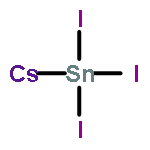
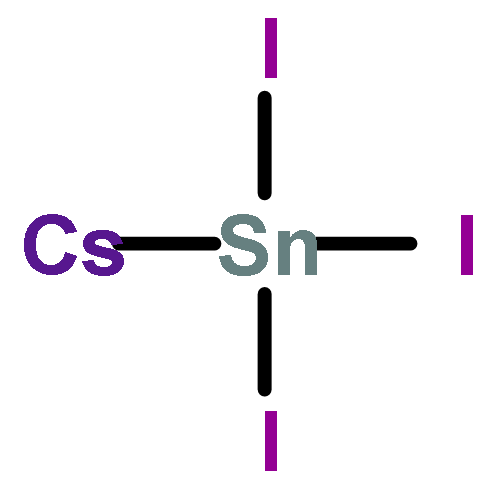
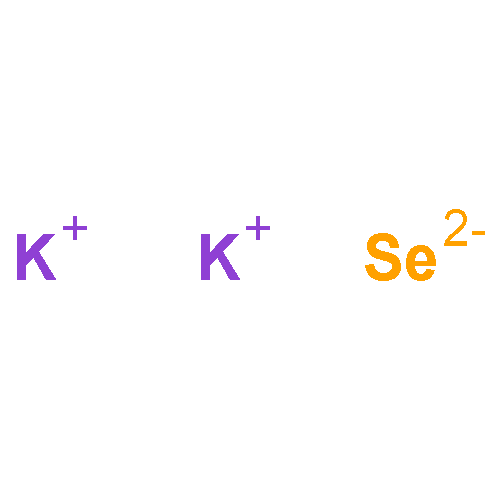Co-reporter:Yuan Yin, Yang Huang, Yelong Wu, Guangde Chen, Wan-Jian Yin, Su-Huai Wei, and Xingao Gong
Chemistry of Materials November 14, 2017 Volume 29(Issue 21) pp:9429-9429
Publication Date(Web):October 15, 2017
DOI:10.1021/acs.chemmater.7b03507
Because of the stability and toxic issue of CH3NH3PbI3, great efforts have been made to search emerging materials beyond perovskite. Most of the explorations are based on ns2-containing compounds, because lone-pair s-orbital-derived antibonding states are believed to play a crucial role in unique properties of CH3NH3PbI3. In this work, we chose skutterudite-structure IrSb3 (Eg ≈ 1.3 eV) as a case study to show that the strong antibonding character at valence band maximum (VBM) can appear without the contribution from lone-pair s orbital. First-principles calculations show that IrSb3 possesses similar electronic properties as CH3NH3PbI3: (i) ambipolar conductivity with much better electron and hole effective masses than that of CH3NH3PbI3; (ii) strong optical absorption (∼1 × 104 cm–1); (iii) shallow dominating defects. More importantly, IrSb3 is much more stable than CH3NH3PbI3. Our work may shed light on searching new promising solar cell materials beyond ns2-containing perovskite.
Co-reporter:Yan Pan, Xiaoying Dai, Stefano de Gironcoli, Xin-Gao Gong, Gian-Marco Rignanese, Aihui Zhou
Journal of Computational Physics 2017 Volume 348(Volume 348) pp:
Publication Date(Web):1 November 2017
DOI:10.1016/j.jcp.2017.07.033
•Propose three parallel orbital-updating based plane-wave basis methods for electronic structure calculations.•These new methods can avoid the generating of large scale eigenvalue problems and then reduce the computational cost.•These new methods allow for two-level parallelization which is particularly interesting for large scale parallelization.•Numerical experiments show that these new methods are reliable and efficient for large scale calculations on modern supercomputers.Motivated by the recently proposed parallel orbital-updating approach in real space method [1], we propose a parallel orbital-updating based plane-wave basis method for electronic structure calculations, for solving the corresponding eigenvalue problems. In addition, we propose two new modified parallel orbital-updating methods. Compared to the traditional plane-wave methods, our methods allow for two-level parallelization, which is particularly interesting for large scale parallelization. Numerical experiments show that these new methods are more reliable and efficient for large scale calculations on modern supercomputers.
