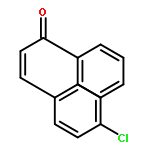
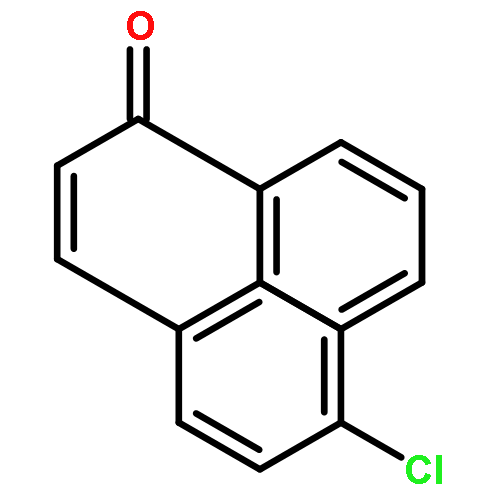
Co-reporter:Xi-Rui Li;Xiang-Yuan Yang;Yongxin Li;Sumod A. Pullarkat
Dalton Transactions 2017 vol. 46(Issue 4) pp:1311-1316
Publication Date(Web):2017/01/24
DOI:10.1039/C6DT04447F
A chiral phosphine auxiliary was generated with excellent ee via catalytic asymmetric hydrophosphination of 3-(naphthalen-1-ylmethylene)pentane-2,4-dione. The subsequent metal complexation of the monophosphine yielded two different coordination complexes depending on the reaction conditions. The ortho-palladation of both coordination complexes resulted in the formation of a single dimeric phosphapalladacycle complex that could be further converted to the monomeric bisacetonitrile derivative. Moreover, the palladium complex exhibits interesting oxophilicity as the stable bisaquo derivative could be isolated and characterized crystallographically. The catalytic potential of the phosphapalladacycle was also demonstrated.
Co-reporter:Wee Shan Tay;Xiang-Yuan Yang;Yongxin Li;Sumod A. Pullarkat
Chemical Communications 2017 vol. 53(Issue 47) pp:6307-6310
Publication Date(Web):2017/06/08
DOI:10.1039/C7CC02044A
A catalytic asymmetric hydroarsination reaction of an activated alkene viz. (E)-nitrostyrene was developed using chiral PCP Pt-, Pd- and Ni-pincer complexes as catalysts. The corresponding chiral tertiary arsine adduct was obtained in ees of up to 80% under mild reaction conditions using the PCP Ni–Cl pincer catalyst. The arsine adduct was furnished with catalyst loadings of 1–5 mol% and the reaction duration ranging from <5 min to 180 min. The subsequent coordination of the hydroarsination product to gold(I) chloride allowed for the confirmation of the stereochemistry of the arsine adduct via crystallographic analysis.
Co-reporter:Xiang-Yuan Yang, Wee Shan Tay, Yongxin Li, Sumod A. Pullarkat and Pak-Hing Leung
Chemical Communications 2016 vol. 52(Issue 22) pp:4211-4214
Publication Date(Web):17 Feb 2016
DOI:10.1039/C6CC00763E
Cyclopalladation of the pyridyl-substituted chiral phosphine sulfide (N–PS) and oxide (N–PO) compounds afforded the asymmetric N–C(sp3)*–S and N–C(sp3)*–O pincer complexes. When applied as catalysts in asymmetric hydrophosphination, the newly developed aliphatic pincer catalyst could be recycled over three runs and obtained in large quantities via a one-pot “self-breeding” catalytic protocol.
Co-reporter:Yu-Xiang Jia, Xiang-Yuan Yang, Wee Shan Tay, Yongxin Li, Sumod A. Pullarkat, Kai Xu, Hajime Hirao and Pak-Hing Leung
Dalton Transactions 2016 vol. 45(Issue 5) pp:2095-2101
Publication Date(Web):07 Oct 2015
DOI:10.1039/C5DT02049B
A 13C{1H} NMR based investigation was conducted to examine the electronic properties of C(aryl)–M bonds and their trans influence in P–C(aryl)–P pincer complexes. A series of structurally related platinum pincer complexes were rationally designed and their corresponding 13C–195Pt coupling constants were systematically examined. By methodical substitution of the ligand trans to the organometallic C(aryl)–Pt bond, this study revealed the significant influence of the ligands on the nature of the C(aryl)–M bonds. The single crystal X-ray analysis of the complexes and computational studies further confirmed the observations that the C–M bond exhibits significant π-character.
Co-reporter:Xiang-Yuan Yang, Yu-Xiang Jia, Wee Shan Tay, Yongxin Li, Sumod A. Pullarkat and Pak-Hing Leung
Dalton Transactions 2016 vol. 45(Issue 34) pp:13449-13455
Publication Date(Web):2016/07/22
DOI:10.1039/C6DT02588A
The impact of the structural attributes of chiral PC- and PCP-palladium catalysts was investigated in the asymmetric hydrophosphination of various heterocycle-functionalized enone substrates. Due to the architecture of the catalysts, they are confronted with potential catalyst deactivation arising from the coordination of the electron-rich heteroatoms (P, O, N and S) to the metal center. A systematic variation of the location and identity of the heteroatoms demonstrated the impact of structural modifications on the substrates, which have a significant influence on both yields (16–99%) and enantioselectivities (0–99%). A detailed discussion on the distinct catalytic mechanisms (intra- vs. inter-molecular addition) provides important information to explain the results obtained.
Co-reporter:Weiwei Yao, Mengtao Ma, Na Zhang, Yongxin Li, Sumod A. Pullarkat, Pak-Hing Leung
Journal of Organometallic Chemistry 2016 Volume 801() pp:1-5
Publication Date(Web):1 January 2016
DOI:10.1016/j.jorganchem.2015.10.021
•Asymmetric hydrophosphination reaction of 2-ethynylpyridine was reported.•A new chiral diphosphine ligand was synthesized.•The organopalladium complex has been successfully used as the chiral template.The organopalladium complex containing ortho-metalated (R)-[1-(dimethylamino) ethyl]naphthalene as the chiral auxiliary has been used as the chiral template to promote the asymmetric hydrophosphination reaction between diphenylphosphine and 2-ethynylpyridine in high selectivity under mild conditions. The double hydrophosphination reaction produces four stereoisomeric products in the ratio of 1:19.4:2.4:1.3. The naphthylamine auxiliary could be removed chemoselectively from the template products by treatment with concentrated hydrochloric acid to form the corresponding chiral dichloro complexes [(P–P)PdCl2]. The molecular structure and absolute configuration of the dichloro complex have been resolved by single-crystal X-ray crystallography. It confirms that the asymmetric diphosphines as P–P bidentate chelate on the chiral naphthylamine palladium templates. Finally, the enantiomerically pure diphosphine ligand with chirality on the carbon backbone can be readily liberated from the dichloro palladium complex as an air-sensitive solid by treatment of the complex with aqueous potassium cyanide in high yield.Asymmetric hydrophosphination reaction between diphenylphosphine and 2-ethynylpyridine was promoted by the palladium complex containing ortho-metalated (R)-[1-(dimethylamino)ethyl]naphthalene as the chiral auxiliary.
Co-reporter:Mengtao Ma, Jia Li, Na Zhang, Weiwei Yao, Sumod A. Pullarkat, Pak-Hing Leung
Journal of Organometallic Chemistry 2016 Volume 824() pp:99-103
Publication Date(Web):1 December 2016
DOI:10.1016/j.jorganchem.2016.10.003
•Asymmetric Diels−Alder reaction of 3,4-dimethyl-1-phenylphosphole was reported.•Two new chiral P−N ligands were synthesized.•The molecular structure was elucidated by single crystal X-ray diffraction.The organoplatinum complex containing ortho-metalated (S)-[1-(dimethylamino)ethyl]naphthalene as the chiral auxiliary has been used to promote the asymmetric cycloaddition reactions between 3,4-dimethyl-1-phenyl phosphole and (E)-1-phenyl-3-pyridin-2-yl-2-propenone or (E)-1-methyl-3-pyridin-2- yl-2-propenoate in high selectivity under mild condition. However, the corresponding palladium complex didn't show similar reactivity. This Diels−Alder reaction generated five new stereogenic centers in a single step. The naphthylamine auxiliary could be removed chemoselectively from the template products by treatment with concentrated hydrochloric acid to form the corresponding dichloro platinum complexes [(P−N)PtCl2]. The molecular structure and absolute configuration of the major dichloro platinum complex have been resolved by single-crystal X-ray diffraction. Finally, the enantiomerically pure P−N ligands can be readily liberated from the corresponding dichloro platinum complexes as air-sensitive solids by treatment with aqueous potassium cyanide in quantitative yield.
