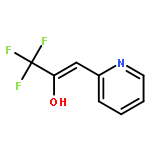
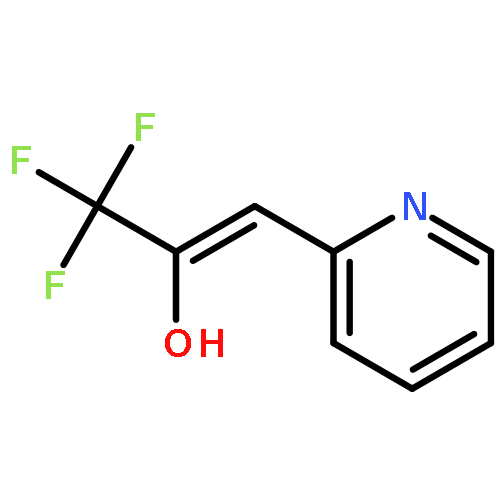
Co-reporter:Mathias Schäfer, Katrin Peckelsen, Mathias Paul, Jonathan Martens, Jos Oomens, Giel Berden, Albrecht Berkessel, and Anthony J. H. M. Meijer
Journal of the American Chemical Society April 26, 2017 Volume 139(Issue 16) pp:5779-5779
Publication Date(Web):March 10, 2017
DOI:10.1021/jacs.6b10348
While hydrogen tunneling at elevated temperatures has, for instance, often been postulated in biochemical processes, spectroscopic proof is thus far limited to cryogenic conditions, under which thermal reactivity is negligible. We report spectroscopic evidence for H-tunneling in the gas phase at temperatures around 320–350 K observed in the isomerization reaction of a hydroxycarbene into an aldehyde. The charge-tagged carbene was generated in situ in a tandem mass spectrometer by decarboxylation of oxo[4-(trimethylammonio)phenyl]acetic acid upon collision induced dissociation. All ion structures involved are characterized by infrared ion spectroscopy and quantum chemical calculations. The charge-tagged phenylhydroxycarbene undergoes a 1,2-H-shift to the corresponding aldehyde with an half-life of about 10 s, evidenced by isomer-selective two-color (IR-IR) spectroscopy. In contrast, the deuterated (OD) carbene analogue showed much reduced 1,2-D-shift reactivity with an estimated half-life of at least 200 s under the experimental conditions, and provides clear evidence for hydrogen atom tunneling in the H-isotopologue. This is the first spectroscopic confirmation of hydrogen atom tunneling governing 1,2-H-shift reactions at noncryogenic temperatures, which is of broad significance for a range of (bio)chemical processes, including enzymatic transformations and organocatalysis.
Co-reporter:Katrin Peckelsen;Jonathan Martens;Lisa Czympiel;Jos Oomens;Giel Berden;Dirk Gründemann;Anthony J. H. M. Meijer;Mathias Schäfer
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 34) pp:23362-23372
Publication Date(Web):2017/08/30
DOI:10.1039/C7CP03843G
L-Ergothioneine (ET) is a sulfur-containing derivative of the amino acid histidine that offers unique antioxidant properties. The enzyme independent redox-chemistry of ET relies on the availability of the thiol tautomer to allow oxidative formation of disulfide bridges, i.e., the tautomeric equilibrium. To study the intrinsic properties of ET the tautomeric equilibrium is studied in the gas-phase by infrared multiphoton dissociation (IRMPD) spectroscopy. The IR ion spectra of isolated molecular ions of ET and of the biosynthetic precursors of ET, i.e., hercynine and Nε-methyl-hercynine are acquired. The analyte structures are independently investigated by density functional theory (DFT) and computed linear IR-spectra of tautomer ion structures are compared with the gas-phase spectra for identification. For the molecular ion of ET the simulated IR spectra of thione and thiol structures match the recorded IRMPD spectrum and that prevents an individual structure assignment. On the other hand, theory suggests that ET adopts a thione tautomer in MeOH solution which could be carried over from the condensed phase to gas phase and could be kinetically trapped after effective electrospray phase transfer and desolvation. Such a non-thermal behavior is also found for the molecular ions of protonated hercynine and Nε-methyl-hercynine. Contrary to that, the sodium complex ions of ET, hercynine and Nε-methyl-hercynine adopt the respective ground structures predicted by theory, which are reliably identified spectroscopically. For ET the thione tautomer is by far the most stable isomer in the sodium complex molecular ion.
