Co-reporter:Qi Su;Petrina A. Boucher; Steven E. Rokita
Angewandte Chemie International Edition 2017 Volume 56(Issue 36) pp:10862-10866
Publication Date(Web):2017/08/28
DOI:10.1002/anie.201703628
AbstractNatural and engineered nitroreductases have rarely supported full reduction of nitroaromatics to their amine products, and more typically, transformations are limited to formation of the hydroxylamine intermediates. Efficient use of these enzymes also requires a regenerating system for NAD(P)H to avoid the costs associated with this natural reductant. Iodotyrosine deiodinase is a member of the same structural superfamily as many nitroreductases but does not directly consume reducing equivalents from NAD(P)H, nor demonstrate nitroreductase activity. However, exchange of its flavin cofactor with a 5-deazaflavin analogue dramatically suppresses its native deiodinase activity and leads to significant nitroreductase activity that supports full reduction to an amine product in the presence of the convenient and inexpensive NaBH4.
Co-reporter:Nattha Ingavat, Jennifer M. KavranZuodong Sun, Steven E. Rokita
Biochemistry 2017 Volume 56(Issue 8) pp:
Publication Date(Web):February 3, 2017
DOI:10.1021/acs.biochem.6b01308
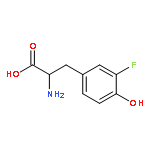
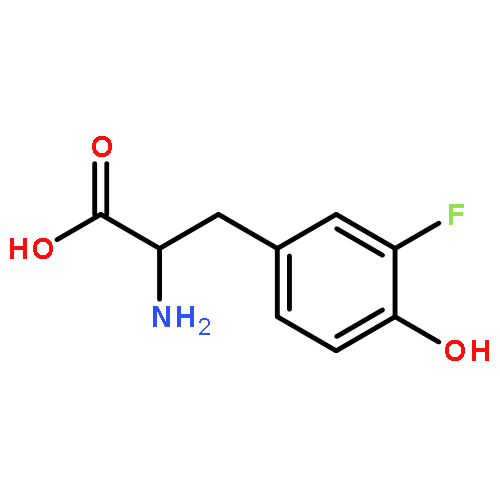
The minimal requirements for substrate recognition and turnover by iodotyrosine deiodinase were examined to learn the basis for its catalytic specificity. This enzyme is crucial for iodide homeostasis and the generation of thyroid hormone in chordates. 2-Iodophenol binds only very weakly to the human enzyme and is dehalogenated with a kcat/Km that is more than 4 orders of magnitude lower than that for iodotyrosine. This discrimination likely protects against a futile cycle of iodinating and deiodinating precursors of thyroid hormone biosynthesis. Surprisingly, a very similar catalytic selectivity was expressed by a bacterial homologue from Haliscomenobacter hydrossis. In this example, discrimination was not based on affinity since 4-cyano-2-iodophenol bound to the bacterial deiodinase with a Kd lower than that of iodotyrosine and yet was not detectably deiodinated. Other phenols including 2-iodophenol were deiodinated but only very inefficiently. Crystal structures of the bacterial enzyme with and without bound iodotyrosine are nearly superimposable and quite similar to the corresponding structures of the human enzyme. Likewise, the bacterial enzyme is activated for single electron transfer after binding to the substrate analogue fluorotyrosine as previously observed with the human enzyme. A cocrystal structure of bacterial deiodinase and 2-iodophenol indicates that this ligand stacks on the active site flavin mononucleotide (FMN) in a orientation analogous to that of bound iodotyrosine. However, 2-iodophenol association is not sufficient to activate the FMN chemistry required for catalysis, and thus the bacterial enzyme appears to share a similar specificity for halotyrosines even though their physiological roles are likely very different from those in humans.
