyl, pentafluorophenyl, or p-cyanophenyl, to test the potential for the distal meso substituents to impart different characteristics to the metal/molecule/Si junction. The methods of interrogation used include ellipsometry, atomic force microscopy, FTIR spectroscopy, Raman spectroscopy, X-ray photoelectron spectroscopy, cyclic voltammetry, and current−voltage measurements. The studies indicate that all of the porphyrin monolayers are robust under the conditions of metal deposition, experiencing no noticeable degradation, and that none of the distal functional groups reacts with the deposited metal, except for the nitrile group of the p-cyanophenyl-substituted porphyrin, which reacts/complexes with Cu. Neither Cu nor Ag penetrates through the monolayer to an appreciable extent to form electrically conductive filaments, whereas Au does penetrate through the porphyrin monolayer and contacts the Si substrate. Collectively, the studies provide a comprehensive assessment of the characteristics of the metal (Cu, Ag, Au)/porphyrin monolayer/Si junctions.
Co-reporter:Hansheng Guo, Francisco Zaera
Surface Science 2003 Volume 547(Issue 3) pp:284-298
Publication Date(Web):20 December 2003
DOI:10.1016/j.susc.2003.10.028
The thermal chemistry of diiodomethane on Ni(1 1 0) single-crystal surfaces was studied by temperature-programmed desorption (TPD) and X-ray photoelectron spectroscopy (XPS). Diiodomethane was chosen as a precursor for the formation of methylene surface species. I 3d and C 1s XPS data indicated that, indeed, adsorbed diiodomethane undergoes the C–I bond dissociations needed for that transformation, and detection of iodomethane production in TPD experiments pointed to the stepwise nature of those reactions. Significant amounts of methane are produced from further thermal activation of the chemisorbed methylene groups. This involves surface hydrogen, both coadsorbed from background gases and produced by dehydrogenation of some of the adsorbed diiodomethane, and can be induced at temperatures as low as about 160 K, right after the C–I bond breaking steps. Unique to this system is the detection of significant amounts, up to 10% of the total CH2I2 adsorbed, of heavier hydrocarbons, including ethene, ethane, propene, propane, and butene. Deuterium labeling experiments were used to provide support for a mechanism where the initial hydrogenation of some adsorbed methylene to methyl moieties is followed by a rate-limiting methylene insertion step to yield ethyl intermediates. Facile subsequent β-hydride elimination and reductive elimination with coadsorbed hydrogen account for the formation of ethene and ethane, respectively, while a second and third methylene insertions lead to C3 and C4 production. Based on the final product distribution, the methylene insertion was estimated to be approximately 20 times slower than the following hydrogenation–dehydrogenation reactions. Normal kinetic isotope effects were observed for most of the hydrogenation and dehydrogenation reactions involved.
Co-reporter:Hansheng Guo, Francisco Zaera
Surface Science 2003 Volume 547(Issue 3) pp:299-314
Publication Date(Web):20 December 2003
DOI:10.1016/j.susc.2003.10.029
The effect of coadsorbed oxygen on the thermal chemistry of diiodomethane on Ni(1 1 0) single-crystal surfaces was studied by temperature-programmed desorption (TPD) and X-ray photoelectron spectroscopy (XPS). I 3d and C 1s XPS data indicated that adsorbed diiodomethane undergoes two sequential C–I bond scission steps to ultimately produce methylene surface species, the same as on clean Ni(1 1 0). Moreover, significant amounts of methane and other heavier hydrocarbons are produced after further thermal activation of those chemisorbed methylene groups. The production of alkanes and alkenes, which is accounted for by a chain growth mechanism where the initial hydrogenation of some adsorbed methylene to methyl moieties is followed by a rate-limiting methylene insertion step to yield ethyl intermediates, is inhibited but not fully blocked by the coadsorbed oxygen. New reaction pathways are also opened up by the presence of oxygen in this system, including a direct coupling of two methylene groups to ethene, the insertion of an oxygen atom into a nickel–methylene group to produce formaldehyde, and a parallel methylene insertion chain growth sequence starting from a CH2Iads intermediate to ultimately yield C3H5 and C4H7 unsaturated gas-phase radicals.
Co-reporter:Ton V.W. Janssens, Francisco Zaera
Surface Science 2002 Volume 501(1–2) pp:16-30
Publication Date(Web):20 March 2002
DOI:10.1016/S0039-6028(01)02024-6
Co-reporter:Ton V.W. Janssens, Francisco Zaera
Surface Science 2002 Volume 501(1–2) pp:1-15
Publication Date(Web):20 March 2002
DOI:10.1016/S0039-6028(01)02023-4
The chemistry of neopentyl iodide on Pt(1 1 1) single-crystal surfaces was characterized by temperature programmed desorption and reflection-absorption infrared spectroscopy. As with many other halo hydrocarbons, the first thermal reaction observed with neopentyl iodide on the platinum surface is the cleavage of the C–I bond, a step that leads to the formation of neopentyl surface species by 170 K. Hydrogenation of those intermediates is quite facile, taking place by 270 K and removing over 90% of the initial surface species. The remaining neopentyl groups undergo stepwise dehydrogenation to form a number of surface intermediates around 235 K, the identity of which is discussed in the following paper. The selectivity towards a given dehydrogenation reaction at that temperature depends on the regiospecificity of the deuterium labeling of the original molecule. Nevertheless, heating of any of those intermediates to 300 K leads to the formation of neopentylidyne. Small amounts of hydrogenolysis products are also detected at 300 and 430 K, presumably resulting from cracking of neopentylidene and neopentylidyne surface species, respectively.
Co-reporter:Francisco Zaera, Celso M. Aldao, Richard M. Lambert, Wilfred T. Tysoe, Giorgio Zgrablich
Journal of Molecular Catalysis A: Chemical 2001 Volume 167(1–2) pp:1
Publication Date(Web):20 February 2001
DOI:10.1016/S1381-1169(00)00483-0
Co-reporter:Francisco Zaera, Demetrius Chrysostomou
Surface Science 2000 Volume 457(1–2) pp:89-108