Co-reporter:Christoph Hage;Christian H. Ihling
Journal of The American Society for Mass Spectrometry 2017 Volume 28( Issue 1) pp:56-68
Publication Date(Web):2017 January
DOI:10.1007/s13361-016-1426-9
We have synthesized a homobifunctional amine-reactive cross-linking reagent, containing a TEMPO (2,2,6,6-tetramethylpiperidine-1-oxy) and a benzyl group (Bz), termed TEMPO-Bz-linker, to derive three-dimensional structural information of proteins. The aim for designing this novel cross-linker was to facilitate the mass spectrometric analysis of cross-linked products by free radical initiated peptide sequencing (FRIPS). In an initial study, we had investigated the fragmentation behavior of TEMPO-Bz-derivatized peptides upon collision activation in (+)-electrospray ionization collision-induced dissociation tandem mass spectrometry (ESI-CID-MS/MS) experiments. In addition to the homolytic NO-C bond cleavage FRIPS pathway delivering the desired odd-electron product ions, an alternative heterolytic NO-C bond cleavage, resulting in even-electron product ions mechanism was found to be relevant. The latter fragmentation route clearly depends on the protonation of the TEMPO-Bz-moiety itself, which motivated us to conduct (−)-ESI-MS, CID-MS/MS, and MS3 experiments of TEMPO-Bz-cross-linked peptides to further clarify the fragmentation behavior of TEMPO-Bz-peptide molecular ions. We show that the TEMPO-Bz-linker is highly beneficial for conducting FRIPS in negative ionization mode as the desired homolytic cleavage of the NO–C bond is the major fragmentation pathway. Based on characteristic fragments, the isomeric amino acids leucine and isoleucine could be discriminated. Interestingly, we observed pronounced amino acid side chain losses in cross-linked peptides if the cross-linked peptides contain a high number of acidic amino acids.
Co-reporter:Mareike C. Holl;Giel Berden;Jos Oomens;Anthony J. H. M. Meijer;Mathias Schäfer;Ryan Gilmour
European Journal of Organic Chemistry 2014 Volume 2014( Issue 26) pp:5675-5680
Publication Date(Web):
DOI:10.1002/ejoc.201402845
Abstract
Herein we report the first application of infrared multiple-photon dissociation (IRMPD) spectroscopy to study noncovalent interactions in organocatalysis. Phenylalanine-derived iminium ions, central to numerous organocatalytic processes, display dynamic conformational behavior as a consequence of stabilizing noncovalent interactions (e.g., CH–π, π–π). Electronic modulation of the aryl ring causes notable variation in the conformation; this can be detected spectroscopically and correlated with enantioselectivity. Given that these interactions, which orchestrate stereoinduction, encode for specific conformers (I, II, or III), a diagnostic IRMPD spectrum is generated: the C=O stretching frequency of the imidazole carbonyl group serves as a diagnostic marker. The calculated conformers and their respective spectra can be compared with experimental data. Consequently, valuable insight into the ubiquitous noncovalent interactions associated with MacMillan-catalyst-derived α,β-unsaturated iminium ions can be obtained in the absence of solvent or counterion effects. A preliminary structure–catalysis correlation is disclosed, thus demonstrating the potential of this approach for studying reactive intermediates and facilitating catalyst design.
Co-reporter:Francesco Falvo, Lukas Fiebig, Mathias Schäfer
International Journal of Mass Spectrometry 2013 Volumes 354–355() pp:26-32
Publication Date(Web):15 November 2013
DOI:10.1016/j.ijms.2013.04.012
•Azo-containing compound used as a CID-labile reagent for chemical cross-linking (XL).•CID of peptide-azo-peptide ions yields N2 loss and open shell peptide product ions.•ESI-MSn of interpeptide and intrapeptide XL demonstrates proof-of-principle.•The distonic nature of peptide radical cations is proved by ion-molecule reactions.4,4′-Azobis[4-cyanopentanoic] acid, a symmetrical free radical initiator, is transformed in the bis-N-succinimidyl-active ester (azoXL) and utilized for peptide cross-linking (XL). The azoXL-reagent reacts with amine functionalities and performs upon collision induced dissociation (CID) exclusively the loss of N2 leading to the formation of two radical sites. (+)ESI-MSn product-ion spectra of the protonated molecular ion of two model peptides (MRFA and RKDVY) interconnected N-terminally by 4,4′-azobis[4-cyanopentanoic] acid show characteristically mass-shifted product ions and neutral losses (N2 = 28 Da; 2-methylacrylonitrile = 67 Da), potentially useful for identification of XL derivatized peptides. Additionally, CID fragmentation reactions lead to the formation of sequence-specific fragment ions, allowing primary structure elucidation. The presented results demonstrate the potential of azo-compounds as CID-labile XL-reagents for tandem MS analysis. The fragmentation behaviour of peptides (MRFA, RKDVY and Substance P) derivatized with the azoXL reagent is studied by MSn, exact ion mass measurements and ion-molecule reactions (IMRs) with allyliodide and dimethyl disulfide.
