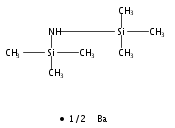
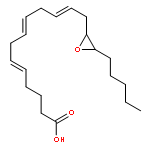
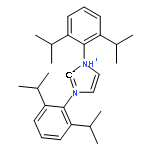
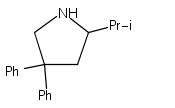
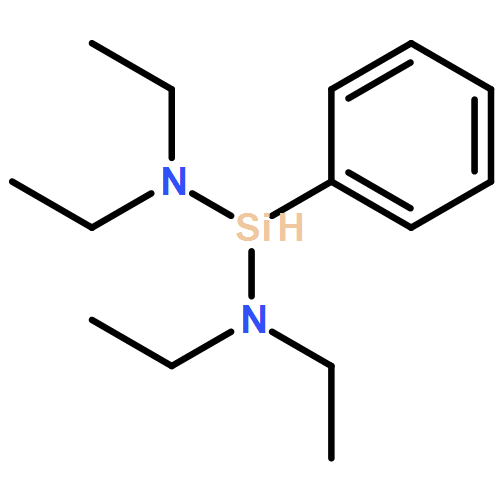
Co-reporter:Lucía García;Khalidah H. M. Al Furaiji;David J. D. Wilson;Jason L. Dutton;Mary F. Mahon
Dalton Transactions 2017 vol. 46(Issue 36) pp:12015-12018
Publication Date(Web):2017/09/19
DOI:10.1039/C7DT03046K
Reaction of phenylsilane with the ring expanded carbene 6-Mes results in facile Si–H oxidative addition to the carbenic carbon at room temperature. Heating the resultant diorganosilane product induces ring expansion through silicon to carbon migration of either the Si–H or Si–Ph bonds.
Co-reporter:Merle Arrowsmith, Brant Maitland, Gabriele Kociok-Köhn, Andreas Stasch, Cameron Jones, and Michael S. Hill
Inorganic Chemistry 2014 Volume 53(Issue 19) pp:10543-10552
Publication Date(Web):September 9, 2014
DOI:10.1021/ic501638v
The highly sterically encumbered chelating β-diketiminate ligand, [HC{C(Me)N(2,6-CHPh2-4-MeC6H2)}2]−, ArL–, has been used to prepare a series of heteroleptic three-coordinate magnesium complexes. Both the bis(imine) and imine-enamine tautomers of the ligand precursor, ArLH, as well as the diethyl ether adduct of the bromide complex [ArLMgBr(OEt2)], the monomeric methyl complex [ArLMgMe], the THF-solvated and unsolvated n-butylmagnesium complexes [ArLMgnBu(THF)] and [ArLMgnBu], and the 1-hexynyl analogue [ArLMgC≡CnBu] have been crystallographically characterized. Both n-butylmagnesium complexes showed remarkable stability in air, both in the solid state and in solution. Single crystals of the highly sensitive magnesium hydride, [ArLMgH], underwent partial hydrolysis by solid-state water diffusion to the isostructural hydroxide compound [ArLMgOH].
Co-reporter:Andrew L. Johnson, Michael S. Hill, Gabriele Kociok-Köhn, Kieran C. Molloy, Anna L. Sudlow
Inorganic Chemistry Communications 2014 Volume 49() pp:8-11
Publication Date(Web):November 2014
DOI:10.1016/j.inoche.2014.09.003
The copper(II) xanthate Cu(S2COEt)2·TMEDA (1) (TMEDA = N,N-tetramethylethylenediamine) has been synthesised and is the first structurally-characterised xanthate of copper in the + 2 oxidation state. 1 has an octahedral cis, cis, cis-ligand arrangement about the metal, in which xanthate chelation is markedly asymmetric. Both bulk thermal decomposition and film growth by aerosol-assisted chemical vapour deposition (AACVD) using 1 as precursor lead to the formation of Cu2S.
Co-reporter:Merle Arrowsmith, Mark R. Crimmin, Michael S. Hill, Sarah L. Lomas, Dugald J. MacDougall, and Mary F. Mahon
Organometallics 2013 Volume 32(Issue 17) pp:4961-4972
Publication Date(Web):August 22, 2013
DOI:10.1021/om400678d
A series of β-diketiminate-supported magnesium and calcium acetylide complexes have been synthesized by σ-bond metathesis of magnesium n-butyl or magnesium and calcium amido precursors and a range of terminal acetylenes. The dimeric complexes have been characterized by NMR spectroscopy and X-ray diffraction analysis. The homoleptic bis(amido) and dialkyl complexes [M{X(SiMe3)2}2(THF)2] (M = Ca, Sr; X = N, CH) have been assessed for the atom-efficient, catalytic head-to-head dimerization of donor-functionalized terminal alkynes into butatrienes and aryl-/silyl-substituted terminal acetylenes into 1,3-enynes. Deuterium labeling studies of the catalytic reactions are suggested to imply that triene formation requires concerted proton delivery and rearrangement via an adjacent methylene group at a bimetallic alkaline-earth species.
Co-reporter:Christine Brinkmann ; Anthony G. M. Barrett ; Michael S. Hill ;Panayiotis A. Procopiou
Journal of the American Chemical Society 2011 Volume 134(Issue 4) pp:2193-2207
Publication Date(Web):December 22, 2011
DOI:10.1021/ja209135t
