Co-reporter:Robert B. Laprairie, Pushkar M. Kulkarni, Jeffrey R. Deschamps, Melanie E. M. Kelly, David R. Janero, Maria G. Cascio, Lesley A. Stevenson, Roger G. Pertwee, Terrence P. Kenakin, Eileen M. Denovan-Wright, and Ganesh A. Thakur
ACS Chemical Neuroscience June 21, 2017 Volume 8(Issue 6) pp:1188-1188
Publication Date(Web):January 19, 2017
DOI:10.1021/acschemneuro.6b00310
The cannabinoid 1 receptor (CB1R) is one of the most widely expressed metabotropic G protein-coupled receptors in brain, and its participation in various (patho)physiological processes has made CB1R activation a viable therapeutic modality. Adverse psychotropic effects limit the clinical utility of CB1R orthosteric agonists and have promoted the search for CB1R positive allosteric modulators (PAMs) with the promise of improved drug-like pharmacology and enhanced safety over typical CB1R agonists. In this study, we describe the synthesis and in vitro and ex vivo pharmacology of the novel allosteric CB1R modulator GAT211 (racemic) and its resolved enantiomers, GAT228 (R) and GAT229 (S). GAT211 engages CB1R allosteric site(s), enhances the binding of the orthosteric full agonist [3H]CP55,490, and reduces the binding of the orthosteric antagonist/inverse agonist [3H]SR141716A. GAT211 displayed both PAM and agonist activity in HEK293A and Neuro2a cells expressing human recombinant CB1R (hCB1R) and in mouse-brain membranes rich in native CB1R. GAT211 also exhibited a strong PAM effect in isolated vas deferens endogenously expressing CB1R. Each resolved and crystallized GAT211 enantiomer showed a markedly distinctive pharmacology as a CB1R allosteric modulator. In all biological systems examined, GAT211’s allosteric agonist activity resided with the R-(+)-enantiomer (GAT228), whereas its PAM activity resided with the S-(−)-enantiomer (GAT229), which lacked intrinsic activity. These results constitute the first demonstration of enantiomer-selective CB1R positive allosteric modulation and set a precedent whereby enantiomeric resolution can decisively define the molecular pharmacology of a CB1R allosteric ligand.Keywords: 7-transmembrane receptor; Allosteric regulation; cannabinoid; cellular signaling; central nervous system; G-protein-coupled receptor; ligand bias; molecular pharmacology; probe dependence; receptor activation; therapeutics discovery;
Co-reporter:Abhijit R. Kulkarni, Sumanta Garai, and Ganesh A. Thakur
The Journal of Organic Chemistry 2017 Volume 82(Issue 2) pp:
Publication Date(Web):December 14, 2016
DOI:10.1021/acs.joc.6b02521
We report a facile, microwave-accelerated, one-pot tandem synthesis of unsymmetrical ureas via a Curtius rearrangement. In this method, one-pot microwave irradiation of commercially available (hetero)aromatic acids and amines in the presence of diphenylphosphoryl azide enabled extremely rapid (1–5 min) construction of an array of unsymmetrical ureas in good to excellent yields. We demonstrate the utility of our method in the efficient, gram-scale synthesis of key biologically active compounds targeting the cannabinoid 1 and α7 nicotinic acetylcholine receptors.
Co-reporter:Pushkar M. Kulkarni; Abhijit R. Kulkarni; Anisha Korde; Ritesh B. Tichkule; Robert B. Laprairie$; Eileen M. Denovan-Wright$; Han Zhou; David R. Janero; Nikolai Zvonok; Alexandros Makriyannis; Maria G. Cascio; Roger G. Pertwee;Ganesh A. Thakur
Journal of Medicinal Chemistry 2016 Volume 59(Issue 1) pp:44-60
Publication Date(Web):November 3, 2015
DOI:10.1021/acs.jmedchem.5b01303
Undesirable side effects associated with orthosteric agonists/antagonists of cannabinoid 1 receptor (CB1R), a tractable target for treating several pathologies affecting humans, have greatly limited their translational potential. Recent discovery of CB1R negative allosteric modulators (NAMs) has renewed interest in CB1R by offering a potentially safer therapeutic avenue. To elucidate the CB1R allosteric binding motif and thereby facilitate rational drug discovery, we report the synthesis and biochemical characterization of first covalent ligands designed to bind irreversibly to the CB1R allosteric site. Either an electrophilic or a photoactivatable group was introduced at key positions of two classical CB1R NAMs: Org27569 (1) and PSNCBAM-1 (2). Among these, 20 (GAT100) emerged as the most potent NAM in functional assays, did not exhibit inverse agonism, and behaved as a robust positive allosteric modulator of binding of orthosteric agonist CP55,940. This novel covalent probe can serve as a useful tool for characterizing CB1R allosteric ligand-binding motifs.