Co-reporter:Lukas Fiebig, Julian Kuttner, Gerhard Hilt, Martin C. Schwarzer, Gernot Frenking, Hans-Günther Schmalz, and Mathias Schäfer
The Journal of Organic Chemistry 2013 Volume 78(Issue 20) pp:10485-10493
Publication Date(Web):September 17, 2013
DOI:10.1021/jo402001g
In situ-formed cobalt(I) complexes are proposed to act as efficient catalysts in regioselective Diels–Alder reactions of unactivated substrates such as 1,3-dienes and alkynes. We report the first experimental evidence for the in situ reduction of CoBr2(dppe) [dppe = 1,2-bis(diphenylphosphino)ethane] by Zn/ZnI2 to [Co(I)(dppe)]+ by means of electrospray MSn experiments. Additionally, the reactivities of Co(II) and Co(I) dppe complexes toward the Diels–Alder substrates isoprene and phenylacetylene were probed in gas-phase ion/molecule reactions (IMRs). Isoprene and phenylacetylene were introduced into the mass spectrometer via the buffer gas flow of a linear ion trap. The IMR experiments revealed a significantly higher substrate affinity of [Co(I)(dppe)]+ compared with [Co(II)Br(dppe)]+. Furthermore, the central intermediate of the solution-phase cobalt-catalyzed Diels–Alder reaction, [Co(I)(dppe)(isoprene)(phenylacetylene)]+, could be generated via IMR and examined in the gas phase. Collision activation of this complex ion delivered evidence for the gas-phase reaction of isoprene with phenylacetylene in the coordination sphere of the cobalt ion. The experimental findings are consistent with the results of quantum-chemical calculations on all of the observed Co(I) dppe complex ions. The results constitute strong analytical evidence for the formation and importance of different cobalt(I) species in regioselective Diels–Alder reactions of unactivated substrates and identify [Co(I)(dppe)]+ as the active Diels–Alder catalyst.
Co-reporter:Lisa Brückmann;Dr. Wiel Tyrra; Dr. Sanjay Mathur;Dr. Giel Berden; Dr. Jos Oomens;Dr. Anthony J. H. M. Meijer;Dr. Mathias Schäfer
ChemPhysChem 2012 Volume 13( Issue 8) pp:2037-2045
Publication Date(Web):
DOI:10.1002/cphc.201200132
Abstract
A series of aluminium complex ions with trifluoromethyl-heteroarylalkenolato (TMHA) ligands are studied by gas-phase infrared multiphoton-dissociation (IRMPD) spectroscopy and computational modelling. The selected series of aluminium TMHA complex ions are promising species for the initial study of intrinsic binding characteristics of AlIII cations in the gas phase as corresponding molecular ions. They are readily available for examination by (+) and (−) electrospray ionization mass spectrometry (ESI-MS) by spraying of [Al3+⋅(L−)3] solutions. The complex ions under investigation contain trivalent Al3+ cations with two chelating anionic enolate ligands, [Al3+⋅(L−)2]+, providing insights in the nature of the heteroatom-Al bonds. Additionally, the structure of a deprotonated benzimidazole ligand, L−, and an anionic complex ion of AlIII with two doubly deprotonated benzimidazole ligands, [Al3+⋅(L2−)2]−, are examined by (−)ESI-IRMPD spectroscopy. Experimental and computational results are highly consistent and allow a reliable identification of the ion structures. In all complex ions examined the planar TMHA ligands are oriented perpendicular to each other around the metal ion, leading to a tetrahedral coordination sphere in which aluminium interacts with the enolate oxygen and heteroaryl nitrogen atoms available in each of the bidentate ligands.