Publication Date(Web):1 June 2000
DOI:10.1016/S0039-6028(00)00337-X
The thermal chemistry of propylene on Pt(111) single-crystal surfaces was studied under vacuum by using temperature-programmed desorption spectroscopy. Besides molecular desorption and hydrogen production from propylene dehydrogenation (first to propylidyne and eventually to surface carbon), a small amount of propane from self-hydrogenation of the olefin is detected at about 280 K. Hydrogen coadsorption weakens the adsorption of propylene on the surface, and enhances the production of propane. In addition to deuteration to propane, deuterium coadsorption with propylene leads to H–D exchange. Multiple exchange is evidenced by the formation of all possible deuterium-substituted propylenes and propanes, including propylene-d6 and propane-d6. A detailed analysis of the data provided support for a mechanism where the slow half-hydrogenation of propylene to propyl species is followed by competitive β-hydride and reductive elimination, to (exchanged) propylene and propane, respectively. The unexpectedly low yields of propylene-d5 and propane-d7 suggest a preferential hydrogenation at the central carbon atom. Most of the propylene that remains on the surface above 350 K dehydrogenates to propylidyne (Pt3CCH2CH3). A small fraction of that species rehydrogenates to propane at 430 K, while the rest undergoes stepwise dehydrogenation to surface carbon. Arguments against the formation of an allylic intermediate are provided, and a comparison with the chemistry of ethylene on the same surface is included.
Co-reporter:Francisco Zaera, Demetrius Chrysostomou
Surface Science 2000 Volume 457(1–2) pp:71-88
Publication Date(Web):1 June 2000
DOI:10.1016/S0039-6028(00)00336-8
The adsorption of propylene on Pt(111) single-crystal surfaces was characterized by reflection-absorption infra-red spectroscopy (RAIRS). The uptake of propylene on the surface at 90 K results in the development of at least four adsorption species as a function of coverage. Significant rehybridization of the CC double bond of propylene takes place at low coverages, so the molecule primarily interacts with the metal via two σ metalcarbon bonds. Below half-saturation, the molecule mainly bonds through the central carbon atom, but at higher coverage, the CC bond becomes flat, and the terminal methyl group tilts towards a more vertical orientation. Further dosing of propylene after saturation of the di-σ state leads to the build-up of a flat π-bonded second layer. Ultimately, a layer of condensed propylene could be grown on the surface under the vacuum conditions of the experiment as long as the temperature was kept below 80 K. Annealing of the low-temperature propylene-saturated Pt(111) surface first induces the desorption of the weakly held π species, and later, between 230 and 250 K, to the dehydrogenation and rearrangement of the remaining di-σ species to propylidyne (Pt3CCH2CH3). The details of the conversion of propylene to propylidyne change somewhat with the conditions under which this transformation is carried out, and appear to involve a stable and identifiable intermediate [2-propyl, CH3CH(Pt)CH3, and/or propylidene, Pt2CHCH2CH3]. Propylene π-bonding is also possible on propylidyne-saturated Pt(111) surfaces under vacuum.
Co-reporter:D. Chrysostomou, J. Flowers, F. Zaera
Surface Science 1999 Volume 439(1–3) pp:34-48
Publication Date(Web):20 September 1999
DOI:10.1016/S0039-6028(99)00458-6
The thermal chemistry of NH3 on Ni(110) surfaces has been investigated using temperature-programmed desorption (TPD). The thermal activation of ammonia-saturated Ni(110) leads primarily to molecular desorption, but some decomposition is identified by the appearance of hydrogen and nitrogen TPD peaks ca 400 and 800 K, respectively. Previous reports have shown that ammonia dehydrogenation is readily induced by mild electron bombardment, but this study found dehydrogenation to persist despite the absence of any electron current at the surface, indicating that both thermal- and electron-induced dissociation processes are possible in the NH3/Ni(110) system. Our experimental data also identified the onset of the thermally-activated ammonia dehydrogenation at about 300 K. Evidence is given to suggest that the surface intermediate formed subsequent to this dehydrogenation is NH2,ads. Further activation of the NH2,ads moiety by the Ni(110) surface results in its dehydrogenation to NHads and Nads around 380 K, but also to some NH2,ads+Hads recombination to NH3(g) ca 360 K. After considering this recombination step, it was calculated that as much as 23% of adsorbed ammonia dissociates by thermal activation, a much greater value than previously reported.
Co-reporter:Francisco Zaera
Israel Journal of Chemistry 1998 Volume 38(Issue 4) pp:293-311
Publication Date(Web):21 NOV 2013
DOI:10.1002/ijch.199800035
The catalytic chemical conversion of hydrocarbons on transition metals is at the heart of a good number of processes of industrial importance, including oil and food processing, pharmacological synthesis, manufacturing of specialty chemicals, and polymer production, to name a few. This is an old field that has advanced in great measure by process development work based on trial and error. It has only been in the last few decades, with the advent of a battery of new surface-sensitive techniques, that it has been possible to address the fundamental questions on the basic chemistry behind those hydrocarbon conversion reactions. In this review we highlight some of the main ideas advanced in this field by modern surface-science studies. Particular emphasis is given here to the pioneering work of Professor Somorjai's group as well as our more recent contributions. A discussion is provided on the state of the art of the knowledge available in terms of the elementary steps involved in the transformations of hydrocarbons on metals, as obtained in experiments performed under well-controlled vacuum conditions; on the connection between that knowledge and the reactions that may prevail under the atmospheric pressures encountered in real catalytic processes; and on the key questions that remain unanswered to date and the possible future directions in this area of research.