Co-reporter:Zuodong Sun, Qi Su, Steven E. Rokita
Archives of Biochemistry and Biophysics 2017 Volume 632(Volume 632) pp:
Publication Date(Web):15 October 2017
DOI:10.1016/j.abb.2017.07.019
Iodotyrosine deiodinase (IYD) is unusual for its reliance on flavin to promote reductive dehalogenation under aerobic conditions. As implied by the name, this enzyme was first discovered to catalyze iodide elimination from iodotyrosine for recycling iodide during synthesis of tetra- and triiodothyronine collectively known as thyroid hormone. However, IYD likely supports many more functions and has been shown to debrominate and dechlorinate bromo- and chlorotyrosines. A specificity for halotyrosines versus halophenols is well preserved from humans to bacteria. In all examples to date, the substrate zwitterion establishes polar contacts with both the protein and the isoalloxazine ring of flavin. Mechanistic data suggest dehalogenation is catalyzed by sequential one electron transfer steps from reduced flavin to substrate despite the initial expectations for a single two electron transfer mechanism. A purported flavin semiquinone intermediate is stabilized by hydrogen bonding between its N5 position and the side chain of a Thr. Mutation of this residue to Ala suppresses dehalogenation and enhances a nitroreductase activity that is reminiscent of other enzymes within the same structural superfamily.Download high-res image (116KB)Download full-size image
Co-reporter:Chengyun Huang
Frontiers of Chemical Science and Engineering 2016 Volume 10( Issue 2) pp:213-221
Publication Date(Web):2016 June
DOI:10.1007/s11705-015-1541-3
Biological application of conjugates derived from oligonucleotides and quinone methides have previously been limited by the slow exchange of their covalent self-adducts and subsequent alkylation of target nucleic acids. To enhance the rates of these processes, a new quinone methide precursor with an electron donating substituent has been prepared. Additionally, this substituent has been placed para to the nascent exo-methylene group of the quinone methide for maximum effect. A conjugate made from this precursor and a 5'-aminohexyloligonucleotide accelerates formation of its reversible self-adduct and alkylation of its complementary DNA as predicted from prior model studies.
Co-reporter:Arnab Mukherjee
Journal of the American Chemical Society 2015 Volume 137(Issue 49) pp:15342-15345
Publication Date(Web):November 30, 2015
DOI:10.1021/jacs.5b07540
A single mutation within a flavoprotein is capable of switching the catalytic activity of a dehalogenase into a nitroreductase. This change in function correlates with a destabilization of the one-electron-reduced flavin semiquinone that is differentially expressed in the nitro-FMN reductase superfamily during redox cycling. The diversity of function within such a superfamily therefore has the potential to arise from rapid evolution, and its members should provide a convenient basis for developing new catalysts with an altered specificity of choice.
Co-reporter:Kostyantyn D. Bobyk, David P. Ballou, and Steven E. Rokita
Biochemistry 2015 Volume 54(Issue 29) pp:4487-4494
Publication Date(Web):July 7, 2015
DOI:10.1021/acs.biochem.5b00410
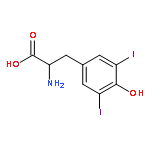
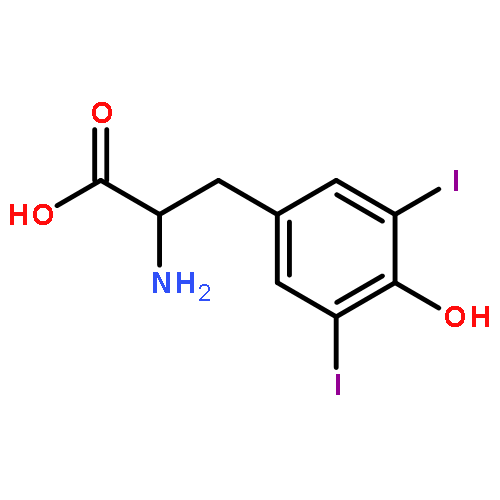
Reductive dehalogenation such as that catalyzed by iodotyrosine deiodinase (IYD) is highly unusual in aerobic organisms but necessary for iodide salvage from iodotyrosine generated during thyroxine biosynthesis. Equally unusual is the dependence of this process on flavin. Rapid kinetics have now been used to define the basic processes involved in IYD catalysis. Time-dependent quenching of flavin fluorescence was used to monitor halotyrosine association to IYD. The substrates chloro-, bromo-, and iodotyrosine bound with similar rate constants (kon) ranging from 1.3 × 106 to 1.9 × 106 M–1 s–1. Only the inert substrate analogue fluorotyrosine exhibited a significantly (5-fold) slower kon (0.3 × 106 M–1 s–1). All data fit a standard two-state model and indicated that no intermediate complex accumulated during closure of the active site lid induced by substrate. Subsequent halide elimination does not appear to limit reactions of bromo- and iodotyrosine since both fully oxidized the reduced enzyme with nearly equivalent second-order rate constants (7.3 × 103 and 8.6 × 103 M–1 s–1, respectively) despite the differing strength of their carbon–halogen bonds. In contrast to these substrates, chlorotyrosine reacted with the reduced enzyme approximately 20-fold more slowly and revealed a spectral intermediate that formed at approximately the same rate as the bromo- and iodotyrosine reactions.