Co-reporter:Renta Jonathan Chew
The Chemical Record 2016 Volume 16( Issue 1) pp:141-158
Publication Date(Web):
DOI:10.1002/tcr.201500220
Co-reporter:Renta Jonathan Chew;Kristel Sepp;Bin-Bin Li;Yongxin Li;Peng-Cheng Zhu;Nguan Soon Tan
Advanced Synthesis & Catalysis 2015 Volume 357( Issue 14-15) pp:3297-3302
Publication Date(Web):
DOI:10.1002/adsc.201500638
Co-reporter:Xiang-Yuan Yang, Jun Hao Gan, Yongxin Li, Sumod A. Pullarkat and Pak-Hing Leung
Dalton Transactions 2015 vol. 44(Issue 3) pp:1258-1263
Publication Date(Web):12 Nov 2014
DOI:10.1039/C4DT02673J
Both PC-cyclometalated and PCP-pincer type palladium catalysts have recently been found to be robust and efficacious catalysts for the asymmetric P–H addition reaction involving activated olefins. Our studies on the asymmetric P–H addition of diphenylphosphine to malonate ester and α,β,γ,δ-alkylidenemalonate ester revealed for the first time that the catalyst choice can have a dramatic impact in terms of reactivity as well as regio- and stereo-control for this asymmetric hydrofunctionalization reaction. Besides showing significantly contrasting reactivity and stereoselectivity in the hydrophosphination reaction involving malonate ester, in the case of α,β,γ,δ-alkylidenemalonate ester, a novel regiodivergent method was developed with the 1,4-adduct being obtained exclusively with the PC-catalyst while the pincer catalyst produced only the 1,6-adduct.
Co-reporter:Bin-Bin Li, Yu-Xiang Jia, Peng-Cheng Zhu, Renta Jonathan Chew, Yongxin Li, Nguan Soon Tan, Pak-Hing Leung
European Journal of Medicinal Chemistry 2015 Volume 98() pp:250-255
Publication Date(Web):15 June 2015
DOI:10.1016/j.ejmech.2015.05.027
•New chiral dinuclear gold(I) phosphine complexes are highly selective and effective against breast cancer.•Multi-methylester group incorporation led to suppression of side-effects and improved anti-cancer efficiency.•Chirality of complexes may function as a sensitive probe for future mechanistic studies.Two chiral (–)-diphosphine-digold(I) complexes containing mono- and di-methylester substituted diphosphine ligands have been prepared and structurally characterized. Both complexes are highly potent against breast cancer cell line MDA-MB-231 but showed much lower cytotoxicity against the normal human breast epithelial cells MCF10A. When compared with its mono-substituted analogue, the di-methylester substituted complex caused markedly lower and relatively insignificant damage to the normal breast cells. The analogous mono- and di-ethylester substituted complexes with the same stereochemistry exhibited similar anti-cancer properties but with noticeably higher cytotoxicity against the MCF10A cells. The enantiomeric complex (+)-diphosphine-digold(I) complexes containing the di-methylester substituted diphosphine ligand exhibited clearly different biological properties from its (–)-enantiomer. Furthermore, a structurally similar diphosphine-digold(I) complex but in the absence of an ester substituent, killed both the cancerous and the healthy cells indiscriminately. The current study thus revealed that the introduction of multi-esters, particularly methylesters, is an efficient approach to suppress the side-effects and to improve the efficiency of potential gold-based anti-cancer reagents. When combined with the biological observations, the chirality of gold complexes may serve as a sensitive probe for the future mechanistic studies.
Co-reporter:Xiang-Yuan Yang, Wee Shan Tay, Yongxin Li, Sumod A. Pullarkat, and Pak-Hing Leung
Organometallics 2015 Volume 34(Issue 20) pp:5196-5201
Publication Date(Web):October 2, 2015
DOI:10.1021/acs.organomet.5b00787
An enantioselective asymmetric 1,4-addition of diarylphosphines to linear α,β,γ,δ-unsaturated dienones was developed. A series of chiral PCP- and PCN-transition-metal (Pd, Pt and Ni) pincers, themselves prepared catalytically via asymmetric hydrophosphination, were sequentially screened to reveal the roles of backbone architecture and metal ion in catalyst design. The selected ester-functionalized PCP-palladium pincer afforded the chiral 1,4-phosphine adducts in excellent yields with up to >99% ee. The same catalyst when utilized for the hydrophosphination of an α,β,γ,δ-unsaturated malonate ester also revealed the critical role played by the ester functionality on the ligand backbone in dictating the enantioselectivity of the 1,6-adduct.
Co-reporter:Xiang-Yuan Yang, Wee Shan Tay, Yongxin Li, Sumod A. Pullarkat, and Pak-Hing Leung
Organometallics 2015 Volume 34(Issue 8) pp:1582-1588
Publication Date(Web):April 16, 2015
DOI:10.1021/acs.organomet.5b00132
A series of chiral C-stereogenic PCP and PCN ligand precursors were prepared in situ from inexpensive achiral starting materials via a simple catalytic asymmetric P–H addition reaction in good overall yields. This facile catalytic method of preparing the ligand backbones renders easy and economical modifications of the electronically crucial para-substituent, chiral functionalities, and donor atoms for different transition metal ions. A one-pot synthetic procedure was used efficiently to prepare the corresponding optically pure pincer complexes. All the new complexes were characterized by NMR and mass spectroscopy. The molecular structures of several selected complexes have also been elucidated by X-ray crystallography. Preliminary studies indicated that minor structural changes on these novel pincer complexes affect their chemical properties significantly when they were applied as catalysts for the reaction between diphenylphosphine and chalcone.
Co-reporter:Mengtao Ma, Na Zhang, Yongxin Li, Sumod A. Pullarkat, and Pak-Hing Leung
Organometallics 2015 Volume 34(Issue 20) pp:5081-5087
Publication Date(Web):September 22, 2015
DOI:10.1021/acs.organomet.5b00682
The orthometalated [1-(dimethylamino)ethyl]naphthalene platinum(II) complex has been used successfully to promote the asymmetric cycloaddition reaction between 3,4-dimethyl-1-phenylphosphole and sulfoxide in high selectivity. The exo-cycloadduct coordinated to the platinum template as bidentate chelates via their phosphorus and sulfur atoms. The dichloro platinum complexes could be crystallized and were stable in the solid state as well as in solution. Optically pure P–S bidentate ligands could be liberated from these dichloro complexes by treatment with aqueous potassium cyanide. The study also highlights the difference in reactivity and mode of substrate activation between an earlier study involving a Pd analogue of the template and the current results.
Co-reporter:Renta Jonathan Chew;Xi-Rui Li;Dr. Yongxin Li;Dr. Sumod A. Pullarkat ;Dr. Pak-Hing Leung
Chemistry - A European Journal 2015 Volume 21( Issue 12) pp:4800-4804
Publication Date(Web):
DOI:10.1002/chem.201406298
Abstract
The palladacycle-catalyzed asymmetric PH addition of 4-oxo-enamides has been developed, which provides efficient access to phosphinocarboxamides and their analogues. Solvent-mediated reversal of stereoselectivity (ee from +96 % to −92 %) was observed, and the underlying mechanism that allows facile access to both enantiomers of the product by judicious choice of solvents is revealed.
Co-reporter:Yinhua Huang ; Yongxin Li ; Pak-Hing Leung ;Tamio Hayashi
Journal of the American Chemical Society 2014 Volume 136(Issue 13) pp:4865-4868
Publication Date(Web):March 17, 2014
DOI:10.1021/ja501007t
The reaction of phenyl(2,4,6-trimethylphenyl)phosphine with a substituted benzoquinone in the presence of a chiral phosphapalladacycle complex as a catalyst and triethylamine in chloroform at −45 °C proceeded in a new type of addition manner to give a high yield of a 4-hydroxyphenyl phenyl(2,4,6-trimethylphenyl)phosphinite with 98% enantioselectivity, which is a versatile intermediate readily convertible into various phosphines and their derivatives with high enantiomeric purity.