Co-reporter:Deniz Bagdas;Jenny L Wilkerson;Abhijit Kulkarni;Wisam Toma;Shakir AlSharari;Zulfiye Gul;Aron H Lichtman;Roger L Papke;Ganesh A Thakur;M Imad Damaj
British Journal of Pharmacology 2016 Volume 173( Issue 16) pp:2506-2520
Publication Date(Web):
DOI:10.1111/bph.13528
Background and Purpose
Orthosteric agonists and positive allosteric modulators (PAMs) of the α7 nicotinic ACh receptor (nAChR) represent novel therapeutic approaches for pain modulation. Moreover, compounds with dual function as allosteric agonists and PAMs, known as ago-PAMs, add further regulation of receptor function.
Experimental Approach
Initial studies examined the α7 ago-PAM, GAT107, in the formalin, complete Freund's adjuvant (CFA), LPS inflammatory pain models, the chronic constriction injury neuropathic pain model and the tail flick and hot plate acute thermal nociceptive assays. Additional studies examined the locus of action of GAT107 and immunohistochemical markers in the dorsal horn of the spinal cord in the CFA model.
Key Results
Complementary pharmacological and genetic approaches confirmed that the dose-dependent antinociceptive effects of GAT107 were mediated through α7 nAChR. However, GAT107 was inactive in the tail flick and hot plate assays. In addition, GAT107 blocked conditioned place aversion elicited by acetic acid injection. Furthermore, intrathecal, but not intraplantar, injections of GAT107 reversed nociception in the CFA model, suggesting a spinal component of action. Immunohistochemical evaluation revealed an increase in the expression of astrocyte-specific glial fibrillary acidic protein and phosphorylated p38MAPK within the spinal cords of mice treated with CFA, which was attenuated by intrathecal GAT107 treatment. Importantly, GAT107 did not elicit motor impairment and continued to produce antinociceptive effects after subchronic administration in both phases of the formalin test.
Conclusions and Implications
Collectively, these results provide the first proof of principle that α7 ago-PAMs represent an effective pharmacological strategy for treating inflammatory and neuropathic pain.