Co-reporter:Cheng-Yan Liu;Zhi-Ming Li;Hong-Yang Gu;Shi-You Chen;Hongjun Xiang
Advanced Energy Materials 2017 Volume 7(Issue 8) pp:
Publication Date(Web):2017/04/01
DOI:10.1002/aenm.201601457
It is well known that sodium at grain boundaries (GBs) increases the photovoltaic efficiencies of CuInSe2 and Cu2ZnSnS4 significantly. However, the mechanism of how sodium influences the GBs is still unknown. Based on the recently proposed self-passivation rule, it is found that the dangling bonds in the GBs can completely be saturated through doping the Na, thus GB states are successfully passivated. It is shown that the Na can easily incorporate into the GB with very low formation energy. Although Cu can also passivate the GB states, it requires a copper rich condition which, however, suppresses the formation of copper vacancies in the bulk and thus decreases the concentration of hole carriers, so copper passivation is practically not as beneficial as sodium. The present work reveals the mechanism about how the Na enhances the photovoltaic performance through passivating the dangling bonds in the GBs of chalcogenide semiconductors, and sheds light on how to passivate dangling bonds in GBs with alterative processes.
Co-reporter:Yue-Yu Zhang, Shiyou Chen, Hongjun Xiang, Xin-Gao Gong
Carbon 2016 Volume 109() pp:246-252
Publication Date(Web):November 2016
DOI:10.1016/j.carbon.2016.08.015
Carbon is a versatile element that has allotropes with both sp2 (graphene) and sp3 (diamond) bonding. However, none of the allotropes can be used as light-absorber materials in solar cells due to either too large or too small band gap. Here, we propose a novel concept that enables a tunable band gap of carbon phases with sp2 carbon atoms within a sp3 carbon structure. The tunability is due to the quantum confinement effect. By embedding the sp2 atoms within the sp3 structure, we can design new carbon allotropes with ideal optical properties for optoelectronic applications. Five carbon allotropes incorporated this structural feature were identified by combining this new concept with our freshly developed multi-objective inverse band structure design approach. They all have proper band gaps for optical absorption, and the simulated photovoltaic efficiency of C10-C is even higher than conventional absorber materials such as GaAs, which indicates that C10-C with mixed sp2sp3 hybridization may have potential application as light-absorber material in electronic and optoelectronic devices.
Co-reporter:Jing Tang, Yueyu Zhang, Biao Kong, Yongcheng Wang, Peimei Da, Jun Li, Ahmed A. Elzatahry, Dongyuan Zhao, Xingao Gong, and Gengfeng Zheng
Nano Letters 2014 Volume 14(Issue 5) pp:2702-2708
Publication Date(Web):April 17, 2014
DOI:10.1021/nl500608w
We report a nitrogen-doped carbon nanodot (N-Cdot)/TiO2 nanowire photoanode for solar-driven, real-time, and sensitive photoelectrochemical probing of the cellular generation of H2S, an important endogenous gasotransmitter based on a tunable interfacial charge carrier transfer mechanism. Synthesized by a microwave-assisted solvothermal method and subsequent surface chemical conjugation, the obtained N-Cdot/TiO2 nanowire photoanode shows much enhanced photoelectrochemical photocurrent compared with pristine TiO2 nanowires. This photocurrent increase is attributed to the injection of photogenerated electrons from N-Cdots to TiO2 nanowires, confirmed by density functional theory simulation. In addition, the charge transfer efficiency is quenched by Cu2+, whereas the introduction of H2S or S2– ions resets the charge transfer and subsequently the photocurrent, thus leading to sensitive photoelectrochemical recording of the H2S level in buffer and cellular environments. Moreover, this N-Cdot-TiO2 nanowire photoanode has been demonstrated for direct growth and interfacing of H9c2 cardiac myoblasts, with the capability of interrogating H2S cellular generation pathways by vascular endothelial growth factor stimulation as well as inhibition.
Co-reporter:Congcong Wang, Shiyou Chen, Ji-Hui Yang, Li Lang, Hong-Jun Xiang, Xin-Gao Gong, Aron Walsh, and Su-Huai Wei
Chemistry of Materials 2014 Volume 26(Issue 11) pp:3411
Publication Date(Web):May 15, 2014
DOI:10.1021/cm500598x
Through element substitution in Cu2ZnSnS4, a class of kesterite-structured I2–II–IV–VI4 semiconductors can be designed as novel functional materials. Using the first-principles calculations, we show that this element-substitution design is thermodynamically limited, that is, although I2–II–IV–VI4 with I = Cu, Ag, II = Zn, Cd, Hg, IV = Si, Ge, Sn, and VI = S, Se, Te are stable quaternary compounds, those with II = Mg, Ca, Sr, Ba, IV =Ti, Zr, Hf, and VI = O are unstable against the phase-separation into the competing binary and ternary compounds. Three main phase-separation pathways are revealed. In general, we show that if the secondary II–VI or I2–IV–VI3 phases prefer to have nontetrahedral structures, then the I2–II–IV–VI4 semiconductors tend to phase separate. This finding can be used as a guideline for future design of new quaternary semiconductors.