Co-reporter:Francesco Falvo, Lukas Fiebig, Frank Dreiocker, Ran Wang, P.B. Armentrout, Mathias Schäfer
International Journal of Mass Spectrometry 2012 s 330–332() pp: 124-133
Publication Date(Web):
DOI:10.1016/j.ijms.2012.06.023
Co-reporter:Ahmad R. Massah, Frank Dreiocker, Richard F. W. Jackson, Barry T. Pickup, Jos Oomens, Anthony J. H. M. Meijer and Mathias Schäfer
Physical Chemistry Chemical Physics 2011 vol. 13(Issue 29) pp:13255-13267
Publication Date(Web):22 Jun 2011
DOI:10.1039/C1CP20747D
An extensive set of organozinc iodides, useful for Negishi-type cross-coupling reactions, are investigated as respective cations after formal loss of iodide in the gas phase. Firstly, two new alkylzinc compounds derived from Tyrosine (Tyr) and Tryptophan (Trp) are closely examined. Secondly, the influence of specific protecting groups on the subtle balance between intra- and intermolecular coordination of zinc in these reagents is probed through trifluoroacetyl (TFA)-derivatized alkylzinc compounds. Finally, the influence of the strongly coordinating bidentate ligand N,N,N′,N′-tetramethylethylenediamine (TMEDA) on the structure of alkylzinc cations is further explored in order to better understand the stability of the respective complexes towards water. A combination of electrospray (ESI)-MS/MS, accurate ion mass measurements, infrared multiple-photon dissociation (IRMPD) spectroscopy and computational modelling allowed the full characterisation of all dimethylformamide (DMF)-solvated and TMEDA-coordinated alkylzinc cations in the gas phase. The calculations indicate that the zinc cation in gas-phase alkylzinc–DMF or TMEDA–complex ions preferentially adopts a tetrahedral coordination sphere with four ligands. Additionally, conformers with only three binding partners bound to zinc but with effectively combined hydrogen-bond interactions are also found. Collision induced dissociation (CID) patterns demonstrate that the zinc–DMF interaction in tetrahedral four-coordinate mono-DMF–zinc complex ions as well as the interaction between TMEDA and zinc in the corresponding complex ions is even stronger than typical covalent bonds. In most cases, all major features of the IRMPD spectra are consistent with only a single major isomer, allowing secured identification and assignment.
Co-reporter:Lukas Fiebig, Hans-Günther Schmalz, Mathias Schäfer
International Journal of Mass Spectrometry 2011 Volume 308(2–3) pp:307-310
Publication Date(Web):1 December 2011
DOI:10.1016/j.ijms.2011.07.013
The Heck reaction of isoprene with (p-iodophenyl)-trimethylammonium iodide was conducted in the gas phase. Relevant species of the catalytic cycle including the ionic product of the Pd-catalyzed coupling reaction were reliably characterized by exact ion mass measurements and by characteristic product ions generated by tandem-MS.Graphical abstractHighlights► Labile Heck coupling reaction intermediates and the coupling product are investigated in the gas phase. ► Charge-tagging, ESI-MSn, ion/molecule reactions and exact ion mass measurements are performed. ► A LTQ-Orbitrap Instrument is modified for the study. ► The results suggest that the Heck reaction proceeds via the accepted mechanism in solution and in the gas phase.
Co-reporter:Mathias Q. Müller, Frank Dreiocker, Christian H. Ihling, Mathias Schäfer and Andrea Sinz
Analytical Chemistry 2010 Volume 82(Issue 16) pp:6958
Publication Date(Web):July 22, 2010
DOI:10.1021/ac101241t
Chemical cross-linking combined with a subsequent enzymatic cleavage of the created cross-linked complex and a mass spectrometric analysis of the resulting cross-linked peptide mixture presents an alternative approach to high-resolution analysis, such as NMR spectroscopy or X-ray crystallography, to obtain low-resolution protein structures and to gain insight into protein interfaces. Here, we describe a novel urea-based cross-linker, which allows distinguishing different cross-linking products by collision-induced dissociation (CID) tandem MS experiments based on characteristic product ions and constant neutral losses. The novel cross-linker is part of our ongoing efforts in developing collision-induced dissociative reagents that allow an efficient analysis of cross-linked proteins and protein complexes. Our innovative analytical concept is exemplified for the Munc13-1 peptide and the recombinantly expressed ligand binding domain of the peroxisome proliferator-activated receptor α, for which cross-linking reaction mixtures were analyzed both by offline nano-HPLC/MALDI-TOF/TOF mass spectrometry and by online nano-HPLC/nano-ESI-LTQ-orbitrap mass spectrometry. The characteristic fragment ion patterns of the novel cross-linker greatly simplify the identification of different cross-linked species, namely, modified peptides as well as intrapeptide and interpeptide cross-links, from complex mixtures and drastically reduce the potential of identifying false-positive cross-links. Our novel urea-based CID cleavable cross-linker is expected to be highly advantageous for analyzing protein 3D structures and protein−protein complexes in an automated manner.