Co-reporter:Robert B. Laprairie, Abhijit R. Kulkarni, Pushkar M. Kulkarni, Dow P. Hurst, Diane Lynch, Patricia H. Reggio, David R. Janero, Roger G. Pertwee, Lesley A. Stevenson, Melanie E. M. Kelly, Eileen M. Denovan-Wright, and Ganesh A. Thakur
ACS Chemical Neuroscience 2016 Volume 7(Issue 6) pp:776
Publication Date(Web):April 4, 2016
DOI:10.1021/acschemneuro.6b00041
One of the most abundant G-protein coupled receptors (GPCRs) in brain, the cannabinoid 1 receptor (CB1R), is a tractable therapeutic target for treating diverse psychobehavioral and somatic disorders. Adverse on-target effects associated with small-molecule CB1R orthosteric agonists and inverse agonists/antagonists have plagued their translational potential. Allosteric CB1R modulators offer a potentially safer modality through which CB1R signaling may be directed for therapeutic benefit. Rational design of candidate, druglike CB1R allosteric modulators requires greater understanding of the architecture of the CB1R allosteric endodomain(s) and the capacity of CB1R allosteric ligands to tune the receptor’s information output. We have recently reported the synthesis of a focused library of rationally designed, covalent analogues of Org27569 and PSNCBAM-1, two prototypic CB1R negative allosteric modulators (NAMs). Among the novel, pharmacologically active CB1R NAMs reported, the isothiocyanate GAT100 emerged as the lead by virtue of its exceptional potency in the [35S]GTPγS and β-arrestin signaling assays and its ability to label CB1R as a covalent allosteric probe with significantly reduced inverse agonism in the [35S]GTPγS assay as compared to Org27569. We report here a comprehensive functional profiling of GAT100 across an array of important downstream cell-signaling pathways and analysis of its potential orthosteric probe-dependence and signaling bias. The results demonstrate that GAT100 is a NAM of the orthosteric CB1R agonist CP55,940 and the endocannabinoids 2-arachidonoylglycerol and anandamide for β-arrestin1 recruitment, PLCβ3 and ERK1/2 phosphorylation, cAMP accumulation, and CB1R internalization in HEK293A cells overexpressing CB1R and in Neuro2a and STHdhQ7/Q7 cells endogenously expressing CB1R. Distinctively, GAT100 was a more potent and efficacious CB1R NAM than Org27569 and PSNCBAM-1 in all signaling assays and did not exhibit the inverse agonism associated with Org27569 and PSNCBAM-1. Computational docking studies implicate C7.38(382) as a key feature of GAT100 ligand-binding motif. These data help inform the engineering of newer-generation, druggable CB1R allosteric modulators and demonstrate the utility of GAT100 as a covalent probe for mapping structure–function correlates characteristic of the druggable CB1R allosteric space.Keywords: 7-transmembrane receptor; Allosteric covalent probe; allosteric site; biased signaling; binding domain; cannabinoid 1 receptor; cysteine; functional selectivity; G protein-coupled receptors; homology modeling; isothiocyanate; ligand bias; ligand-binding motif; negative allosteric modulator; signal transduction; therapeutics discovery
Co-reporter:Ganesh A. Thakur ; Abhijit R. Kulkarni ; Jeffrey R. Deschamps ;Roger L. Papke
Journal of Medicinal Chemistry 2013 Volume 56(Issue 21) pp:8943-8947
Publication Date(Web):October 3, 2013
DOI:10.1021/jm401267t
An expeditious microwave-assisted synthesis of 4BP-TQS, its enantiomeric separation, and their functional evaluation is reported. Electrophysiological characterization in Xenopus oocytes revealed that activity exclusively resided in the (+)-enantiomer 1b (GAT107) and (−)-enantiomer 1a did not affect its activity when coapplied. X-ray crystallography studies revealed the absolute stereochemistry of 1b to be 3aR,4S,9bS. 1b represents the most potent ago-PAM of α7 nAChRs available to date and is considered for further in vivo evaluation.
Co-reporter:Ganesh A. Thakur ; Shama Bajaj ; Carol Paronis ; Yan Peng ; Anna L. Bowman ; Lawrence S. Barak ; Marc G. Caron ; Demon Parrish ; Jeffrey R. Deschamps ;Alexandros Makriyannis
Journal of Medicinal Chemistry 2013 Volume 56(Issue 10) pp:3904-3921
Publication Date(Web):April 26, 2013
DOI:10.1021/jm4000775
In previous studies, compound 1 (AM411), a 3-(1-adamantyl) analogue of the phytocannabinoid (−)-Δ8-tetrahydrocannabinol (Δ8-THC), was shown to have improved affinity and selectivity for the CB1 receptor. In this work, we further explored the role of the 1-adamantyl group at the C-3 position in a series of tricyclic cannabinoid analogues modified at the 9-northern aliphatic hydroxyl (NAH) position. Of these, 9-hydroxymethyl hexahydrocannabinol 11 (AM4054) exhibited high CB1 affinity and full agonist profile. In the cAMP assay, the 9-hydroxymethyl cannabinol analogue 24 (AM4089) had a partial agonist profile, with high affinity and moderate selectivity for rCB1 over hCB2. In vivo results in rat models of hypothermia and analgesia were congruent with in vitro data. Our in vivo data indicate that 3-(1-adamantyl) substitution, within NAH cannabinergics, imparts improved pharmacological profiles when compared to the corresponding, traditionally used 3-dimethylheptyl analogues and identifies 11 and 24 as potentially useful in vivo CB1 cannabinergic probes.