The heavier group 2 complexes [M{N(SiMe3)2}2]2(1, M = Ca; 2, M = Sr) and [M{CH(SiMe3)2}2(THF)2] (3, M = Ca; 4, M = Sr) are shown to be effective precatalysts for the intermolecular hydroamination of vinyl arenes and dienes under mild conditions. Initial studies revealed that the amide precatalysts, 1 and 2, while compromised in terms of absolute activity by a tendency toward transaminative behavior, offer greater stability toward polymerization/oligomerization side reactions. In every case the strontium species, 2 and 4, were found to outperform their calcium congeners. Reactions of piperidine with para-substituted styrenes are indicative of rate-determining alkene insertion in the catalytic cycle while the ease of addition of secondary cyclic amines was found to be dependent on ring size and reasoned to be a consequence of varying amine nucleophilicity. Hydroamination of conjugated dienes yielded isomeric products via η3-allyl intermediates and their relative distributions were explained through stereoelectronic considerations. The ability to carry out the hydroamination of internal alkynes was found to be dramatically dependent upon the identity of the alkyne substituents while reactions employing terminal alkynes resulted in the precipitation of insoluble and unreactive group 2 acetylides. The rate law for styrene hydroamination with piperidine catalyzed by [Sr{N(SiMe3)2}2]2 was deduced to be first order in [amine] and [alkene] and second order in [catalyst], while large kinetic isotope effects and group 2 element-dependent ΔS⧧ values implicated the formation of an amine-assisted rate-determining alkene insertion transition state in which there is a considerable entropic advantage associated with use of the larger strontium center.
Co-reporter:Mike Hill
Applied Organometallic Chemistry 2011 Volume 25( Issue 10) pp:
Publication Date(Web):
DOI:10.1002/aoc.1840
No abstract is available for this article.
Co-reporter:Michael S. Hill, Mary F. Mahon and Thomas P. Robinson
Chemical Communications 2010 vol. 46(Issue 14) pp:2498-2500
Publication Date(Web):28 Jan 2010
DOI:10.1039/B923172B
Reactions of triphenylphosphine oxide and diphenylphosphine oxide with calcium alkyls and amides in the presence of PhSiH3 occur to give P–C bond cleavage, P(V) to P(III) reduction and P–P coupling.
Co-reporter:Mark R. Crimmin, Michael S. Hill, Peter B. Hitchcock and Mary F. Mahon
New Journal of Chemistry 2010 vol. 34(Issue 8) pp:1572-1578
Publication Date(Web):26 Mar 2010
DOI:10.1039/C0NJ00042F
Reaction of the β-diketiminato calcium amide [{ArNC(Me)CHC(Me)NAr}Ca{N(SiMe3)2}(OEt2)] (Ar = 2,6-di-iso-propylphenyl) with triethylaluminium yields a novel calcium aluminate complex, [κ3-{ArNC(Me)C(AlEt3)HC(Me)NAr}Ca{Et4Al}], in which coordination at calcium is provided by not only bridging interactions with the ethyl groups of the aluminate anion but also a tripodal κ3-N,N,C-coordinated ligand derived from further reaction of the ‘spectator’ β-diketiminate ligand with triethylaluminium. In the solid state, this compound exists as a 1 ∶ 1 mixture of coordination isomers in which the Et4Al anion binds to calcium via either μ2- or μ3-coordination modes. Although persistent, and rapidly interconverting, at room temperature in solution, variable temperature NMR studies suggest that these two isomers undergo dissociative loss of triethylaluminium from the diketiminate ligand at low temperature.
Co-reporter:DavidJ. Liptrot;MichaelS. Hill Dr.;MaryF. Mahon Dr. ;DugaldJ. MacDougall Dr.
Chemistry - A European Journal 2010 Volume 16( Issue 28) pp:8508-8515
Publication Date(Web):
DOI:10.1002/chem.201000127
Abstract