Co-reporter:Renta Jonathan Chew, Kai Yuan Teo, Yinhua Huang, Bin-Bin Li, Yongxin Li, Sumod A. Pullarkat and Pak-Hing Leung
Chemical Communications 2014 vol. 50(Issue 63) pp:8768-8770
Publication Date(Web):26 Jun 2014
DOI:10.1039/C4CC01610F
An enantioselective hydrophosphination of β,γ-unsaturated α-ketoesters and amides has been developed using a chiral palladacycle catalyst. Adducts can be obtained in excellent yields and enantioselectivities, providing direct access to chiral tertiary phosphines which are synthetically useful intermediates in the preparation of bidentate ligands.
Co-reporter:Jeanette See Leng Yap, Bin Bin Li, Jonathan Wong, Yongxin Li, Sumod A. Pullarkat and Pak-Hing Leung
Dalton Transactions 2014 vol. 43(Issue 15) pp:5777-5784
Publication Date(Web):29 Jan 2014
DOI:10.1039/C4DT00062E
A novel amine ligand, 1-(2,5-dichlorophenyl)-N,N-dimethylethanamine, was synthesized from 1-(2,5-dichlorophenyl)ethanone via a three step synthetic route. Direct ortho-palladation of the amine ligand with Pd(OAc)2 gave the racemic dimeric complex in high yield. This racemic palladacycle was efficiently resolved through the formation of its (S)-prolinato derivatives. The resulting diastereomeric complexes were separated efficiently by column chromatography. In the solid state, the structure and absolute configuration of the two optically resolved palladium complexes were determined by single crystal X-ray crystallography. In solution, their absolute conformations were also investigated by the 2D 1H–1H rotating frame nuclear Overhauser enhancement (ROESY) NMR spectroscopy. Both (R,R) and (S,S)-di-μ-chloro dimeric palladium complexes could be obtained chemoselectively by treating the corresponding prolinato derivatives with dilute hydrochloric acid. The amine auxiliary could be subsequently removed from the palladium center by treatment with concentrated hydrochloric acid. The enantiomerically pure palladacycle was used to promote the asymmetric hydrophosphination reaction between diphenylphosphine and dimethyl acetylenedicarboxylate. The 31P{1H} NMR spectroscopy indicated that only one stereo-isomeric product was formed.
Co-reporter:Jeanette See Leng Yap;Yi Ding;Xiang-Yuan Yang;Jonathan Wong;Yongxin Li;Sumod A. Pullarkat
European Journal of Inorganic Chemistry 2014 Volume 2014( Issue 29) pp:5046-5052
Publication Date(Web):
DOI:10.1002/ejic.201402547
Abstract
Two structurally isomeric substituted N,N-dimethylethanamines have been prepared. Treatment of the 2,4-di-tert-butylphenyl isomer with PdII ions generated the ortho-metalated complexes. On the other hand, treatment of the 2,5-di-tert-butylphenyl-substituted amine resulted in the unexpected chemoselective cleavage of one of the three N–C bonds, thus generating the corresponding secondary amine. The N-demethylation process could be catalyzed at room temperature by palladium(II) catalysts such as PdCl2 or Pd(OAc)2. Furthermore, treatment with a stoichiometric amount of PdII ions gave a metal complex in which both secondary amines were bound to Pd in an N-monodentate fashion. When triethylamine was introduced, one of the N-ethyl groups in NEt3 was cleaved, and an unexpected heteroamine complex was produced. The products generated were isolated and characterized by X-ray crystallography. Mechanistic insights into the cyclometalation and C–N cleavage observed are discussed.
Co-reporter:Yu-Xiang Jia, Bin-Bin Li, Yongxin Li, Sumod A. Pullarkat, Kai Xu, Hajime Hirao, and Pak-Hing Leung
Organometallics 2014 Volume 33(Issue 21) pp:6053-6058
Publication Date(Web):October 7, 2014
DOI:10.1021/om500662q
The stereoelectronic properties of palladium(II) and platinum(II) complexes containing ortho-metalated (1-(diphenylphosphino)ethyl)naphthalene were examined by X-ray crystallography in the solid state and by NMR spectroscopy in solution. The studies revealed that the aromatic carbon induced a significantly stronger electronic trans-influence than the phosphorus donor within the same five-membered chelate. Both the organopalladium and organoplatinum chelate complexes were formed with classical hard oxygen and soft phosphorus donor atoms. Computational studies further confirmed the observations in the solid state and solution that the C–metal bond in the trans C–Pt–O moiety has a stronger π-character than that in the trans C–Pt–P moiety. Both organometallic units are efficient catalysts for the asymmetric P–H addition reaction. They showed almost identical efficiency in dichloromethane, but their reactivity in THF is markedly different.
Co-reporter:Renta Jonathan Chew;Dr. Yunpeng Lu;Yu-Xiang Jia;Bin-Bin Li;Esther Hui Yen Wong;Rosanne Goh;Dr. Yongxin Li;Dr. Yinhua Huang;Dr. Sumod Appukuttan Pullarkat;Dr. Pak-Hing Leung
Chemistry - A European Journal 2014 Volume 20( Issue 44) pp:14514-14517
Publication Date(Web):
DOI:10.1002/chem.201403885
Abstract
The first asymmetric phospha-Michael addition of diarylphosphines to N-enoyl phthalimides has been developed in the presence of a chiral palladacycle catalyst. A library of free chiral tertiary phosphine adducts were directly obtained with excellent yields and enantioselectivities. Products can be subsequently functionalized to afford β-phosphinoamides, the direct preparation of which from cinnamides has been notoriously challenging.
Co-reporter:Jeanette See Leng Yap, Houguang Jeremy Chen, Yongxin Li, Sumod A. Pullarkat, and Pak-Hing Leung
Organometallics 2014 Volume 33(Issue 4) pp:930-940
Publication Date(Web):February 5, 2014
DOI:10.1021/om401044z
A novel racemic tertiary amine, 1-(2,5-diisopropylphenyl)-N,N-dimethylethanamine, was synthesized from 2,5-diisopropylbenzaldehyde via a multistep approach in high overall yield. The ortho palladation of this ligand was found to be sensitive to the reaction conditions and the palladating reagents employed. The metal complexation process could thus generate a cyclopalladated complex in high yield, lead to an unexpected N-demethylated amine palladium(II) complex, or both. Both products have been isolated and characterized crystallographically in the solid state and spectroscopically in solution. The racemic cyclopalladated complex could be efficiently resolved via the formation of (S)-prolinato derivatives. The absolute stereochemistries of the resolved diastereomeric complexes were determined by single-crystal X-ray crystallography in the solid state and by 1H–1H rotating frame Overhauser effect (ROESY) NMR spectroscopy in solution. An evaluation of the sterically hindered resolved cyclopalladated units as chiral auxiliaries was conducted in the endo-cycloaddition reaction between 3,4-dimethyl-1-phenylphosphole (DMPP) and ethyl vinyl ketone. The two expected phosphanorbornene adducts were generated with moderate stereoselectivity.
Co-reporter:Renta Jonathan Chew;Yinhua Huang;Yongxin Li;Sumod A. Pullarkat
Advanced Synthesis & Catalysis 2013 Volume 355( Issue 7) pp:1403-1408
Publication Date(Web):
DOI:10.1002/adsc.201300164
Abstract
An enantioselective Michael addition reaction of diphenylphosphine to substituted alkenylisoxazoles has been developed. The reaction proceeds efficiently under mild conditions with high yields (up to 99%) and moderate to excellent enantioselectivities (up to 92%) thus providing a hitherto unavailable direct access to a library of chiral tertiary phosphine-functionalized isoxazoles.
Co-reporter:Shuli Chen;Jun Xuan Chiew;Sumod A. Pullarkat;Yongxin Li
European Journal of Inorganic Chemistry 2013 Volume 2013( Issue 31) pp:5487-5494
Publication Date(Web):
DOI:10.1002/ejic.201300834
Abstract
Phenylbis(phenylethynyl)phosphane PhP(C≡CPh)2 coordinates regiospecifically to the α-methyl-chiral ortho-platinated and -palladated naphthylamine units at the positions trans to the nitrogen donors. The PPt coordination bond is kinetically inert, whereas the PPd bond is labile. Upon heating of these phosphane complexes at 70 °C, one of the C≡C bonds in the coordinated PhP(C≡CPh)2 was activated towards an intermolecular Pd–C bond insertion reaction with an external ortho-palladated naphthylamine ring. No intramolecular insertion reaction occurred. In contrast to its palladium analogue, the ortho-platinated ring is not reactive towards coordinated PhP(C≡CPh)2, although it can promote the Pd–C bond insertion reaction. However, despite the high kinetic stability of the PPt coordination, the organoplatinum unit is a noticeably weaker activator than its organopalladium counterpart. The chirality of the reacting ortho-metallated naphthylamine ligand exhibited high stereochemical influence on the formation of the new stereogenic phosphorus center during the course of these C–C bond-formation reactions. The coordination chemistry and the absolute stereochemistry of the dimetallic products were determined by single-crystal X-ray crystallographic analysis.