Co-reporter:Abhijit R. Kulkarni, Ganesh A. Thakur
Tetrahedron Letters 2013 Volume 54(Issue 48) pp:6592-6595
Publication Date(Web):27 November 2013
DOI:10.1016/j.tetlet.2013.09.107
We report here an efficient and expeditious microwave-assisted synthesis of cyclopentadiene ring-fused tetrahydroquinolines using the three-component Povarov reaction catalyzed by indium (III) chloride. This method has an advantage of shorter reaction time (10–15 min) with high and reproducible yields (up to 90%) and is suitable for parallel library synthesis. The optimization process is reported and the results from the microwave route are compared with those of the conventional synthetic route. In almost all cases, the microwave acceleration consistently provided improved yields favoring the cis-diastereomer.

![1-(4-Chloro-phenyl)-3-[3-(6-pyrrolidin-1-yl-pyridin-2-yl)-phenyl]-urea](http://img.cochemist.com/ccimg/877300/877202-74-9.png)
![1-(4-Chloro-phenyl)-3-[3-(6-pyrrolidin-1-yl-pyridin-2-yl)-phenyl]-urea](http://img.cochemist.com/ccimg/877300/877202-74-9_b.png)
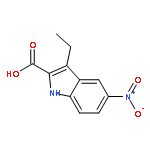
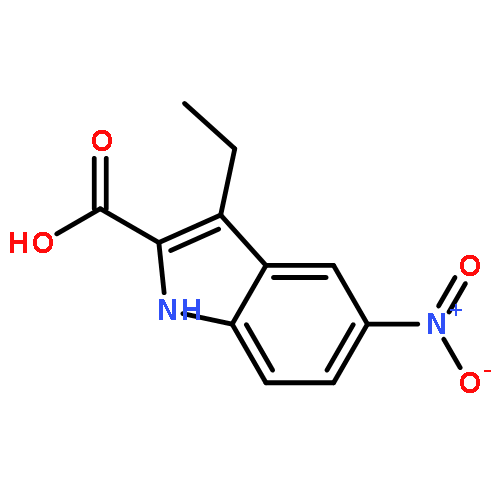


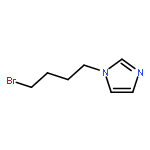
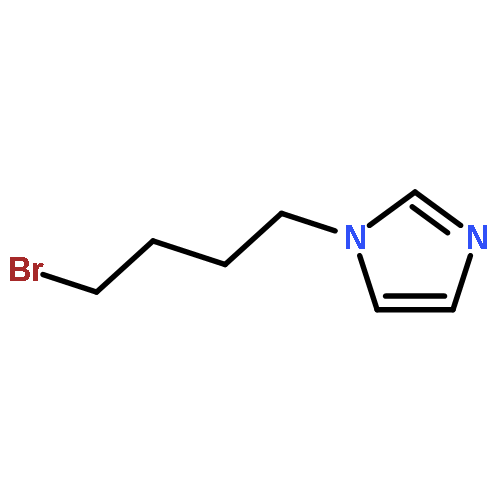


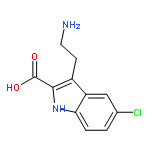
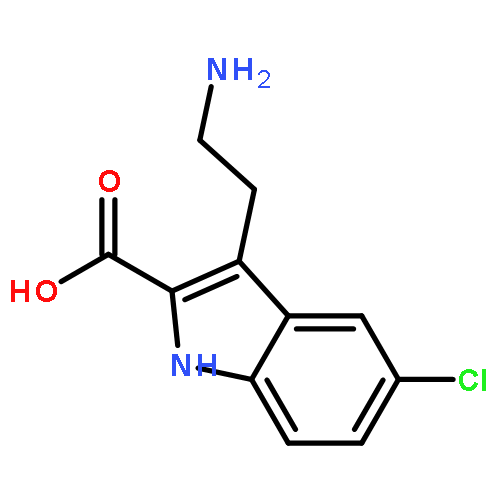
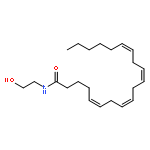