![N-{[(2-METHYL-2-PROPANYL)OXY]CARBONYL}-L-METHIONYL-L-WEI -ASPARTYL-L<WBR />-PHENYLALANINAMIDE](http://img.cochemist.com/ccimg/3700/3681-29-6.png)
![N-{[(2-METHYL-2-PROPANYL)OXY]CARBONYL}-L-METHIONYL-L-WEI -ASPARTYL-L<WBR />-PHENYLALANINAMIDE](http://img.cochemist.com/ccimg/3700/3681-29-6_b.png)
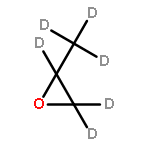
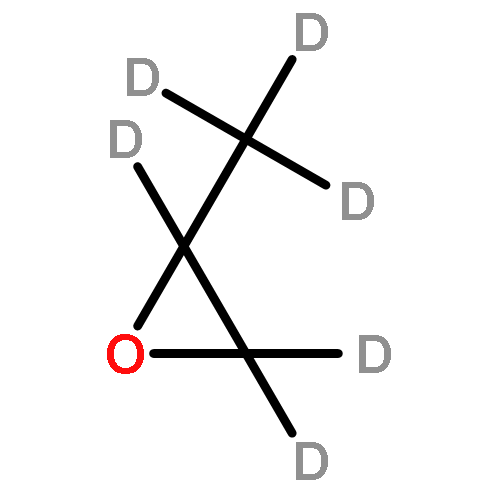
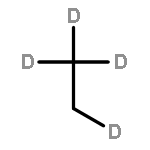
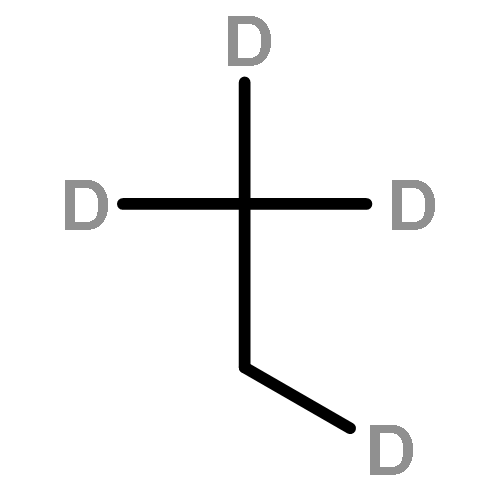