Co-reporter:Deepa Krishnan, Meiyi Wu, Minyi Chiang, Yongxin Li, Pak-Hing Leung, and Sumod A. Pullarkat
Organometallics 2013 Volume 32(Issue 8) pp:2389-2397
Publication Date(Web):April 3, 2013
DOI:10.1021/om400110t
A series of five-membered N-heterocyclic carbene C,S palladium(II) π-allyl complexes were successfully developed and characterized. Structural analyses of these complexes revealed that the organopalladium chelates adopt a skew-envelope conformation with a trans disposition of the substituents on the metal chelate rings. Utilizing these C,S palladium(II) π-allyl complexes as catalysts, a catalytic system for the allylic amination reaction has been developed. A series of C–N bond formations between amines and unsymmetrically substituted allylic carbonates could be catalyzed efficiently by complex (±)-9 in a regioselective manner.
Co-reporter:Dr. Deepa Krishnan;Dr. Sumod A. Pullarkat;Meiyi Wu;Dr. Yongxin Li ; Pak-Hing Leung
Chemistry - A European Journal 2013 Volume 19( Issue 17) pp:5468-5475
Publication Date(Web):
DOI:10.1002/chem.201204320
Abstract
Palladium and platinum complexes containing a sulfur-functionalised N-heterocyclic carbene (S-NHC) chelate ligand have been synthesised. The absolute conformations of these novel organometallic S-NHC chelates were determined by X-ray structural analyses and solution-phase 2D 1H–1H ROESY NMR spectroscopy. The structural studies revealed that the phenyl substituents on the stereogenic carbon atoms invariably take up the axial positions on the Pd-C-S coordination plane to afford a skewed five-membered ring structure. All of the chiral complexes are structurally rigid and stereochemically locked in a chiral ring conformation that is either (Rs,S,R)-λ or (Ss,R,R)-δ in both the solid state and solution.
Co-reporter:Ke Chen;Yongxin Li;Sumod A. Pullarkat
Advanced Synthesis & Catalysis 2012 Volume 354( Issue 1) pp:83-87
Publication Date(Web):
DOI:10.1002/adsc.201100424
Abstract
An efficient methodology involving the predominant formation of CC bonds is described for the first direct synthesis of 2-allylanilines from allylic alcohols via a one-pot tandem allylic amination/allylation protocol catalyzed by a palladacycle under mild conditions without the requirement for additional activators.
Co-reporter:Yinhua Huang ; Sumod A. Pullarkat ; Yongxin Li
Inorganic Chemistry 2012 Volume 51(Issue 4) pp:2533-2540
Publication Date(Web):January 30, 2012
DOI:10.1021/ic202472f
A highly reactive and stereoselective hydrophosphination of enones catalyzed by palladacycles for the synthesis of C*- and P*-chiral tertiary phosphines has been developed. When Ph2PH was employed as the hydrophosphinating reagent, a series of C*-chiral tertiary phosphines were synthesized (C*–P bond formation) in high yields with excellent enantioselectivities, and a single recrystallization provides access to their enantiomerically pure forms. When racemic secondary phosphines rac-R3(R4)PH were utilized, a series of tertiary phosphines containing both C*- and P*-chiral centers were generated (C*–P* bond formation) in high yields with good diastereo- and enantioselectivities. The stereoelectronic factors involved in the catalytic cycle have been revealed.
Co-reporter:Ke Chen, Sumod A. Pullarkat, Mengtao Ma, Yongxin Li and Pak-Hing Leung
Dalton Transactions 2012 vol. 41(Issue 17) pp:5391-5400
Publication Date(Web):09 Feb 2012
DOI:10.1039/C2DT12379G
A series of enantiomerically pure 1,2-diester substituted P,N-ligands incorporating two chiral carbons in the backbone were generated in high yields and high stereoselectivity from acetylenedicarboxylate via initial hydrophosphination using diphenylphosphine followed by hydroamination with various primary and secondary amines. The reactions were activated and stereochemically controlled by the organopalladium complex containing ortho-palladated (S)-(1-(dimethylamino)ethyl)naphthalene under mild conditions. The absolute stereochemistry and the coordination chemistry of P,N-products were determined by the single crystal X-ray diffraction analysis. All the chiral P,N-ligands could be liberated from the palladium template without loss of optical purity. Subsequent recomplexation to selected chiral palladium centers confirmed the optical purity of the new functionalized chiral P,N-ligands.
Co-reporter:Shuli Chen;Sumod A. Pullarkat;Yongxin Li
European Journal of Inorganic Chemistry 2012 Volume 2012( Issue 11) pp:1823-1831
Publication Date(Web):
DOI:10.1002/ejic.201101343
Abstract
A series of chiral ortho-platinated and -palladated complexes derived from N,N,α-trimethylbenzylamine has been prepared. The dialkynylphosphane PhP(C≡CMe)2 coordinated regiospecifically as a PM monodentate ligand to these cyclometallated units in the positions trans to the nitrogen donors of the metallacycles. Upon introduction of selected chirality on the benzylamine unit and subsequent monitoring by using NMR spectroscopy, the PPt bond was found to be thermodynamically stable and kinetically inert. The PPd bond must be kinetically labile since the palladium complexes readily undergo a facile ligand redistribution process. Nevertheless, both ortho-metallated units activated one of the C≡C bonds towards an intermolecular Pd–C bond insertion reaction into another cyclopalladated ring. No intramolecular insertion reaction occurred within the same molecule. Coordination of PhP(C≡CMe)2 to the ortho-palladated unit also protected the organopalladium ring from intermolecular attack by other coordinated dialkynylphosphanes. The Pt–C bonds in cycloplatinated rings are not reactive toward the insertion reaction, although they are good reaction activators for coordinated dialkynylphosphanes.
Co-reporter:Kien-Wee Tan, Xiang-Yuan Yang, Yongxin Li, Yinhua Huang, Sumod A. Pullarkat, and Pak-Hing Leung
Organometallics 2012 Volume 31(Issue 23) pp:8407-8413
Publication Date(Web):November 12, 2012
DOI:10.1021/om300976y
The monomeric ortho-platinated complexes [Pt{R1CH(1-C6H4)NMe2-C,N}{Ph2PC≡CR2}Cl] (R1 = Me, Et; R2 = Me, Ph) with trans-N,P geometries were obtained regiospecifically from the reaction between the dimeric [Pt2(μ-Cl)2{R1CH(1-C6H4)NMe2-C,N}2] and the corresponding alkynylphosphines in high yields. The phosphine complexes are highly stable in the solid state and in solution. However, in the presence of additional Pt(II) ions, an intramolecular coupling reaction occurred in which a new carbon–carbon bond was formed between the aromatic γ-carbon of the ortho-platinated chiral phenylamine and the α-carbon of the (Ph2P)–Cα≡Cβ–(R2) ligand. The (Ph2P) moiety migrated to the neighboring β-carbon during the coupling reaction. By the judicious selection of the substituents on the alkynylphoshine along with deliberate introduction of selected chirality on the ortho-platinated phenylamine, the coupling reaction and the (Ph2P) migration were found to proceed via an associative intramolecular mechanism that involves a Pt-vinylidene intermediate.
Co-reporter:Chang Xu, Gan Jun Hao Kennard, Felix Hennersdorf, Yongxin Li, Sumod A. Pullarkat, and Pak-Hing Leung
Organometallics 2012 Volume 31(Issue 8) pp:3022-3026
Publication Date(Web):March 20, 2012
DOI:10.1021/om201115n
A chiral palladacycle-promoted enantioselective asymmetric hydrophosphination of substituted methylidenemalonate esters using diphenylphosphine that provides direct access to chiral tertiary phosphines is reported. Screening of three easily accessible C,N and C,P palladacycles as catalysts for this synthetic scenario provided insights into critical factors in catalyst design that influence the activation and stereochemistry in Pd(II)-catalyzed asymmetric P–H addition reactions involving such activated substrates.