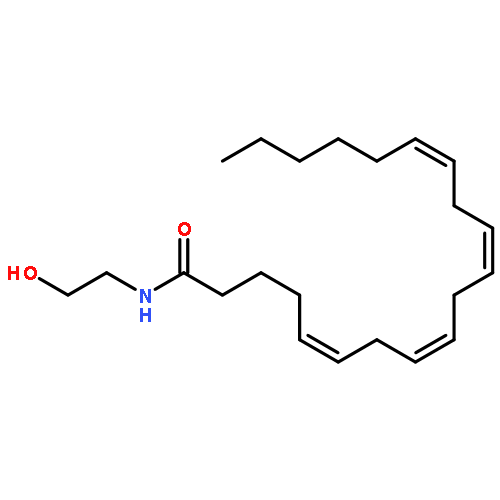
![Phenol, 5-(1,1-dimethylheptyl)-2-[(1R,2R,5R)-5-hydroxy-2-(3-hydroxypropyl)cyclohexyl]-, rel-](/data/chemimg/1338600/83003-12-7.png)
![Phenol, 5-(1,1-dimethylheptyl)-2-[(1R,2R,5R)-5-hydroxy-2-(3-hydroxypropyl)cyclohexyl]-, rel-](/data/chemimg/1338600/83003-12-7_b.png)




![1H-Indole-2-carboxylic acid,5-chloro-3-[2-(1,3-dihydro-1,3-dioxo-2H-isoindol-2-yl)ethyl]-, ethyl ester](http://img.cochemist.com/ccimg/55800/55747-56-3.png)
![1H-Indole-2-carboxylic acid,5-chloro-3-[2-(1,3-dihydro-1,3-dioxo-2H-isoindol-2-yl)ethyl]-, ethyl ester](http://img.cochemist.com/ccimg/55800/55747-56-3_b.png)
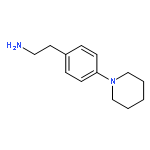



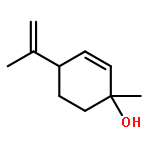
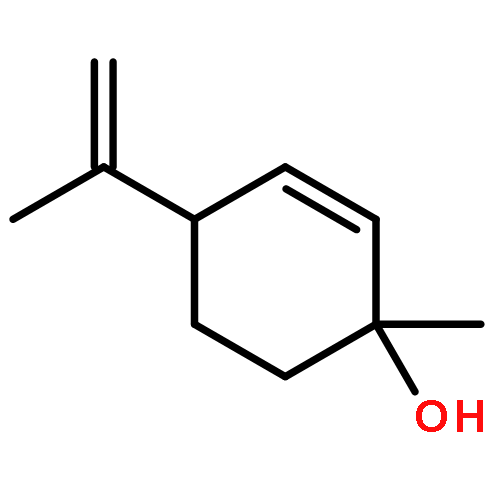
![6-chloro-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indol-1-one](http://img.cochemist.com/ccimg/18000/17952-83-9.png)
![6-chloro-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indol-1-one](http://img.cochemist.com/ccimg/18000/17952-83-9_b.png)
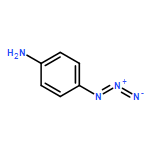

![9-Octadecenoic acid(9Z)-,1,1'-[(1R)-1-[[[(2-aminoethoxy)hydroxyphosphinyl]oxy]methyl]-1,2-ethanediyl]ester](http://img.cochemist.com/ccimg/4100/4004-05-1.png)
![9-Octadecenoic acid(9Z)-,1,1'-[(1R)-1-[[[(2-aminoethoxy)hydroxyphosphinyl]oxy]methyl]-1,2-ethanediyl]ester](http://img.cochemist.com/ccimg/4100/4004-05-1_b.png)