Co-reporter:Miriam K. Drayß, P.B. Armentrout, Jos Oomens, Mathias Schäfer
International Journal of Mass Spectrometry 2010 Volume 297(1–3) pp:18-27
Publication Date(Web):1 November 2010
DOI:10.1016/j.ijms.2010.04.010
Gas-phase structures of alkali metal cationized (Li+, Na+, K+, Rb+, and Cs+) proline (Pro) and N-methyl alanine have been investigated using infrared multiple photon dissociation (IRMPD) spectroscopy utilizing light generated by a free electron laser and computational modeling. Measured IRMPD spectra are compared to spectra calculated at the B3LYP/6-311++G(2d,2p) level of theory to identify individual conformers. Calculations indicate that the stability of the salt bridge (SB; zwitterionic) conformer relative to the most stable canonical structure with a single formal charge site (charge solvation; CS) of aliphatic amino acids (e.g., Pro, N-methyl alanine, N-methyl glycine, and glycine) does not increase with size and polarizability of the alkali metal cations, in contrast to the trend commonly found for functionalized amino acids. In fact, the relative stability of SB over CS conformers reaches a maximum at [amino acid + Na]+. A uniform SB structure and two characteristic CS conformers are identified by theory to be relevant for alkali metalized Pro, N-methyl alanine, and N-methyl glycine. For CS structures, the alkali metal cation is either coordinated to the nitrogen and the carbonyl oxygen of the acid functionality (Li+, Na+) or is solely interacting with the carboxylic acid oxygens (K+, Rb+, and Cs+).The IRMPD spectra exhibit clearly distinguishable bands for the CO stretching modes of the carboxylic acid moiety in CS structures and for the carboxylate moiety in SB structures, allowing reliable structure assignments for all complexes investigated. The IRMPD spectra clearly exhibit the presence of mixed populations of SB and CS structures with the contribution of CS increasing toward the larger metal cations, in good agreement with the predictions from computational modeling. The special trend regarding formation and stability of individual gas-phase ion structures of aliphatic amino acids, lacking functionalized α-side chains, can be rationalized with the concept of hard and soft Lewis acids and bases. Furthermore, calculations show that the trends with metal cation size found for aliphatic amino acids with secondary amines are similar for ordinary aliphatic amino acids (Gly, Ala).Stability trend of charge-solvated versus salt-bridge structures in alkali metal cationization is reversed for aliphatic versus functionalized amino acids as shown by IRMPD-spectroscopy and computational modeling.
Co-reporter:Miriam K. Drayß, Dirk Blunk, Jos Oomens, Nick Polfer, Carsten Schmuck, Bing Gao, Thomas Wyttenbach, Michael T. Bowers, Mathias Schäfer
International Journal of Mass Spectrometry 2009 Volume 281(1–2) pp:97-100
Publication Date(Web):15 March 2009
DOI:10.1016/j.ijms.2008.12.011
Sodium and lithium adduct ions of a synthetic guanidiniocarbonylpyrrole-derivative are examined in the gas phase. A wavelength tunable free electron laser (FEL) was used for photo-dissociation spectroscopy experiments in the infrared (1400–1800 cm−1). The photo-dissociation spectra are compared to calculated IR spectra of structures identified by theory. All photo-dissociation spectra acquired are strikingly similar, indicating that all ions adopt analogous gas-phase structures. Additionally, the respective sodium adduct ions were examined with ion mobility mass spectrometry (IMS). Although computational efforts succeeded in finding a charge solvated conformer that matched both the photo-dissociation spectra and the IMS data, the predicted global minimum was a salt-bridge structure.
Co-reporter:Mathias Schäfer, Miriam K. Drayss, Dirk Blunk, Jeremiah M. Purcell, Christopher L. Hendrickson, Alan G. Marshall, Abhigya Mookherjee and P. B. Armentrout
The Journal of Physical Chemistry A 2009 Volume 113(Issue 27) pp:7779-7783
Publication Date(Web):June 16, 2009
DOI:10.1021/jp903232y
Dissociation kinetics of the K+ loss reaction of three potassiated tertiary amino acids (Scheme 1) were studied by infrared multiple photon dissociation (IRMPD) in a Fourier transform ion cyclotron resonance (FT ICR)-MS instrument. The aim of the study was to probe if a kinetic study by IRMPD can yield useful information on the ion structure of the precursor ion species. The measured activation energy values determined by IRMPD are related to the potassium affinity, ΔHK+, of N-methyl proline determined by threshold collision-induced dissociation experiments. By appropriate scaling with this reference value, the experimentally determined activation energy values for the K+ loss are transformed into respective potassium affinities, ΔHK+IRMPD. These values match the calculated potassium affinity values for salt bridge (SB) structures, ΔHK+SB, substantially better than those for canonical structures with a single formal charge site (charge solvation (CS)), thereby allowing structure identification. This conclusion is consistent with other spectroscopic data, which yielded unambiguous evidence of these tertiary amino acids adopting SB structures in the gas phase. This study demonstrates that IRMPD can be applied to determine individual ion structures in the gas phase, given that adequate reference values are available for proper scaling.