Co-reporter:Yuen Lin Cheow, Sumod A. Pullarkat, Yongxin Li, Pak-Hing Leung
Journal of Organometallic Chemistry 2012 696(26) pp: 4215-4220
Publication Date(Web):
DOI:10.1016/j.jorganchem.2011.09.016
Co-reporter:Yinhua Huang, Sumod A. Pullarkat, Siewping Teong, Renta Jonathan Chew, Yongxin Li, and Pak-Hing Leung
Organometallics 2012 Volume 31(Issue 13) pp:4871-4875
Publication Date(Web):June 19, 2012
DOI:10.1021/om300405h
A palladacycle-catalyzed diastereo- and enantioselective stepwise double hydrophosphination of bis(enones) with PhPH2 has been developed, allowing intermolecular construction of chiral tertiary bulky P-heterocycles in one pot in high yields. A catalytic cycle for the reaction is proposed as well.
Co-reporter:Yinhua Huang, Renta Jonathan Chew, Sumod A. Pullarkat, Yongxin Li, and Pak-Hing Leung
The Journal of Organic Chemistry 2012 Volume 77(Issue 16) pp:6849-6854
Publication Date(Web):July 27, 2012
DOI:10.1021/jo300893s
A highly reactive, chemo- and enantioselective addition of diphenylphosphine to α,β-unsaturated imines catalyzed by a palladacycle has been developed, thus providing the access to a series of chiral tertiary enaminophosphines in high yields. A putative catalytic cycle has also been proposed.
Co-reporter:Yinhua Huang, Renta Jonathan Chew, Yongxin Li, Sumod A. Pullarkat, and Pak-Hing Leung
Organic Letters 2011 Volume 13(Issue 21) pp:5862-5865
Publication Date(Web):October 10, 2011
DOI:10.1021/ol202480r
A highly diastereo- and enantioselective Pd(II)-catalyzed hydrophosphination of dienones with Ph2PH involving formation of double C*–P bonds has been developed, providing a series of chiral tertiary diphosphines (chiral PCP pincer ligands) in high yields. A catalytic cycle for the reaction was proposed.
Co-reporter:Shuli Chen;Sumod A. Pullarkat;Yongxin Li
European Journal of Inorganic Chemistry 2011 Volume 2011( Issue 20) pp:3111-3121
Publication Date(Web):
DOI:10.1002/ejic.201100208
Abstract
Cyclopalladated and cycloplatinated complexes, which incorporated the N,N-dimethylbenzylamine and N,N-dimethylnaphthylamine motifs, were successfully employed to promote a series of intermolecular monoinsertion reactions of diphenyl-1-propynylarsane, Ph2AsC≡CMe, into the Pd–C bond of the chiral α-methyl N,N-dimethylbenzylamine palladacycles. The precursor complexes were prepared by means of the coordination of the Ph2AsC≡CMe moiety onto the metal center trans to the benzylamine– or naphthylamine–N donor atom of the cyclometallated complexes. Subsequently a series of monoinsertion reactions were carried out between these precursor complexes and the enantiomerically pure N,N-dimethyl benzylamine palladacycles. These insertion reactions showed high regioselectivity under mild conditions and a variety of homo- and heterobimetallic arsenic functionalized complexes were formed. The coordination chemistry and the absolute stereochemistry of the monoinsertion products were determined by X-ray crystallography.
Co-reporter:Yi Zhang, Yi Ding, Sumod A. Pullarkat, Yongxin Li, Pak-Hing Leung
Journal of Organometallic Chemistry 2011 696(3) pp: 709-714
Publication Date(Web):
DOI:10.1016/j.jorganchem.2010.09.057
Co-reporter:Shuli Chen, Sumod A. Pullarkat, Yongxin Li, and Pak-Hing Leung
Organometallics 2011 Volume 30(Issue 6) pp:1530-1550
Publication Date(Web):February 17, 2011
DOI:10.1021/om101078p
Subsequent to their coordination onto chiral cyclopalladated/platinated [1-(dimethylamino)ethyl]naphthalene templates, a series of asymmetric monoinsertions of the carbon−carbon triple bond of dialkynylphosphines into the Pd−C bond of chiral α-methyl N,N-dimethyl benzylamine palladacycles have been demonstrated. These insertion reactions exhibited high regioselectivity and moderate stereoselectivity under mild conditions, and a variety of chiral homo- or heterobimetallic complexes incorporating a newly generated P-stereogenic center were formed. In some instances, the monoinsertion product would subsequently undergo a series of transformations during their purification via column chromatography or upon stirring them with H2O to generate a zwitterionic complex incorporating an additional four-membered ring system with a newly generated C-stereogenic center. The coordination chemistry and the absolute stereochemistry of the monoinsertion products and the transformation products were determined by single-crystal X-ray crystallographic analysis.
Co-reporter:Mingjun Yuan, Sumod A. Pullarkat, Yongxin Li, Pak-Hing Leung
Journal of Organometallic Chemistry 2011 696(4) pp: 905-912
Publication Date(Web):
DOI:10.1016/j.jorganchem.2010.10.029
Co-reporter:Yinhua Huang, Sumod A. Pullarkat, Yongxin Li and Pak-Hing Leung
Chemical Communications 2010 vol. 46(Issue 37) pp:6950-6952
Publication Date(Web):23 Aug 2010
DOI:10.1039/C0CC00925C
Chiral tertiary phosphines were synthesized by asymmetric hydrophosphination of aromatic enones catalyzed by an organopalladium complex with high yields and stereoselectivity. The procedure offers practical access to chiral tertiary phosphines.
Co-reporter:Mingjun Yuan ; Na Zhang ; Sumod A. Pullarkat ; Yongxin Li ; Fengli Liu ; Phuong-Tu Pham
Inorganic Chemistry 2010 Volume 49(Issue 3) pp:989-996
Publication Date(Web):December 30, 2009
DOI:10.1021/ic9018053
Aldehyde, ester- and keto-functionalized monophosphine palladium complexes containing the ortho-metalated (R)-(1-(dimethylamino)ethyl)naphthalene as the chiral auxiliary and reaction promoter were synthesized via hydrophosphination of acrolein and the subsequent Wittig reactions in a one-pot process. Under very mild conditions, the second-stage hydrophosphination of the monophosphine substrates gave the corresponding ester-, keto-, and hydroxyl-functionalized chiral 1,3-bis(diphenylphosphino)propane palladium complexes with good yields and stereoselectivities. The coordination properties and absolute configurations of the novel 1,3-diphosphine complexes were established by single crystal X-ray crystallography. The enantiomerically pure functionalized diphosphine ligands with ester and keto functionalities could be subsequently liberated stereospecifically by treatment of the corresponding dichloro palladium complexes with aqueous potassium cyanide in high yields.
Co-reporter:Mengtao Ma, Ruifeng Lu, Sumod A. Pullarkat, Weiqiao Deng and Pak-Hing Leung
Dalton Transactions 2010 vol. 39(Issue 23) pp:5453-5461
Publication Date(Web):17 May 2010
DOI:10.1039/B924613D
The asymmetric cycloaddition reactions of 3,4-dimethyl-1-phenylarsole and (Z/E)-diphenyl-1-propenylphosphine/diphenyl-1-styrylphosphine promoted by a chiral organopalladium(II) complex derived from (S)-[1-(dimethylamino)ethyl]naphthalene proceeded stereoselectively to generate different exo/endo-products. The reactions involving the (Z/E)-methyl substituted phosphines gave the individual optically pure exo- and endo-cycloadducts in very high stereoselectivity (33:1). However, when the methyl group was replaced by a Ph moiety both (Z/E)-phenyl substituted phosphines produced the same endo-cycloadduct in a stereoselectivity of 15:1. Every reaction produced five new chiral centers (four of them are sterically independent) in a single step and all three optically pure As–P heterobidentate ligands were obtained in high yields. The mechanism involved in the conversion of exo- to endo-product was investigated via density functional theory calculations. Computational results were consistent with the experimentally observed endo/exo-selectivity.