Both homo- and heteroleptic alkyl and amide complexes of the Group 2 elements Mg and Ca are shown to be active for the catalytic dehydrocoupling of Me2NH⋅BH3. Reactions of either magnesium dialkyls or the β-diketiminate complex [HC{(Me)CN(Dipp)}2MgnBu] with four or two equivalents of Me2NHBH3, respectively, produce compounds containing the [H3BNMe2BH2Me2N]− ion, which coordinates to the magnesium centers through MgN and Mg⋅⋅⋅HB interactions in both the solution and solid states. Thermolysis of these compounds at 60 °C produces the cyclic product [(H2BNMe2)2] and, it is proposed, magnesium hydrido species by an unprecedented δ-hydride elimination process. Calcium-based species, although less reactive than their magnesium-based counterparts, are found to engage in similar dehydrocoupling reactivity and to produce a similar distribution of products under thermally promoted catalytic conditions. A mechanism for these observations is presented that involves initial production and insertion of H2BNMe2 into polarized MN bonds as the major BN bond-forming step. The efficacy of this insertion and subsequent β- or δ-hydride elimination steps is proposed to be dependent upon the charge density and polarizing capability of the participating Group 2 center, providing a rationale for the observed differences in reactivity between magnesium and calcium.
Co-reporter:Merle Arrowsmith, Michael S. Hill, and Gabriele Kociok-Köhn
Organometallics 2010 Volume 29(Issue 19) pp:4203-4206
Publication Date(Web):September 10, 2010
DOI:10.1021/om100649z
We report that reaction of group 2 dialkyl compounds, [M{CH(SiMe3)2}2(THF)n] [M = Mg, Ca, n = 2; M = Sr, Ba, n = 3] with a bis(imino)pyridine ligand ultimately results in deprotonation of both methyl groups attached to the imine carbon center. The resulting compounds are mononuclear for M = Mg or Ca, but assemble into cyclic hexameric arrays when M = Sr or Ba. In all four cases deprotonation appears to occur by a common pathway involving pyridine dearomatization and subsequent deprotonation of a single imine-bound methyl substituent. Monitoring of each reaction has revealed that the efficacy of the transformation, as well as the stability of each intermediate, is dependent upon the identity, ionic radius, and resultant charge density of the alkaline earth reagent employed.
Co-reporter:Merle Arrowsmith;MichaelS. Hill Dr.;DugaldJ. MacDougall Dr. ;MaryF. Mahon Dr.
Angewandte Chemie 2009 Volume 121( Issue 22) pp:4073-4076
Publication Date(Web):
DOI:10.1002/ange.200900878
Co-reporter:Merle Arrowsmith;MichaelS. Hill Dr.;DugaldJ. MacDougall Dr. ;MaryF. Mahon Dr.
Angewandte Chemie International Edition 2009 Volume 48( Issue 22) pp:4013-4016
Publication Date(Web):
DOI:10.1002/anie.200900878
Co-reporter:Naveed Alam, Michael S. Hill, Gabriele Kociok-Köhn, Matthias Zeller, Muhammad Mazhar and Kieran C. Molloy
Chemistry of Materials 2008 Volume 20(Issue 19) pp:6157
Publication Date(Web):September 18, 2008
DOI:10.1021/cm801330v
Compounds of the type [Ni(S2COnBu)2·(py)2] (py = pyridine derivative) have been synthesized and studied as single-source precursors for the fabrication of phase-pure thin films of rhombohedral (Millerite) NiS by aerosol-assisted chemical vapor deposition. The films have been characterized by XRD, SEM, EDX, and AFM and have been found to display a substrate-dependent morphology that is effectively independent of the precise identity of the precursor complex, regardless of minor variations in thermal stability.
Co-reporter:Anthony G. M. Barrett, Mark R. Crimmin, Michael S. Hill, Gabriele Kociok-Köhn, Jennifer R. Lachs and Panayiotis A. Procopiou
Dalton Transactions 2008 (Issue 10) pp:1292-1294
Publication Date(Web):04 Feb 2008
DOI:10.1039/B717402K
Protonolysis of the β-diketiminate calcium bis(trimethylsilyl)amide [{ArNC(Me)CHC(Me)NAr}Ca{N(SiMe3)2}(THF)] with benzylamine is reversible and forms a quantifiable equilibrium mixture.
Co-reporter:Anthony G. M. Barrett, Mark R. Crimmin, Michael S. Hill, Peter B. Hitchcock and Panayiotis A. Procopiou
Dalton Transactions 2008 (Issue 33) pp:4474-4481
Publication Date(Web):11 Jul 2008
DOI:10.1039/B717383K