Co-reporter:Miriam K. Drayβ, Dirk Blunk, Jos Oomens, Bing Gao, Thomas Wyttenbach, Michael T. Bowers and Mathias Schäfer
The Journal of Physical Chemistry A 2009 Volume 113(Issue 34) pp:9543-9550
Publication Date(Web):July 28, 2009
DOI:10.1021/jp903036t
The gas-phase structures of a series of potassiated tertiary amino acids have been systematically investigated using infrared multiple photon dissociation (IRMPD) spectroscopy utilizing light generated by a free electron laser, ion mobility spectrometry (IMS), and computational modeling. The examined analytes comprise a set of five linear N,N-dimethyl amino acids derived from N,N-dimethyl glycine and three cyclic N-methyl amino acids including N-methyl proline. The number of methylene groups in either the alkyl chain of the linear members or in the ring of the cyclic members of the series is gradually varied. The spectra of the cyclic potassiated molecular ions are similar and well resolved, whereas the clear signals in the respective spectra of the linear analytes increasingly overlap with longer alkyl chains. Measured IRMPD spectra are compared to spectra calculated at the B3LYP/6-311++G(2d,2p) level of theory to identify the structures present in the experimental studies. On the basis of these experiments and calculations, all potassiated molecular ions of this series adopt salt bridge structures in the gas phase, involving bidentate coordination of the potassium cation to the carboxylate moiety. The assigned salt bridge structures are predicted to be the global minima on the potential energy surfaces. IMS cross-section measurements of the potassiated systems show a monotonic increase with growing system size, suggesting that the precursor ions adopt the same type of structure and comparisons between experimental and theoretical cross sections are consistent with salt bridge structures and the IRMPD results.
Co-reporter:Miriam K. Drayss, Dirk Blunk, Jos Oomens and Mathias Schäfer
The Journal of Physical Chemistry A 2008 Volume 112(Issue 47) pp:11972-11974
Publication Date(Web):November 4, 2008
DOI:10.1021/jp809111b
The structure of proline in [proline + K]+ has been investigated in the gas phase using high level DFT and MP2 calculations and infrared photo dissociation spectroscopy with a free electron laser (FELIX). The respective FELIX spectrum of [proline + K]+ matches convincingly the calculated spectra of two structurally closely related and nearly iso-energetic zwitterionic salt bridge (SB) structures. An additional unresolved band at ∼1725 cm−1 matching with the characteristic CO stretching mode of charge solvation (CS) structures points toward the presence of a minor population of these conformers of proline in [proline + K]+. However, theory predicts a significant energy gap of 18.9 kJmol−1 (B3LYP/6-311++G(2d,2p)) or 15.6 kJ mol−1 (MP2) between the lowest CS conformer of proline and the clearly favored SB structure.