Co-reporter:Mengtao Ma;Sumod A. Pullarkat;Yongxin Li
European Journal of Inorganic Chemistry 2010 Volume 2010( Issue 12) pp:1865-1871
Publication Date(Web):
DOI:10.1002/ejic.200901203
Abstract
The cycloaddition reaction between 3,4-dimethyl-1-phenylarsole and diphenylvinylphosphane sulfide was promoted by chiral palladium and platinum complexes containingortho-metalated (S)-[1-(dimethylamino)ethyl]naphthalene. They exhibited similar stereoselectivity; the palladium cycloadducts could not be separated via column chromatography and fractional crystallization, however, the corresponding platinum complexes could be successfully converted into their enantiomerically pure counterpart. A formal arsanylidene elimination reaction was observed on the liberated free As/P=S bidentate ligand.
Co-reporter:Yi Ding;Yi Zhang;Yongxin Li;Sumod A. Pullarkat;Paul Andrews
European Journal of Inorganic Chemistry 2010 Volume 2010( Issue 28) pp:
Publication Date(Web):
DOI:10.1002/ejic.201090085
Abstract
The cover picture shows a novel chiral palladacycle, which was readily prepared from p-xylene and then resolved through the separation of its (S)-prolinate diastereomeric derivatives. The catalytic ability of the newly synthesized palladacycle was demonstrated in the preparation of a new diester-substituted diphosphane ligand by an asymmetric hydrophosphanation reaction, which proceeded with excellent selectivity. Details are discussed in the article by P.-H. Leung et al. on p. 4427 ff. The key molecule is depicted as superimposed over the image of the Chinese Heritage Centre building at the Nanyang Technological University, Singapore, and is meant to represent the contributions of Asian authors/readers to the European Journal of Inorganic Chemistry.
Co-reporter:Minyi Chiang;Yongxin Li;Deepa Krishnan;Pullarkat Sumod;Kim Hong Ng
European Journal of Inorganic Chemistry 2010 Volume 2010( Issue 9) pp:1413-1418
Publication Date(Web):
DOI:10.1002/ejic.200901142
Abstract
A novel chiral six-membered bidentate pyridine-N-heterocyclic carbene palladacycle was prepared via optical resolution of the racemic palladacycle. This is the first successful demonstration of the synthesis of a chiral carbene palladacycle via fractional crystallization of its chiral amino acid derivatives. Upon formation of (RC,SC) and (SC,SC)-phenylalanate derivatives, the (RC,SC)-phenylalanate diastereomer crystallized out spontaneously. Optically active (R)- and (S)-dichloropalladacycles can then be achieved by subsequent cleavage of the chiral amino acid auxiliary in the presence of aqueous HCl from their respective diastereomers. The absolute configurations of both the (R)- and (S)-palladacycles were determined by X-ray diffraction studies.
Co-reporter:Yi Ding;Yi Zhang;Yongxin Li;Sumod A. Pullarkat;Paul Andrews
European Journal of Inorganic Chemistry 2010 Volume 2010( Issue 28) pp:4427-4437
Publication Date(Web):
DOI:10.1002/ejic.201000471
Abstract
A novel amine ligand, 1-(2,5-dimethylphenyl)-N,N,2,2-tetramethylpropan-1-amine, was synthesized in six steps from commercially available p-xylene. Direct ortho-palladation of this amine ligand proceeded readily to form the racemic dimeric complex. The palladacycle structure and ring conformations of its triphenylphosphane derivative were thoroughly investigated by X-ray structural analysis in the solid state and 2D 1H–1H ROESY NMR spectroscopy in solution. This racemic palladacycle was efficiently resolved through the formation of its (S)-prolinato derivatives and an efficient separation of the resulting diastereomeric complexes was achieved by slow crystallization with the use of different solvent systems. The structure and absolute configuration of the two optically resolved palladium complexes were determined by X-ray diffraction. Both the (R,R) and (S,S)-di-μ-chlorido dimeric palladium complexes could be obtained chemoselectively by treating the corresponding prolinato derivatives with 1 M hydrochloric acid. The asymmetric hydrophosphanation reaction between diphenylphosphane and diethyl acetylenedicarboxylate was promoted by this newly synthesized palladacycle and resulted in the formation of a single isomer according to the 31P NMR spectrum. The amine auxiliary could be subsequently removed chemoselectively from the palladium center by treatment with concentrated HCl. An optically pure C2-symmetrical diphosphane ligand containing two ester functional groups at the two chiral carbon stereogenic centers was prepared by ligand displacement.
Co-reporter:Shuli Chen, Joseph Kok-Peng Ng, Sumod A. Pullarkat, Fengli Liu, Yongxin Li and Pak-Hing Leung
Organometallics 2010 Volume 29(Issue 15) pp:3374-3386
Publication Date(Web):July 2, 2010
DOI:10.1021/om100358g
A chiral palladacycle has been used successfully to promote the asymmetric hydrophosphination reactions between the racemic secondary phosphine ethylphenylphosphine and (E)/(Z)-diphenyl-1-propenylphosphine or 2-vinylpyridine in high regio- and stereoselectivities under mild conditions. Hydrophosphination of (E)-diphenyl-1-propenylphosphine with ethylphenylphosphine gave the asymmetric diphosphine chelate containing one stereogenic phosphorus donor with the R absolute configuration and one neighboring chiral S-carbon center as the major product in 60% yield. Using the same chiral metal template, the corresponding hydrophosphination reaction with (Z)-diphenyl-1-propenylphosphine gave the diastereomeric diphosphine in 40% yield with a chiral R-carbon center, but the controlled formation of the R-phosphorus configuration was not affected by the different stereochemistry in the carbon chain. A pair of separable diastereomeric palladium templates containing the naphthylamine auxiliary and the enantiomeric forms of (Rp/Sp)-[1-ethylphenylphosphino-2-(2-pyridine)]ethane were also generated in the ratio 1.5:1 via hydrophosphination of 2-vinylpyridine with ethylphenylphosphine. The optically pure diphosphine ligands and P, N ligands could be stereospecifically liberated from the template complexes. The coordination chemistry and the absolute stereochemistry of the template complexes and dichloro complexes were determined by X-ray crystallography.
Co-reporter:Yinhua Huang, Sumod A. Pullarkat, Mingjun Yuan, Yi Ding, Yongxin Li and Pak-Hing Leung
Organometallics 2010 Volume 29(Issue 3) pp:536-542
Publication Date(Web):December 16, 2009
DOI:10.1021/om900829t
Organopalladium complexes containing ortho-metalated (R)-(1-(dimethylamino)ethyl)naphthalene and (R)-[1-(1-(dimethylamino)-2,2-dimethylpropyl]-2,5-dimethylbenzene as the chiral auxiliaries were used to promote the asymmetric hydrophosphination reactions between diphenylphosphine and ester- and keto-functionalized allenes in high regio- and stereoselectivites under mild conditions. The hydrophosphination reactions generated the 1,2-diphosphine ligands with chirality residing on the carbon backbone as bidentate chelates on the chiral organopalladium templates. The major isomers were isolated in appreciable yields in configurationally pure forms and characterized by means of single-crystal X-ray crystallography. The chiral auxiliaries could be removed chemoseletively by treatment with concentrated hydrochloric acid to form the corresponding optically pure neutral dichloro complexes. Subsequently, the dichloro complexes underwent ligand displacement with aqueous cyanide to generate their corresponding optically pure diphosphine ligands in high yields.
Co-reporter:Mingjun Yuan, Sumod A. Pullarkat, Yongxin Li, Zhi-Yi Lee and Pak-Hing Leung
Organometallics 2010 Volume 29(Issue 16) pp:3582-3588
Publication Date(Web):July 28, 2010
DOI:10.1021/om100505w
A novel cyano-functionalized monophosphine palladium substrate containing the ortho-metalated (R)-(1-(dimethylamino)ethyl)naphthalene as the chiral auxiliary was synthesized from 3-chloropropionaldehyde diethylacetal via a one-pot process. The asymmetric hydrophosphination reactions between diphenylphosphine and the trans- or cis-monophosphine substrates were carried out under mild conditions, which gave the corresponding cyano-substituted chiral 1,3-bis(diphenylphosphino)propane palladium complexes with good yields and stereoselectivities. Subsequent functional group transformation reactions were conducted by successive treatment of the hydrophosphination products with Dibal-H and chemoselectively yielded the formyl- and hydroxyl-functionalized chiral 1,3-diphosphine complexes. The absolute configurations and coordination information of the novel 1,3-diphosphine complexes were analyzed by X-ray crystallography. The optically pure 1,3-bis(diphenylphosphino)propane ligands with cyano, formyl, and hydroxyl functionalities could be liberated in high yields from the corresponding dihalo palladium complexes by treatment with aqueous potassium cyanide.