A series of heteroleptic β-diketiminate-stabilised calcium amides of the form [{ArNC(Me)CHC(Me)NAr}Ca{NR1R2}(THF)] (Ar = 2,6-diisopropylphenyl; R1 = H, R2 = Ar; R1 = H, R2 = CH2CH2OMe; R1 = R2 = Ph) react with 1,3-dialkylcarbodiimides, R3NCNR3 (R3 = Cy, iPr), to yield the corresponding insertion products [{ArNC(Me)CHC(Me)NAr}Ca{(R3N)2CNR1R2}(THF)] at room temperature in hydrocarbon solutions. These latter compounds contain both β-diketiminate and guanidinate ligands bound to calcium. Solid-state data are consistent with the guanidinate ligands adopting a number of binding modes including κ2 through κ3 coordination, with varying degrees of delocalisation of the non-bound guanidinate nitrogen lone-pair across the π-framework of the ligand. DFT computational studies have been conducted to address these variations in coordination behaviour.
Co-reporter:Michael S. Hill, Peter B. Hitchcock and Ruti Pongtavornpinyo
Dalton Transactions 2008 (Issue 21) pp:2854-2860
Publication Date(Web):08 Apr 2008
DOI:10.1039/B801160E
Oxidative insertion of the In(I) ‘carbene analogues’, [In{N(Dipp)C(Me)}2CH] (Ar = Dipp = 2,6-iPr2C6H3; Ar = Mes = 2,4,6-Me3C6H2) into the Fe–I bond of [CpFe(CO)2I] occurred cleanly and under mild conditions to yield the In(III) compounds [CH{(CH3)2CN-2,6-iPr2C6H3}2In(I)FeCp(CO)2] and [CH{(CH3)2CN-2,4,6-Me3C6H3}2In(I)FeCp(CO)2], which have been fully characterised in solution and the solid state. Attempts to abstract the iodide anion from [CH{(CH3)2CN-2,6-iPr2C6H3}2In(I)FeCp(CO)2] to form cationic species containing a coordinated indium diyl were unsuccessful and resulted in a complex mixture of products from which two ionic species were isolated. Neither cation was found to contain indium by X-ray crystallographic analysis. These observations were indicative of ill-defined decomposition pathways as have been noted by previous workers. A further attempt to form a cationic iron species containing a coordinated [In{N(Dipp)C(Me)}2CH] fragment resulted in oxidation of the iron centre from Fe(II) to Fe(III), with deposition of indium metal, and the isolation of a cationic Fe(III) β-diketiminate complex.
Co-reporter:Mark R. Crimmin, Anthony G. M. Barrett, Michael S. Hill, Peter B. Hitchcock and Panayiotis A. Procopiou
Organometallics 2008 Volume 27(Issue 4) pp:497-499
Publication Date(Web):February 1, 2008
DOI:10.1021/om7011198
Amides of the heavier group 2 elements Ca, Sr, and Ba are effective precatalysts for the atom-efficient addition of phosphine P−H bonds to carbodiimides. A number of intermediates within the catalytic cycle have been identified by in situ NMR methods and by stoichiometric synthesis.
Co-reporter:Anthony G. M. Barrett, Mark R. Crimmin, Michael S. Hill, Peter B. Hitchcock, Sarah L. Lomas, Mary F. Mahon, Panayiotis A. Procopiou and Kogularamanan Suntharalingam
Organometallics 2008 Volume 27(Issue 23) pp:6300-6306
Publication Date(Web):November 5, 2008
DOI:10.1021/om800738r
The β-diketiminate-stabilized calcium amide [{ArNC(Me)CHC(Me)NAr}Ca{N(SiMe3)2}(THF)] (1) reacts with terminal acetylenes in hydrocarbon solvents to yield the corresponding calcium acetylide complexes [{ArNC(Me)CHCN(Me)Ar}Ca{C≡CR1}]2 (R1 = n-Bu, t-Bu, Ph, 4-MeC6H4, ferrocenyl, Ar = 2,6-di-isopropylphenyl, 2a−e). Although in all instances solid and solution state data were consistent with the reaction products existing as dimeric species with aggregation occurring via three-center−two-electron bridging acetylide units, a further reaction of 1 with HC≡CSi(iPr)3 demonstrated that both monomeric solvated [{ArNC(Me)CHC(Me)NAr}Ca{C≡CSi(iPr)3}(THF)2] (3b) or dimeric acetylide [{ArNC(Me)CHC(Me)NAr}Ca{C≡CSi(iPr)3}]2 (3a) species could be isolated from the reaction depending upon the exact conditions of the crystallization of the reaction product from solution. Further solution studies demonstrated the presence of a monomer−dimer equilibrium in solution. A van’t Hoff analysis allowed ΔG°(298 K) for the dimerization reaction to be calculated as +27.0 kJ mol−1. The reaction of these hydrocarbon-soluble kinetically stabilized calcium acetylides