Co-reporter:Peng Xu, Shiyou Chen, Hong-Jun Xiang, Xin-Gao Gong, and Su-Huai Wei
Chemistry of Materials 2014 Volume 26(Issue 20) pp:6068
Publication Date(Web):September 23, 2014
DOI:10.1021/cm503122j
CsSnI3 is a prototype inorganic halide perovskite that has recently been proposed as a strong candidate for photovoltaic applications because of its unique semiconductor properties. Through first-principle calculations, we show that the concentration control of intrinsic defects is critical for optimizing the photovoltaic properties of CsSnI3. Under a Sn-poor condition, a high concentration of acceptor defects, such as Sn or Cs vacancies, can form easily and produce a high p-type conductivity and deep-level defects that can become electron–hole recombination centers, all with high energy. This condition is optimal for growing CsSnI3 as hole-transport material in solar cells. In contrast, when Sn becomes richer, the concentration of acceptor defects decreases; therefore, the p-type conductivity may drop to a moderate level, which can increase the shunt resistance and, thus, the efficiency of the solar cells with CsSnI3 as the light absorber material (LAM). However, under the Sn-rich condition, the concentration of a deep-level donor defect SnI will increase, causing electron traping and non-radiative electron–hole recombination. Therefore, we propose that a moderately Sn-rich condition is optimal when CsSnI3 is used as the LAM. The defect properties of CsSnI3 are general, and the underlying chemistry is expected to be applicable to other halide perovskite semiconductors.
Co-reporter:Zheng-Lu Li, Zhi-Ming Li, Hai-Yuan Cao, Ji-Hui Yang, Qiang Shu, Yue-Yu Zhang, H. J. Xiang and X. G. Gong
Nanoscale 2014 vol. 6(Issue 8) pp:4309-4315
Publication Date(Web):03 Feb 2014
DOI:10.1039/C3NR06823D
We have developed a new global optimization method for the determination of the interface structure based on the differential evolution algorithm. Here, we applied this method to search for the ground state atomic structures of the grain boundary (GB) between armchair and zigzag oriented graphene. We find two new grain boundary structures with a considerably lower formation energy of about 1 eV nm−1 than those of the previously widely used structural models. We also systematically investigate the symmetric GBs with the GB angle ranging from 0° to 60°, and find some new GB structures. Surprisingly, for an intermediate GB angle, the formation energy does not depend monotonically on the defect concentration. We also discovered an interesting linear relationship between the GB density and the GB angle. Our new method provides an important novel route for the determination of GB structures and other interface structures, and our comprehensive study on GB structures could provide new structural information and guidelines to this area.
Co-reporter:Hou-Zun Chen ; Yue-Yu Zhang ; Xingao Gong ;Hongjun Xiang
The Journal of Physical Chemistry C 2014 Volume 118(Issue 5) pp:2333-2337
Publication Date(Web):January 14, 2014
DOI:10.1021/jp411437f
TiO2 has been extensively studied due to the possible application in solar cells and photoelectrochemical (PEC) water-splitting. However, the energy conversion efficiency is rather low because of the large band gaps (larger than 3.0 eV) of rutile and anatase TiO2. Here we introduce the multiobjective differential evolution (MODE) method as a novel global optimization algorithm to predict new polymorphs of bulk TiO2 with better optical properties than rutile and anatase TiO2. The band gaps of the new PI (Pnma) and CI (C2) phases are found to be 1.95 and 2.64 eV. The calculation of formation energy, phonon dispersions, and thermal stability shows that the two novel phases are dynamically and thermally stable. These new TiO2 polymorphs with better electronic and optical properties may pave a new way for high-efficiency solar energy conversion.
Co-reporter:Xin Zhao ; Qiang Shu ; Manh Cuong Nguyen ; Yangang Wang ; Min Ji ; Hongjun Xiang ; Kai-Ming Ho ; Xingao Gong ;Cai-Zhuang Wang
The Journal of Physical Chemistry C 2014 Volume 118(Issue 18) pp:9524-9530
Publication Date(Web):April 15, 2014
DOI:10.1021/jp5010852
Information about the atomic structures at solid–solid interfaces is crucial for understanding and predicting the performance of materials. Due to the complexity of the interfaces, it is very challenging to resolve their atomic structures using either experimental techniques or computer simulations. In this paper, we present an efficient first-principles computational method for interface structure prediction based on an adaptive genetic algorithm. This approach significantly reduces the computational cost, while retaining the accuracy of first-principles prediction. The method is applied to the investigation of both stoichiometric and nonstoichiometric SrTiO3 Σ3(112)[1̅10] grain boundaries with unit cell containing up to 200 atoms. Several novel low-energy structures are discovered, which provide fresh insights into the structure and stability of the grain boundaries.