Co-reporter:Ding Luo, Yongxin Li, Sumod A. Pullarkat, Kirsty E. Cockle and Pak-Hing Leung
Organometallics 2010 Volume 29(Issue 4) pp:893-903
Publication Date(Web):January 15, 2010
DOI:10.1021/om900958p
A class of bifunctionalized phosphine complexes bearing a stereogenic center at phosphorus have been synthesized through stepwise insertion and hydration of the two alkynyl groups of a prochiral dialkynylphosphine complex, [Ru(η6-benzene){PPh(C≡CCH3)2}Cl2]. One of the two alkyne moieties of this complex inserted into the Pd−C bond of cyclopalladated benzylamine complexes to yield bimetallic complexes containing two stereogenic centers at ruthenium and phosphorus with high stereoselectivity and yield. Subsequently, the remaining free alkynyl group underwent hydration in a mixed solvent system of DCM/acetone/H2O (2:10:1), which resulted in the formation of bimetallic zwitterions bearing anionic palladium(II) and cationic ruthenium(II) centers. The carbon−carbon triple bond was converted into a ketonyl group during the hydration, and the absolute configuration at phosphorus was observed to remain unchanged in the products. All structures of the insertion and hydration products have been identified by X-ray analyses or 2D ROESY NMR studies.
Co-reporter:Yi Zhang, Sumod A. Pullarkat, Yongxin Li and Pak-Hing Leung
Inorganic Chemistry 2009 Volume 48(Issue 12) pp:5535-5539
Publication Date(Web):May 15, 2009
DOI:10.1021/ic900461t
An organopalladium complex containing ortho-metalated (R)-(1-(dimethylamino) ethyl)naphthalene as the chiral auxiliary has been used to promote the asymmetric hydrophosphination reaction between diphenylphosphine and phenyldi[(Z)-prop-1-enyl]phosphine in high regio- and stereoselectivity under mild conditions. The hydrophosphination reaction generated only two diastereomers in a ratio of 1:1. The two hydrophosphination products contained both phosphorus and carbon stereogenic centers and were subsequently isolated by fractional crystallization. Their absolute stereochemistries were analyzed by X-ray crystallography. The naphthylamine auxiliary could be removed chemoselectively from the template products by treatment with concentrated hydrochloric acid to form the corresponding optically pure neutral dichloro complexes. Subsequently, the dichloro complexes underwent ligand displacement with aqueous cyanide to generate the optically pure diphosphine ligands in high yields.
Co-reporter:Fengli Liu ; Sumod A. Pullarkat ; Kien-Wee Tan ; Yongxin Li
Inorganic Chemistry 2009 Volume 48(Issue 23) pp:11394-11398
Publication Date(Web):November 4, 2009
DOI:10.1021/ic9014543
The organoplatinum complex containing ortho-metalated (R)-(1-(dimethylamino)ethyl)-naphthalene as the chiral auxiliary has been used efficiently to promote the asymmetric [4 + 2] Diels−Alder reaction between diphenylvinylphosphine and 3-diphenylphosphinofuran to generate two chelating diphosphine endocycloadducts in the ratio 17:1. The absolute configurations of the three newly generated stereocenters have been assigned by single-crystal X-ray analysis.
Co-reporter:Mingjun Yuan, Sumod A. Pullarkat, Crystal Huixian Yeong, Yongxin Li, Deepa Krishnan and Pak-Hing Leung
Dalton Transactions 2009 (Issue 19) pp:3668-3670
Publication Date(Web):26 Mar 2009
DOI:10.1039/B904090K
Optically pure P-chiral 1,2- and 1,3-diphosphines were synthesized chemoselectively and stereoselectively from the chiral organopalladium complex promoted asymmetric Diels–Alder reaction between 3,4-dimethyl-1-phenylphosphole (DMPP) and cis- and trans-ester substituted allylic phosphines.
Co-reporter:Sumod A. Pullarkat;Yuen Lin Cheow;Yongxin Li
European Journal of Inorganic Chemistry 2009 Volume 2009( Issue 16) pp:2375-2382
Publication Date(Web):
DOI:10.1002/ejic.200900095
Abstract
Enantiomerically pure, alcohol-functionalized diphosphane ligands carrying one phosphorus and three carbon stereogenic centers were generated from the Diels–Alder reactions of phosphane-functionalized terminal alkenols [3-(diphenylphosphanyl)but-3-en-1-ol and 2-(diphenylphosphanyl)prop-2-en-1-ol] with 3,4-dimethyl-1-phenyl-1H-phosphole. The reactions were promoted and controlled by the organoplatinum complex containing ortho-metalated (R)-[1-(dimethylamino)ethyl]naphthalene, and both cycloadditions showed excellent regio- and stereoselectivity under mild conditions with only one enantiomer being formed. The products were isolated in high yield and were characterized by single-crystal X-ray diffraction analysis. Their structures in solution were analyzed by 2D 1H–1H ROESY NMR spectroscopy. Subsequent decomplexation and repreparation of the products proved the optical purity of the alcohol-functionalized, chiral diphosphanes formed.(© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2009)
Co-reporter:Yi Ding;Yongxin Li;Sumod A. Pullarkat;See Leng Yap
European Journal of Inorganic Chemistry 2009 Volume 2009( Issue 2) pp:267-276
Publication Date(Web):
DOI:10.1002/ejic.200800923
Abstract
A novel palladacycle was designed and prepared by direct cyclopalladation of the ligand 1-(3,6-dimethylnaphthalen-1-yl)-N,N-dimethylethanamine, which was synthesized by a multistep sequence starting from 2,7-dimethylnaphthalene. The palladacycle structure and ring conformations of its triphenylphosphane derivative was investigated by X-ray structural analysis in the solid state and 2D 1H–1H ROESY NMR spectroscopy in solution. The ortho-palladated ring is stereochemically rigid, and the five-membered ring conformation is locked by repulsive interactions between the methyl group on the prochiral carbon atom and its neighboring naphthylene proton. The racemic cyclopalladated complex could be efficiently resolved through the formation of its (S)-prolinato derivatives, and efficient separation of the resulting diastereomeric complexes was achieved by simple silica-gel chromatography. The structures and absolute configurations of the two optically resolved palladium complexes were determined by X-ray crystallography. Both the (R,R)- and (S,S)-di-μ-chloride dimeric palladium complexes could be obtained chemoselectively by treating the corresponding prolinato derivatives with 1 M hydrochloric acid. Despite the severe interchelate steric constraints within this new organopalladium complex, the bulky monodentate ligand 3,4-dimethyl-1-phenylphosphole (dmpp) was coordinated regiospecifically to the ortho-palladated amine unit trans to the NMe2 group. In comparison to its naphthylamine analogue, the new palladacycle showed a much higher stereoselectivity in the chiral template-promoted asymmetric Diels–Alder reaction of coordinated dmpp and N,N-dimethylacrylamide.(© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2009)
Co-reporter:Fengli Liu;Sumod A. Pullarkat;Yongxin Li;Shuli Chen
European Journal of Inorganic Chemistry 2009 Volume 2009( Issue 27) pp:4134-4140
Publication Date(Web):
DOI:10.1002/ejic.200900460
Abstract
The asymmetric hydroarsanation reactions between diphenylarsane and (E)-1-phenyl-3-(pyridin-2-yl)-2-propenone and (E)-1-methyl-3-(pyridin-2-yl)-2-propenoate have been achieved by use of the organopalladium complex containing ortho-metalated (R)-[1-(dimethylamino)ethyl]naphthalene as the chiral reaction template in high regio- and stereoselectivities under mild conditions. Hydroarsanation of (E)-1-phenyl-3-(pyridin-2-yl)-2-propenone with diphenylarsane generated two stereoisomeric products in the ratio of 3:1 as five-membered As–N bidentate chelates on the chiral naphthylamine palladium template. Using the same chiral metal template, the corresponding hydroarsanation reaction with (E)-1-methyl-3-(pyridin-2-yl)-2-propenoate gave only one product as a six-membered As–N bidentate chelate. The naphthylamine auxiliary could be removed chemoselectively by treatment with concentrated hydrochloric acid to form the corresponding optically pure neutral complexes. Subsequent ligands displacement from the palladium using aqueous potassium cyanide generated the optically pure keto- and ester-functionalized chiral pyridylarsane ligands. The absolute configuration and the coordination properties of the pyridylarsanes have been established by single-crystal X-ray analysis.(© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2009)
Co-reporter:Yi Zhang, Lulu Tang, Sumod A. Pullarkat, Fengli Liu, Yongxin Li, Pak-Hing Leung
Journal of Organometallic Chemistry 2009 694(21) pp: 3500-3505
Publication Date(Web):
DOI:10.1016/j.jorganchem.2009.06.024
Co-reporter:Mengtao Ma, Sumod A. Pullarkat, Ke Chen, Yongxin Li, Pak-Hing Leung
Journal of Organometallic Chemistry 2009 694(12) pp: 1929-1933
Publication Date(Web):
DOI:10.1016/j.jorganchem.2009.01.044
Co-reporter:Mingjun Yuan, Sumod A. Pullarkat, Mengtao Ma, Yi Zhang, Yinhua Huang, Yongxin Li, Akash Goel and Pak-Hing Leung
Organometallics 2009 Volume 28(Issue 3) pp:780-786
Publication Date(Web):January 15, 2009
DOI:10.1021/om8009188
Ester- and keto-functionalized allylic monophosphine palladium complexes containing ortho-metalated (R)-(1-(dimethylamino)ethyl)naphthalene as the chiral auxiliary were synthesized via a versatile one-pot process. Subsequent asymmetric hydrophosphination of the coordinated allylic substrates promoted by the chiral auxiliary gave the corresponding functionalized chiral 1,2-bis(diphenylphosphino)ethane ligands in high yields under mild conditions. The coordination properties and absolute configurations of the resultant diphosphine ligands were established by single-crystal X-ray crystallography. The coordinated 1,2-diphosphine ligands could be subsequently liberated stereospecifically to yield enantiomerically pure functionalized diphosphines in high yields.