with 1,3-dialkylcarbodiimides gave the corresponding heteroleptic calcium C-propargyl amidinate complexes [{ArNC(Me)CHCN(Me)Ar}Ca{(R2N)2CC≡CR1}(THF)n] (R1 = 4-MeC6H4, n = 0, 4a; 4-MeC6H4, n = 1, 4a·THF; R2 = iPr; R1 = Si(iPr3), R2 = Cy, n = 1, 4b·THF) via insertion of the carbodiimide into the calcium−carbon σ-bond. The latter complexes have been characterized in both solution and the solid state including single-crystal X-ray analysis of 4a·THF. Extension of this reactivity to catalytic systems has allowed the application of amide 1 (5 mol %) to the catalytic hydroacetylenation of 1,3-di-isopropylcarbodiimide with phenylacetylene, yielding the corresponding propargyl amidine in 59% yield following crystallization from hexane solution.
Co-reporter:Michael S. Hill, Mary F. Mahon and Thomas P. Robinson
Chemical Communications 2010 - vol. 46(Issue 14) pp:NaN2500-2500
Publication Date(Web):2010/01/28
DOI:10.1039/B923172B
Reactions of triphenylphosphine oxide and diphenylphosphine oxide with calcium alkyls and amides in the presence of PhSiH3 occur to give P–C bond cleavage, P(V) to P(III) reduction and P–P coupling.
Co-reporter:Anthony G. M. Barrett, Mark R. Crimmin, Michael S. Hill, Gabriele Kociok-Köhn, Jennifer R. Lachs and Panayiotis A. Procopiou
Dalton Transactions 2008(Issue 10) pp:NaN1294-1294
Publication Date(Web):2008/02/04
DOI:10.1039/B717402K
Protonolysis of the β-diketiminate calcium bis(trimethylsilyl)amide [{ArNC(Me)CHC(Me)NAr}Ca{N(SiMe3)2}(THF)] with benzylamine is reversible and forms a quantifiable equilibrium mixture.
Co-reporter:Michael S. Hill, Peter B. Hitchcock and Ruti Pongtavornpinyo
Dalton Transactions 2008(Issue 21) pp:NaN2860-2860
Publication Date(Web):2008/04/08
DOI:10.1039/B801160E
Oxidative insertion of the In(I) ‘carbene analogues’, [In{N(Dipp)C(Me)}2CH] (Ar = Dipp = 2,6-iPr2C6H3; Ar = Mes = 2,4,6-Me3C6H2) into the Fe–I bond of [CpFe(CO)2I] occurred cleanly and under mild conditions to yield the In(III) compounds [CH{(CH3)2CN-2,6-iPr2C6H3}2In(I)FeCp(CO)2] and [CH{(CH3)2CN-2,4,6-Me3C6H3}2In(I)FeCp(CO)2], which have been fully characterised in solution and the solid state. Attempts to abstract the iodide anion from [CH{(CH3)2CN-2,6-iPr2C6H3}2In(I)FeCp(CO)2] to form cationic species containing a coordinated indium diyl were unsuccessful and resulted in a complex mixture of products from which two ionic species were isolated. Neither cation was found to contain indium by X-ray crystallographic analysis. These observations were indicative of ill-defined decomposition pathways as have been noted by previous workers. A further attempt to form a cationic iron species containing a coordinated [In{N(Dipp)C(Me)}2CH] fragment resulted in oxidation of the iron centre from Fe(II) to Fe(III), with deposition of indium metal, and the isolation of a cationic Fe(III) β-diketiminate complex.
Co-reporter:Anthony G. M. Barrett, Mark R. Crimmin, Michael S. Hill, Peter B. Hitchcock and Panayiotis A. Procopiou
Dalton Transactions 2008(Issue 33) pp:NaN4481-4481
Publication Date(Web):2008/07/11
DOI:10.1039/B717383K
A series of heteroleptic β-diketiminate-stabilised calcium amides of the form [{ArNC(Me)CHC(Me)NAr}Ca{NR1R2}(THF)] (Ar = 2,6-diisopropylphenyl; R1 = H, R2 = Ar; R1 = H, R2 = CH2CH2OMe; R1 = R2 = Ph) react with 1,3-dialkylcarbodiimides, R3NCNR3 (R3 = Cy, iPr), to yield the corresponding insertion products [{ArNC(Me)CHC(Me)NAr}Ca{(R3N)2CNR1R2}(THF)] at room temperature in hydrocarbon solutions. These latter compounds contain both β-diketiminate and guanidinate ligands bound to calcium. Solid-state data are consistent with the guanidinate ligands adopting a number of binding modes including κ2 through κ3 coordination, with varying degrees of delocalisation of the non-bound guanidinate nitrogen lone-pair across the π-framework of the ligand. DFT computational studies have been conducted to address these variations in coordination behaviour.