Co-reporter:Ji-Hui Yang ; Yingteng Zhai ; Hengrui Liu ; Hongjun Xiang ; Xingao Gong ;Su-Huai Wei
Journal of the American Chemical Society 2012 Volume 134(Issue 30) pp:12653-12657
Publication Date(Web):July 9, 2012
DOI:10.1021/ja303892a
First-principles calculations were performed to study the structural and optoelectronic properties of the newly synthesized nonisovalent and lattice-matched (Si2)0.6(AlP)0.4 alloy (Watkins, T.; et al. J. Am. Chem. Soc.2011, 133, 16212). We found that the most stable structure of Si3AlP is a superlattice along the ⟨111⟩ direction with separated AlP and Si layers, which has a similar optical absorption spectrum to silicon. The ordered C1c1-Si3AlP is found to be the most stable one among all structures with a basic unit of one P atom surrounded by three Si atoms and one Al atom, in agreement with experimental suggestions.(1) We predict that C1c1-Si3AlP has good optical properties, i.e., it has a larger fundamental band gap and a smaller direct band gap than Si; thus, it has much higher absorption in the visible light region. The calculated properties of Si3AlP suggest that it is a promising candidate for improving the performance of the existing Si-based solar cells. The understanding on the stability and band structure engineering obtained in this study is general and can be applied for future study of other nonisovalent and lattice-matched semiconductor alloys.
Co-reporter:Hai-Yuan Cao, Hongjun Xiang, Xin-Gao Gong
Solid State Communications 2012 Volume 152(Issue 19) pp:1807-1810
Publication Date(Web):October 2012
DOI:10.1016/j.ssc.2012.07.013
We have investigated the lattice thermal transport across the asymmetric tilt grain boundary between armchair and zigzag graphene by nonequilibrium molecular dynamics (NEMD). We have observed significant temperature drop and ultra-low temperature-dependent thermal boundary resistance. More importantly, we find an unexpected thermal rectification phenomenon. The thermal conductivity and Kapitza conductance is direction-dependent. The effect of thermal rectification could be amplified by increasing the difference of temperature imposed on two sides. Our results propose a promising kind of thermal rectifier and phonon diodes based on polycrystalline graphene without delicate manipulation of the atomic structure.Highlights► We investigate thermal transport across asymmetric grain boundary using NEMD. ► Ultra-low temperature-dependent thermal boundary resistance was observed. ► Unexpected ultra-high thermal rectification was discovered. ► The thermal rectification could be tuned by changing temperature difference. ► A new method to design nanoscale thermal rectifier was proposed.
Co-reporter:Yingteng Zhai ; Alessandro Laio ; Erio Tosatti
Journal of the American Chemical Society 2011 Volume 133(Issue 8) pp:2535-2540
Publication Date(Web):February 3, 2011
DOI:10.1021/ja1076316
We introduce an approach for the accurate calculation of thermal properties of classical nanoclusters. On the basis of a recently developed enhanced sampling technique, replica exchange metadynamics, the method yields the true free energy of each relevant cluster structure, directly sampling its basin and measuring its occupancy in full equilibrium. All entropy sources, whether vibrational, rotational anharmonic, or especially configurational, the latter often forgotten in many cluster studies, are automatically included. For the present demonstration, we choose the water nonamer (H2O)9, an extremely simple cluster, which nonetheless displays a sufficient complexity and interesting physics in its relevant structure spectrum. Within a standard TIP4P potential description of water, we find that the nonamer second relevant structure possesses a higher configurational entropy than the first, so that the two free energies surprisingly cross for increasing temperature.
Co-reporter:Xiao Gu ; Jin-long Liu ; Ji-hui Yang ; Hong-jun Xiang ; Xin-gao Gong ;Yong-yao Xia
The Journal of Physical Chemistry C 2011 Volume 115(Issue 25) pp:12672-12676
Publication Date(Web):June 3, 2011
DOI:10.1021/jp202846p
The electrochemical stability of layer-structured LiCoO2 in a Li+-containing aqueous electrolyte solution is critically dependent on the solution pH. The capacity fades upon cycling in electrolyte solutions below pH 11. We have investigated the detailed atomic-scale mechanism of the failure of LiCoO2 in the presence of H+ using first-principles methods. In layer-structured LiCoO2, lithium ion diffusion paths are two-dimensional channels between the cobalt–oxygen layers. However, in an aqueous electrolyte solution containing a considerable number of H+ ions, H+ will be transported into the cathodes to replace the Li+ ions. Our calculations show that once the H+ ions are intercalated into the LixCoO2 cathode, they may covalently bond to the oxygen ions, thereby decreasing the capacity of the cathodes. We have also found that such hydrogen intercalation increases barriers to the diffusion of lithium ions. Therefore, the channels would be blocked after a sufficient number of H+ ions have intercalated, typically after a few cycles.