Co-reporter:Shalini Saha, Wei Li, Barbara Gerratana, Steven E. Rokita
Bioorganic & Medicinal Chemistry 2015 23(3) pp: 449-454
Publication Date(Web):
DOI:10.1016/j.bmc.2014.12.024
Co-reporter:Neil P. Campbell and Steven E. Rokita
Organic & Biomolecular Chemistry 2014 vol. 12(Issue 7) pp:1143-1148
Publication Date(Web):06 Jan 2014
DOI:10.1039/C3OB42433B
Covalent conjugation is typically used to fix a potential charge donor to a chosen site for studying either hole or excess electron transport in duplex DNA. A model system based on oligonucleotides containing an abasic site and BrdU was previously developed to provide a rapid method of screening new donors without the need of synthetic chemistry. While this strategy is effective for discovering important lead compounds, it is not appropriate for establishing extensive correlations between molecular structure and donor efficiency as demonstrated with a series of closely related electron donors based on diaminonaphthalene. The non-covalent system accurately identified the ability of the donors to reduce a distal BrdU in DNA, but their varying efficiencies were not recapitulated when attached covalently to an equivalent sequence of DNA. Reduction within the covalent system was not sensitive to the strong donor potentials as consistent with charge recombination dominating the net migration of charge.
Co-reporter:Abhishek Phatarphekar, Jennifer M. Buss and Steven E. Rokita
Molecular BioSystems 2014 vol. 10(Issue 1) pp:86-92
Publication Date(Web):17 Oct 2013
DOI:10.1039/C3MB70398C
Iodide is required for thyroid hormone synthesis in mammals and other vertebrates. The role of both iodide and iodinated tyrosine derivatives is currently unknown in lower organisms, yet the presence of a key enzyme in iodide conservation, iodotyrosine deiodinase (IYD), is suggested by genomic data from a wide range of multicellular organisms as well as some bacteria. A representative set of these genes has now been expressed, and the resulting enzymes all catalyze reductive deiodination of diiodotyrosine with kcat/Km values within a single order of magnitude. This implies a physiological presence of iodotyrosines (or related halotyrosines) and a physiological role for their turnover. At least for Metazoa, IYD should provide a new marker for tracing the evolutionary development of iodinated amino acids as regulatory signals through the tree of life.
Co-reporter:Michael P. McCrane, Mark A. Hutchinson, Omer Ad, and Steven E. Rokita
Chemical Research in Toxicology 2014 Volume 27(Issue 7) pp:1282
Publication Date(Web):June 4, 2014
DOI:10.1021/tx500152d
ortho-Quinone methides (ortho-QM) and para-quinone methides are generated by xenobiotic metabolism of numerous compounds including environmental toxins and therapeutic agents. These intermediates are highly electrophilic and have the potential to alkylate DNA. Assessing their genotoxicity can be difficult when all or some of their resulting adducts form reversibly. Stable adducts are most easily detected but are not necessarily the most prevalent products formed initially as DNA repair commences. Selective oxidation of ortho-QM-DNA adducts by bis[(trifluoroacetoxy)iodo]benzene (BTI) rapidly quenches their reversibility to prevent QM regeneration and allows for observation of the kinetic products. The resulting derivatives persist through standard enzymatic digestion, chromatography, and mass spectral analysis. The structural standards required for this approach have been synthesized and confirmed by two-dimensional NMR spectroscopy. The adducts of dA N6, dG N1, dG N2, and guanine N7 are converted to the expected para-quinol derivatives within 5 min after addition of BTI under aqueous conditions (pH 7). Concurrently, the adduct of dA N1 forms a spiro derivative comparable to that characterized previously after oxidation of the corresponding dC N3 adduct. By application of this oxidative quenching strategy, the dC N3 and dA N1 adducts have been identified as the dominant products formed by both single- and double-stranded DNA under initial conditions. As expected, however, these labile adducts dissipate within 24 h if not quenched with BTI. Still, the products favored by kinetics are responsible for inducing the first response to ortho-QM exposure in cells, and hence, they are also key to establishing the relationship between biological activity and molecular structure.