![[1,1'-Biphenyl]-4-carbonitrile, 4'-[3-(triphenylstannyl)propoxy]-](http://img.cochemist.com/ccimg/649000/648930-64-7.png)
![[1,1'-Biphenyl]-4-carbonitrile, 4'-[3-(triphenylstannyl)propoxy]-](http://img.cochemist.com/ccimg/649000/648930-64-7_b.png)


![2-Azaspiro[4.5]decane, 3-methyl-2-(phenylmethyl)-](http://img.cochemist.com/ccimg/592500/592478-45-0.png)
![2-Azaspiro[4.5]decane, 3-methyl-2-(phenylmethyl)-](http://img.cochemist.com/ccimg/592500/592478-45-0_b.png)







![BENZENEMETHANAMINE, N-[[1-(2-PROPENYL)CYCLOHEXYL]METHYL]-](http://img.cochemist.com/ccimg/592500/592478-44-9.png)
![BENZENEMETHANAMINE, N-[[1-(2-PROPENYL)CYCLOHEXYL]METHYL]-](http://img.cochemist.com/ccimg/592500/592478-44-9_b.png)
![[1,1'-Biphenyl]-4-carbonitrile, 4'-(2-propenyloxy)-](http://img.cochemist.com/ccimg/112000/111928-38-2.png)
![[1,1'-Biphenyl]-4-carbonitrile, 4'-(2-propenyloxy)-](http://img.cochemist.com/ccimg/112000/111928-38-2_b.png)