Co-reporter:Miriam Drayß;Mathias Schäfer;Andreas Springer;Philipp Zacharias;Klaus Meerholz
European Journal of Organic Chemistry 2007 Volume 2007(Issue 31) pp:5162-5174
Publication Date(Web):28 AUG 2007
DOI:10.1002/ejoc.200700199
The ion formation mechanism in electrospray MS is reviewed, with special focus on the electrochemical red/ox reactions responsible for the formation of radical molecular ions. Prerequisites influencing the likelihood of formation and observation of a particular compound as an open-shell molecular species in ESI-MS (i.e., the structure and the oxidation potential of the analyte, the solvent and additives) are evaluated. For illustration of the ESI phenomena governing radical cation formation, an ESI-MS study of tetra(aryl)benzidine compounds is presented. The facile formation of abundant radical molecular cations in ESI-MS demonstrates imposingly that the basicity of the analyte's nitrogen atoms is strongly overcompensated by the ability to stabilize unpaired electrons. ESI-MSn spectra of the tetra(aryl)benzidine molecular ions exhibit a characteristic feature in the loss of radicals. This process is the major fragmentation pathway of open-shell molecular precursor ions in their MS2 spectra, and also that of even-electron ions in sequential MSn spectra. The collision-induced dissociation (CID) behaviour suggests a general assumption: easily oxidizable compounds (e.g., hydrocarbon polyenes, polycyclic aromatic hydrocarbons, porphyrins etc.) generating predominantly molecular radical cations in ESI-MS contradict the even electron rule in ESI-MSn experiments. The strong ability to stabilize unpaired electrons is preserved in product ions and makes the formation of open-shell species energetically less demanding. A selection of solution-phase reaction mechanistic studies in which open-shell intermediates were detected and structurally characterized by ESI-MS and ESI-MS/MS, respectively, is presented. The merits of ESI-MS and ESI-MSn for mechanistic studies of chemical reaction are critically discussed.(© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2007)
Co-reporter:Mathias Schäfer, Carsten Schmuck, Lars Geiger, Michael J. Chalmers, Christopher L. Hendrickson, Alan G. Marshall
International Journal of Mass Spectrometry 2004 Volume 237(Issue 1) pp:33-45
Publication Date(Web):September 2004
DOI:10.1016/j.ijms.2004.07.001
The gas-phase structures of a series of monomeric, homo- and heterodimeric sodium adduct ions of structurally related synthetic compounds Mn [Gua+–NH–(CH2)n–COO−] with n = 1, 2, 3, 5 and Gua = guanidiniocarbonyl pyrrole were investigated by various MS techniques. The compounds Mn are zwitterions in solution and have a strong tendency to aggregate in polar solvents.First, quadrupole ion trap (QIT) collision induced dissociation (CID) product ion experiments with [Mn + Na]+ ions (n = 1, 2, 3, 5) and [arginine + Na]+ were conducted. The fragmentation behavior of the sodium adduct ions provides indirect evidence for a change in structure varying from predominantly charge-solvation of non-ionic molecules (M1, M2 and arginine), to salt-bridge interactions of zwitterionic structures of Mn for n = 3, 5.Second, the sodium affinities (ΔHNa+HNa+) of the compounds Mn were related to the known literature value of arginine by examination of the CID fragmentation behavior of heterodimer ions [Mn + arginine + Na]+ and [Mn + Mm + Na]+ (n ≠ m and n, m = 2, 3, 5) in a QIT. The relative ordering of sodium affinities (ΔHNa+HNa+): M5 ≥ arginine > M3 > M2 can be deduced from the relative abundances of [Mn,m + Na]+ and [arg + Na]+ product ions. The maximum sodium affinity of M5 relative to the reference value of arginine strongly supports the assumption of a gas-phase zwitterionic structure.Third, the dimeric sodium adduct ions [2Mn + Na]+ of M2, M3 and M5 dissociate upon IR activation in FT-ICR MS exclusively into the respective monomeric sodium adduct ion [Mn + Na]+. Hence, the establishment of a relative ordering of the gas-phase dissociation energy barriers Ealaser for the disruption of the non-covalent bond of the complexes by IRMPD-FT-ICR MS was conducted. We find the dimeric complex ion [2M3 + Na]+ more stable than the respective complexes of M2 and M5. Hence, the stability of the examined complex ions [2Mn + Na]+ is obviously strongly determined by the various possible non-covalent interactions between the two respective molecules Mn. The MS study supports the assumption that Mn molecules with n ≥ 3 are able to conserve zwitterionic structures in the gas phase.
Co-reporter:Mathias Schäfer, Carsten Schmuck, Martin Heil, Helen J Cooper, Christopher L Hendrickson, Michael J Chalmers, Alan G Marshall
Journal of the American Society for Mass Spectrometry 2003 Volume 14(Issue 11) pp:1282-1289
Publication Date(Web):November 2003
DOI:10.1016/S1044-0305(03)00576-2
The activation energy for the unimolecular dissociation of a non-covalent supramolecular complex between an Artificial Cationic Receptor A ([Gua-Val-Val-Val-Amide]+, in which Gua is guanidiniocarbonyl pyrrole) and an Anionic Tetrapeptide B ([N-Acetyl-Val-Val-Ile-Ala]−) has been determined by measurement of the dissociation rate constant as a function of infrared CO2 laser power density. Singly-charged quasimolecular [A + B + H]+ ions are isolated, stored in a Fourier transform ion cyclotron resonance (FT-ICR) mass spectrometer, and irradiated by IR photons. The rate constant for dissociation of the non-covalent complex is determined at five different laser power densities. A plot of the natural logarithm of the first-order rate constant versus the natural logarithm of the laser power density yields a straight line, the slope of which provides an approximate measure of the activation energy (Ealaser) for dissociation. Ealaser is calculated by a relationship derived earlier by Dunbar and with a newly proposed equation by Paech et al. The results of the two approaches deliver significantly different activation energy values for the unimolecular dissociation of the non-covalent complex. We obtain EaIlaser = 0.67 eV (Dunbar approximation) and EaIIlaser = 1.12 eV (Paech et al. approximation). Differences between the two approaches are discussed with respect to non-covalent complexes.