Co-reporter:Amethist S. Finch, William B. Davis and Steven E. Rokita
Photochemical & Photobiological Sciences 2013 vol. 12(Issue 8) pp:1474-1482
Publication Date(Web):17 Jun 2013
DOI:10.1039/C3PP50147G
Photochemical cyclobutane dimerization of adjacent thymines generates the major lesion in DNA caused by exposure to sunlight. Not all nucleotide sequences and structures are equally susceptible to this reaction or its potential to create mutations. Photostationary levels of the cyclobutane thymine dimer have now been quantified in homogenous samples of DNA reconstituted into nucleosome core particles to examine the basis for previous observations that such structures could induce a periodicity in dimer yield when libraries of heterogeneous sequences were used. Initial rate studies did not reveal a similar periodicity when a homogenous core particle was analyzed, but this approach examined only formation of this photochemically reversible cyclobutane dimer. Photostationary levels result from competition between dimerization and reversion and, as described in this study, still express none of the periodicity within two alternative core particles that was evident in heterogeneous samples. Such periodicity likely arises from only a limited set of sequences and structural environments that are not present in the homogeneous and well-characterized assemblies available to date.
Co-reporter:Neil P. Campbell and Steven E. Rokita
Organic & Biomolecular Chemistry 2014 - vol. 12(Issue 7) pp:NaN1148-1148
Publication Date(Web):2014/01/06
DOI:10.1039/C3OB42433B
Covalent conjugation is typically used to fix a potential charge donor to a chosen site for studying either hole or excess electron transport in duplex DNA. A model system based on oligonucleotides containing an abasic site and BrdU was previously developed to provide a rapid method of screening new donors without the need of synthetic chemistry. While this strategy is effective for discovering important lead compounds, it is not appropriate for establishing extensive correlations between molecular structure and donor efficiency as demonstrated with a series of closely related electron donors based on diaminonaphthalene. The non-covalent system accurately identified the ability of the donors to reduce a distal BrdU in DNA, but their varying efficiencies were not recapitulated when attached covalently to an equivalent sequence of DNA. Reduction within the covalent system was not sensitive to the strong donor potentials as consistent with charge recombination dominating the net migration of charge.
.jpg)
![Ethanamine, 2-[2-[2-(2-azidoethoxy)ethoxy]ethoxy]-](/data/chemimg/114300/134179-38-7.png)
![Ethanamine, 2-[2-[2-(2-azidoethoxy)ethoxy]ethoxy]-](/data/chemimg/114300/134179-38-7_b.png)






![Benzo[g]pteridine-2,4(1H,3H)-dione](http://img.cochemist.com/ccimg/500/490-59-5.png)
![Benzo[g]pteridine-2,4(1H,3H)-dione](http://img.cochemist.com/ccimg/500/490-59-5_b.png)
![Phenol, 2-[bis(2-pyridinylmethyl)amino]-4-(1,1-dimethylethyl)-](http://img.cochemist.com/ccimg/911500/911437-28-0.png)
![Phenol, 2-[bis(2-pyridinylmethyl)amino]-4-(1,1-dimethylethyl)-](http://img.cochemist.com/ccimg/911500/911437-28-0_b.png)



![2,6-BIS[BIS(PYRIDIN-2-YLMETHYL)AMINO]-4-TERT-BUTYLPHENOL](http://img.cochemist.com/ccimg/548800/548756-41-8.png)
![2,6-BIS[BIS(PYRIDIN-2-YLMETHYL)AMINO]-4-TERT-BUTYLPHENOL](http://img.cochemist.com/ccimg/548800/548756-41-8_b.png)