![9-BORABICYCLO[3.3.1]NONAN-9-AMINE, N-PHENYL-](http://img.cochemist.com/ccimg/120800/120789-33-5.png)
![9-BORABICYCLO[3.3.1]NONAN-9-AMINE, N-PHENYL-](http://img.cochemist.com/ccimg/120800/120789-33-5_b.png)
![9-BORABICYCLO[3.3.1]NONAN-9-AMINE, N-(1,1-DIMETHYLETHYL)-](http://img.cochemist.com/ccimg/100700/100639-37-0.png)
![9-BORABICYCLO[3.3.1]NONAN-9-AMINE, N-(1,1-DIMETHYLETHYL)-](http://img.cochemist.com/ccimg/100700/100639-37-0_b.png)




![Stannane, [5-([1,1'-biphenyl]-4-yloxy)pentyl]diiodophenyl-](http://img.cochemist.com/ccimg/649000/648930-69-2.png)
![Stannane, [5-([1,1'-biphenyl]-4-yloxy)pentyl]diiodophenyl-](http://img.cochemist.com/ccimg/649000/648930-69-2_b.png)



![Stannane, [5-([1,1'-biphenyl]-4-yloxy)pentyl]dibromophenyl-](http://img.cochemist.com/ccimg/649000/648930-67-0.png)
![Stannane, [5-([1,1'-biphenyl]-4-yloxy)pentyl]dibromophenyl-](http://img.cochemist.com/ccimg/649000/648930-67-0_b.png)
![Stannane, [5-([1,1'-biphenyl]-4-yloxy)pentyl]dibromobutyl-](http://img.cochemist.com/ccimg/649000/648930-70-5.png)
![Stannane, [5-([1,1'-biphenyl]-4-yloxy)pentyl]dibromobutyl-](http://img.cochemist.com/ccimg/649000/648930-70-5_b.png)
![5,8-Decadienoic acid, 10-[3-(2Z)-2-octenyloxiranyl]-, (5Z,8Z)-](http://img.cochemist.com/ccimg/201000/200960-01-6.png)
![5,8-Decadienoic acid, 10-[3-(2Z)-2-octenyloxiranyl]-, (5Z,8Z)-](http://img.cochemist.com/ccimg/201000/200960-01-6_b.png)




![Benzyl-[2-(4-methoxy-phenyl)-ethyl]-amine](http://img.cochemist.com/ccimg/51800/51713-72-5.png)
![Benzyl-[2-(4-methoxy-phenyl)-ethyl]-amine](http://img.cochemist.com/ccimg/51800/51713-72-5_b.png)

![Potassium, [bis(trimethylsilyl)methyl]-](http://img.cochemist.com/ccimg/118200/118111-55-0.png)
![Potassium, [bis(trimethylsilyl)methyl]-](http://img.cochemist.com/ccimg/118200/118111-55-0_b.png)
![Stannane, [5-([1,1'-biphenyl]-4-yloxy)pentyl]butyldiphenyl-](http://img.cochemist.com/ccimg/649000/648930-65-8.png)
![Stannane, [5-([1,1'-biphenyl]-4-yloxy)pentyl]butyldiphenyl-](http://img.cochemist.com/ccimg/649000/648930-65-8_b.png)



![Stannane, [4-([1,1'-biphenyl]-4-yloxy)butyl]triphenyl-](http://img.cochemist.com/ccimg/649000/648930-62-5.png)
![Stannane, [4-([1,1'-biphenyl]-4-yloxy)butyl]triphenyl-](http://img.cochemist.com/ccimg/649000/648930-62-5_b.png)


![Stannane, [5-([1,1'-biphenyl]-4-yloxy)pentyl]triphenyl-](http://img.cochemist.com/ccimg/649000/648930-63-6.png)
![Stannane, [5-([1,1'-biphenyl]-4-yloxy)pentyl]triphenyl-](http://img.cochemist.com/ccimg/649000/648930-63-6_b.png)
![Stannane, [3-([1,1'-biphenyl]-4-yloxy)propyl]triphenyl-](http://img.cochemist.com/ccimg/649000/648930-61-4.png)
![Stannane, [3-([1,1'-biphenyl]-4-yloxy)propyl]triphenyl-](http://img.cochemist.com/ccimg/649000/648930-61-4_b.png)












![Benzene, 1,1'-[(1E)-1-buten-3-yne-1,4-diyl]bis-](http://img.cochemist.com/ccimg/13400/13343-79-8.png)
![Benzene, 1,1'-[(1E)-1-buten-3-yne-1,4-diyl]bis-](http://img.cochemist.com/ccimg/13400/13343-79-8_b.png)









![1,3,2-Dioxaborolane, 2,2'-[1,4-butanediylbis(oxy)]bis[4,4,5,5-tetramethyl-](/data/chemimg/1170600/26621-57-8.png)
![1,3,2-Dioxaborolane, 2,2'-[1,4-butanediylbis(oxy)]bis[4,4,5,5-tetramethyl-](/data/chemimg/1170600/26621-57-8_b.png)





