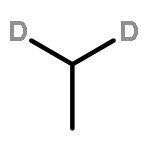
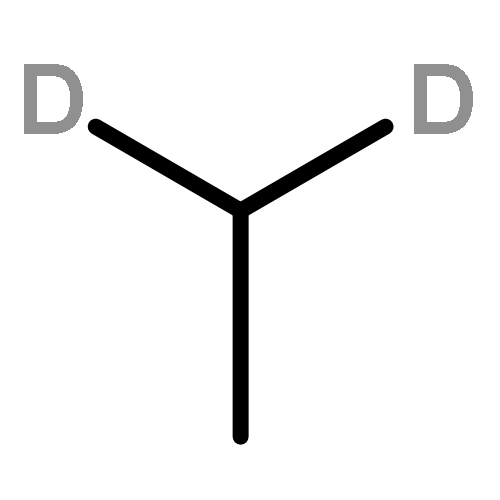






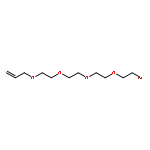
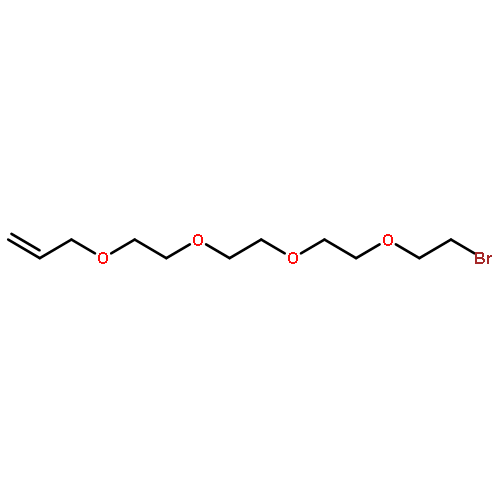