Co-reporter:Mathias Schäfer Dr.
Angewandte Chemie 2003 Volume 115(Issue 17) pp:
Publication Date(Web):30 APR 2003
DOI:10.1002/ange.200201627
Der Diazomalonsäurediester von [18]Krone-6-Methanol bildet mit zweifach protoniertem 1,6-Diaminohexan einen nichtkovalenten Komplex (siehe Bild), der intakt in die Gasphase überführt werden kann. Nach kollisionsinduzierter Abspaltung des Stickstoffs reagiert das intermediär gebildete Carben als molekulare Mausefalle. Die tatsächliche Bildung einer intermolekularen kovalenten Bindung zwischen Wirt- und Gastmolekül kann mithilfe von MS/MS-Experimenten belegt werden.
Co-reporter:Mathias Schäfer Dr.
Angewandte Chemie International Edition 2003 Volume 42(Issue 17) pp:
Publication Date(Web):30 APR 2003
DOI:10.1002/anie.200201627
The diazomalonic acid diester of [18]crown-6-methanol forms a noncovalent complex (see picture) with doubly protonated 1,6-diaminohexane which can be transferred intact into the gas phase. After collision-induced dissociation, the reactive carbene intermediate reacts as a molecular mousetrap. The successful formation of an intermolecular covalent coupling between the host and the guest molecule can be observed by subsequent MS/MS experiments.
Co-reporter:Katrin Peckelsen, Jonathan Martens, Giel Berden, Jos Oomens, Robert C. Dunbar, Anthony J.H.M. Meijer, Mathias Schäfer
Journal of Molecular Spectroscopy (February 2017) Volume 332() pp:38-44
Publication Date(Web):February 2017
DOI:10.1016/j.jms.2016.10.008
Co-reporter:Ahmad R. Massah, Frank Dreiocker, Richard F. W. Jackson, Barry T. Pickup, Jos Oomens, Anthony J. H. M. Meijer and Mathias Schäfer
Physical Chemistry Chemical Physics 2011 - vol. 13(Issue 29) pp:NaN13267-13267
Publication Date(Web):2011/06/22
DOI:10.1039/C1CP20747D
An extensive set of organozinc iodides, useful for Negishi-type cross-coupling reactions, are investigated as respective cations after formal loss of iodide in the gas phase. Firstly, two new alkylzinc compounds derived from Tyrosine (Tyr) and Tryptophan (Trp) are closely examined. Secondly, the influence of specific protecting groups on the subtle balance between intra- and intermolecular coordination of zinc in these reagents is probed through trifluoroacetyl (TFA)-derivatized alkylzinc compounds. Finally, the influence of the strongly coordinating bidentate ligand N,N,N′,N′-tetramethylethylenediamine (TMEDA) on the structure of alkylzinc cations is further explored in order to better understand the stability of the respective complexes towards water. A combination of electrospray (ESI)-MS/MS, accurate ion mass measurements, infrared multiple-photon dissociation (IRMPD) spectroscopy and computational modelling allowed the full characterisation of all dimethylformamide (DMF)-solvated and TMEDA-coordinated alkylzinc cations in the gas phase. The calculations indicate that the zinc cation in gas-phase alkylzinc–DMF or TMEDA–complex ions preferentially adopts a tetrahedral coordination sphere with four ligands. Additionally, conformers with only three binding partners bound to zinc but with effectively combined hydrogen-bond interactions are also found. Collision induced dissociation (CID) patterns demonstrate that the zinc–DMF interaction in tetrahedral four-coordinate mono-DMF–zinc complex ions as well as the interaction between TMEDA and zinc in the corresponding complex ions is even stronger than typical covalent bonds. In most cases, all major features of the IRMPD spectra are consistent with only a single major isomer, allowing secured identification and assignment.