![2,6-Bis[1-(2,6-di-i-propylphenylimino)ethyl]pyridine](/data/chemimg/6300/204203-14-5.png)
![2,6-Bis[1-(2,6-di-i-propylphenylimino)ethyl]pyridine](/data/chemimg/6300/204203-14-5_b.png)

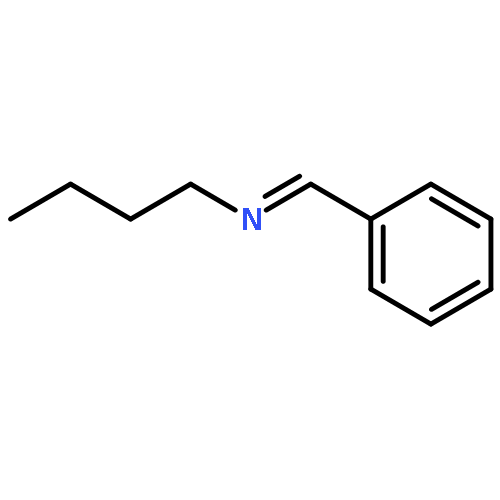
![Silanediamine, N,N'-bis[2,6-bis(1-methylethyl)phenyl]-1-phenyl-](/data/chemimg/139900/1414859-92-9.png)
![Silanediamine, N,N'-bis[2,6-bis(1-methylethyl)phenyl]-1-phenyl-](/data/chemimg/139900/1414859-92-9_b.png)


![Silanamine, N-[2,6-bis(1-methylethyl)phenyl]-1,1-diphenyl-](/data/chemimg/139900/1414859-89-4.png)
![Silanamine, N-[2,6-bis(1-methylethyl)phenyl]-1,1-diphenyl-](/data/chemimg/139900/1414859-89-4_b.png)
![Benzenamine,N-[2,6-bis(1-methylethyl)phenyl]-2-[[[2,6-bis(1-methylethyl)phenyl]imino]methyl]-](http://img.cochemist.com/ccimg/330700/330666-08-5.png)
![Benzenamine,N-[2,6-bis(1-methylethyl)phenyl]-2-[[[2,6-bis(1-methylethyl)phenyl]imino]methyl]-](http://img.cochemist.com/ccimg/330700/330666-08-5_b.png)
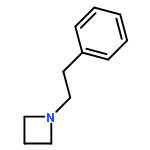



![2-Azaspiro[4.5]decane, 3-methyl-](http://img.cochemist.com/ccimg/592500/592478-37-0.png)
![2-Azaspiro[4.5]decane, 3-methyl-](http://img.cochemist.com/ccimg/592500/592478-37-0_b.png)


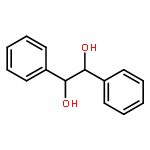
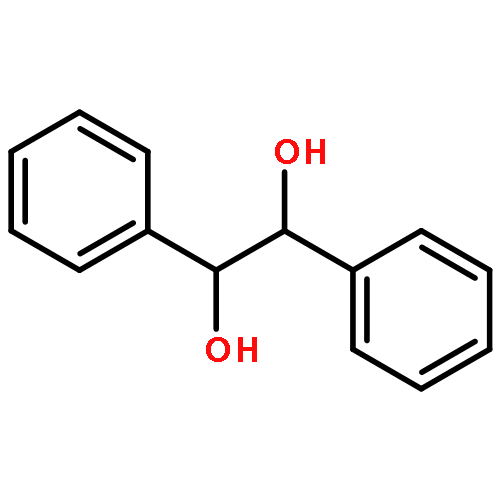
![Tris[N,N-bis(trimethylsilyl)amide]yttrium (III)](http://img.cochemist.com/ccimg/41900/41836-28-6.png)
![Tris[N,N-bis(trimethylsilyl)amide]yttrium (III)](http://img.cochemist.com/ccimg/41900/41836-28-6_b.png)















![Benzenamine, N-[3-[[2,6-bis(1-methylethyl)phenyl]amino]-1-methyl-2-buten-1-ylidene]-2,6-bis(1-methylethyl)-](/data/chemimg/120200/181708-81-6.png)
![Benzenamine, N-[3-[[2,6-bis(1-methylethyl)phenyl]amino]-1-methyl-2-buten-1-ylidene]-2,6-bis(1-methylethyl)-](/data/chemimg/120200/181708-81-6_b.png)