Co-reporter:Wan-Jian Yin, Xiao Gu, Xin-Gao Gong
Solid State Communications 2008 Volume 147(7–8) pp:323-326
Publication Date(Web):August 2008
DOI:10.1016/j.ssc.2008.05.039
Motivated by the recent discovery of cage-like metal clusters, a shell jellium model is proposed to study the stability of cage-like clusters, using the density functional theory (DFT) with local density approximation (LDA). Based on the shell jellium model, it is found that certain metal clusters of a special number of electrons are even more stable than the sphere-like clusters described by the conventional spherical jellium model. The result shows two new magic numbers 32 and 90. These results provide us with a straightforward explanation for the stability of 32-electron clusters.
Co-reporter:Wei Huang, Min Ji, Chuan-Ding Dong, Xiao Gu, Lei-Ming Wang, Xin Gao Gong and Lai-Sheng Wang
ACS Nano 2008 Volume 2(Issue 5) pp:897
Publication Date(Web):May 1, 2008
DOI:10.1021/nn800074b
The atomic structures of bare gold clusters provide the foundation to understand the enhanced catalytic properties of supported gold nanoparticles. However, the richness of diverse structures and the strong relativistic effects have posed considerable challenges for a systematic understanding of gold clusters with more than 20 atoms. We use photoelectron spectroscopy of size-selected anions, in combination with first principles calculations, to elucidate the structures of gold nanoclusters in a critical size regime from 55 to 64 atoms (1.1−1.3 nm in diameter). Au55− is found to be a nonicosahedral disordered cluster as a result of relativistic effects that induce strong surface contractions analogous to bulk surface reconstructions, whereas low-symmetry core−shell-type structures are found for Au56− to Au64−. Au58 exhibits a major electron-shell closing and is shown to possess a low-symmetry, but nearly spherical structure with a large energy gap. Clear spectroscopic and computational evidence has been observed, showing that Au58− is a highly robust cluster and additional atoms are simply added to its surface from Au59− to Au64− without inducing significant structural changes. The unique low-symmetry structures characteristic of gold nanoclusters due to the strong relativistic effects allow abundant surface defects sites, providing a key structure−function relationship to understand the catalytic capabilities of gold nanoparticles.Keywords: density functional theory; electronic structure; gold clusters; photoelectron spectroscopy; structure−function relationship
Co-reporter:Min Ji, Xiao Gu, Xi Li, Xingao Gong, Jun Li,Lai-Sheng Wang
Angewandte Chemie International Edition 2005 44(43) pp:7119-7123
Publication Date(Web):
DOI:10.1002/anie.200502795
Co-reporter:Min Ji;Xiao Gu;Xi Li;Xingao Gong Dr.;Jun Li Dr.;Lai-Sheng Wang Dr.
Angewandte Chemie 2005 Volume 117(Issue 43) pp:
Publication Date(Web):11 OCT 2005
DOI:10.1002/ange.200502795
Ein Herz aus Gold: Ein Vergleich photoelektronenspektroskopischer Ergebnisse mit denen von Rechnungen deckte die Struktur des Au32−-Clusters auf. Zwar legten DFT-Rechnungen eine hochsymmetrische leere Käfigstruktur (Ih, links) als Energieminimum bei 0 K nahe, das berechnete Spektrum für einen wenig symmetrischen, verzerrten Käfig (C1, rechts), in den drei Au-Atome eingeschlossen sind, stimmt jedoch am besten mit den experimentellen Daten überein.
Co-reporter:X.M Duan, X.G Gong
Computational Materials Science 2003 Volume 27(Issue 3) pp:375-380
Publication Date(Web):May 2003
DOI:10.1016/S0927-0256(03)00042-9
The hyper molecular dynamics method (Hyper-MD) with a local bias potential can be used in massive simulations on infrequent events with a very small computation overhead. In this paper, we demonstrate the validity of the local bias potential in simulation of various systems, and study how the results depend on the locality of the bias potential. For an adatom diffusion on surface or interstitial diffusion in bulk, we find that a local bias potential only related to the neighbors of the interesting atoms is good enough. Our studies also show that the Hyper-MD with a local bias potential can be used to study the surface diffusion with exchange mechanism.