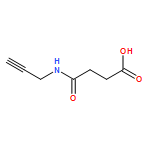
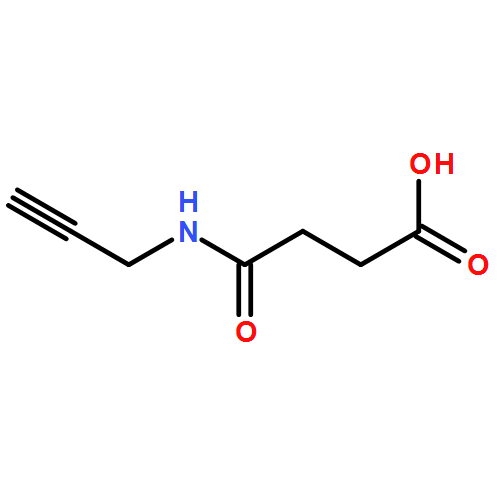

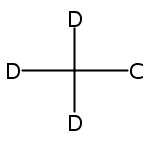


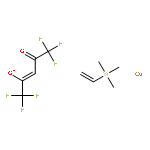
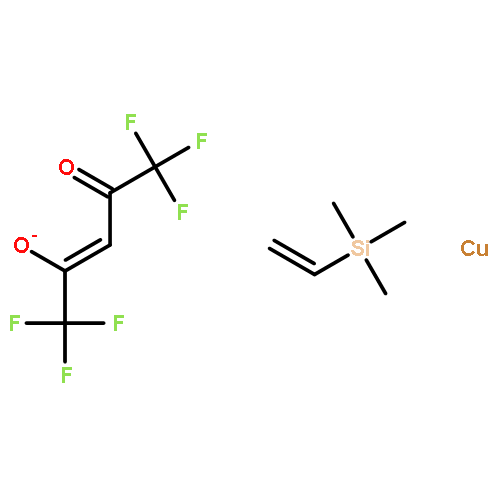






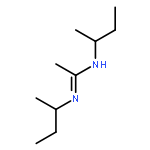
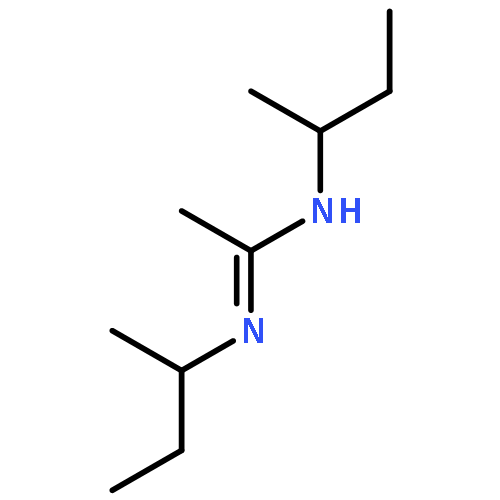
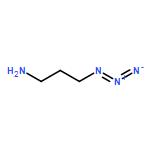


![CYCLOHEXANONE, 3-[[4-(1,1-DIMETHYLETHYL)PHENYL]THIO]-, (R)-](http://img.cochemist.com/ccimg/64900/64888-90-0.png)
![CYCLOHEXANONE, 3-[[4-(1,1-DIMETHYLETHYL)PHENYL]THIO]-, (R)-](http://img.cochemist.com/ccimg/64900/64888-90-0_b.png)



![1-[4-(3-METHYL-BUTOXY)-PHENYL]-ETHANONE](http://img.cochemist.com/ccimg/30300/30237-26-4.png)
![1-[4-(3-METHYL-BUTOXY)-PHENYL]-ETHANONE](http://img.cochemist.com/ccimg/30300/30237-26-4_b.png)







![Zinc, [5,10,15,20-tetraphenyl-21H,23H-porphinato(2-)-κN21,κN22,κN23,κN24]-, (SP-4-1)-](/data/chemimg/478800/14074-80-7.png)
![Zinc, [5,10,15,20-tetraphenyl-21H,23H-porphinato(2-)-κN21,κN22,κN23,κN24]-, (SP-4-1)-](/data/chemimg/478800/14074-80-7_b.png)