Co-reporter:Fengli Liu, Sumod A. Pullarkat, Yongxin Li, Shuli Chen, Mingjun Yuan, Zhi Yi Lee and Pak-Hing Leung
Organometallics 2009 Volume 28(Issue 13) pp:3941-3946
Publication Date(Web):June 1, 2009
DOI:10.1021/om900305u
The organopalladium complex containing ortho-metalated (R)-(1-(dimethylamino)ethyl)naphthalene as the chiral auxiliary has been used to promote the asymmetric hydrophosphination reactions of diphenylphosphine with (E)-1-phenyl-3-pyridin-2-yl-2-propenone and (E)-1-methyl-3-pyridin-2-yl-2- propenoate in high regio- and stereoselectivities under mild conditions. Hydrophosphination of (E)-1-phenyl-3-pyridin-2-yl-2-propenone with diphenylphosphine generated two stereoisomeric products in a ratio of 8:1 as five-membered 2-pyridylphosphine P−N bidentate chelates on the chiral naphthylamine palladium template. Using the same chiral metal template, the corresponding hydrophosphination reaction of (E)-1-methyl-3-pyridin-2-yl-2-propenoate gave only one product as a six-membered P−N bidentate chelate. The naphthylamine auxiliary could be removed chemoselectively from the template product by treatment with concentrated hydrochloric acid to form the corresponding optically pure neutral complexes. Subsequent ligand displacement from the palladium achieved using aqueous potassium cyanide generated the optically pure keto- and ester-functionalized chiral 2-pyridylphosphines ligands in high yields. The absolute configuration and the coordination properties of the 2-pyridylphosphines have been established by single-crystal X-ray analysis.
Co-reporter:Yi Ding, Minyi Chiang, Sumod A. Pullarkat, Yongxin Li and Pak-Hing Leung
Organometallics 2009 Volume 28(Issue 15) pp:4358-4370
Publication Date(Web):July 17, 2009
DOI:10.1021/om900423f
A novel five-membered dimeric phosphapalladacycle was prepared using palladium(II) acetate as the ortho-palladation source. A successful optical resolution of the palladacycle was achieved through the separation of its (S)-prolinate derivatives by fractional crystallization using different solvent systems. Both the (R,R)- and (S,S)-di-μ-chloro dimeric palladium complexes can be obtained by treating the corresponding prolinato derivatives with 1 M hydrochloric acid, and the absolute configurations of both optically active dimers were concluded from the X-ray diffration studies. Two phosphine ligands, triphenylphosphine (PPh3) and 3,4-dimethyl-1-phenylphosphole (dmpp), were able to coordinate with the dimeric palladium complex, and thus the palladacycle’s coordination characteristics were studied by both 31P NMR and X-ray diffraction. The phosphapalladacycle’s conformational behavior was further studied by the 2D 1H−1H ROESY NMR of its acetylacetonate derivative, which indicated that the α-methyl group was axially located and the palladacycle was conformationally rigid in solution. Moreover, the Pd−C bond in the palladacycle and the dmpp derivative was found to be ruptured immediately in the presence of concentrated HCl.
Co-reporter:Mengtao Ma, Sumod A. Pullarkat, Mingjun Yuan, Na Zhang, Yongxin Li and Pak-Hing Leung
Organometallics 2009 Volume 28(Issue 16) pp:4886-4889
Publication Date(Web):July 31, 2009
DOI:10.1021/om900530q
The organopalladium complex containing ortho-metalated (S)-[1-(dimethylamino)ethyl]naphthalene as the chiral auxiliary has been used successfully to promote the asymmetric cycloaddition reaction between 3,4-dimethyl-1-phenylarsole and diphenylvinylphosphine oxide in high stereoselectivity (only one isomer). However, the neutral dichloro palladium complex obtained upon removal of the auxiliary is very unstable and instantly decomposed to produce the arsenic elimination reaction product (3,4-dimethyl-2,4-cyclohexadienyl)diphenylphosphine oxide. However when the same reaction was promoted by the analogous platinum complex, only one stereoisomer was again obtained, but the resulting dichloro platinum complex is very stable in the solid state as well as in solution and coordinates to the platinum center as a As−O bidentate ligand, as confirmed by 1H NMR and single-crystal X-ray analysis. The enantiomerically pure As ͡P═O hetero-bidentate ligand could be readily liberated from the dichloro platinum complex as an air-sensitive solid by treatment of the complex with aqueous potassium cyanide.
Co-reporter:Ding Luo, Yongxin Li, Kien-Wee Tan and Pak-Hing Leung
Organometallics 2009 Volume 28(Issue 21) pp:6266-6274
Publication Date(Web):October 14, 2009
DOI:10.1021/om900502a
Stepwise functionalization of the two alkyne moieties in a dialkynylphosphine complex has been studied. The two alkynyl groups underwent stepwise hydrophosphination and insertion to yield two different substituents on the stereogenic phosphorus. Coordination of the dialkynylphosphine ligand PPh(C≡CCH3)2 to the ruthenium center generated the complex [Ru(η6-benzene){PPh(C≡CCH3)2}Cl2]. Removal of one Cl atom by AgPF6 followed by coordination of HPPh2 to ruthenium promoted the hydrophosphination reaction with high stereoselectivity. The hydrophosphination products then underwent insertion into the Pd−C bond of a cyclopalladated complex to give a bimetallic complex bearing a stereogenic phosphorus center with expected substituents. The product contains also a tridentate ligand chelating to palladium, which is believed to have been generated through a proton exchange process aided by palladium. Furthermore, this complex exists as two interconvertable conformations in a ratio of 3:1. The structures of complexes were confirmed by X-ray crystallographic analyses and 2D ROESY NMR studies.
Co-reporter:Fengli Liu, Sumod A. Pullarkat, Kien-Wee Tan, Yongxin Li and Pak-Hing Leung
Organometallics 2009 Volume 28(Issue 21) pp:6254-6259
Publication Date(Web):October 9, 2009
DOI:10.1021/om900586q
The asymmetric [4+2] Diels−Alder reaction involving 3-diphenylphosphinofuran as the dienophile and 3,4-dimethyl-1-phenylphosphole, DMPP, as the cyclic diene was carried out by utilizing the platinum(II) template complex containing ortho-metalated (R)-(1-(dimethylamino)ethyl)naphthalene as the chiral auxiliary. The cycloaddition reaction proceeded at 40 °C via an intramolecular mechanism in which the cyclic diene and the dienophile were coordinated simultaneously on the chiral platinum template. Two (SP)-exo cycloadducts were obtained with high stereoselectivity as a P−P bidentate chelate on the platinum template. The chiral naphthylamine auxiliary was subsequently removed by treatment with concentrated hydrochloric acid to form three dichloroplatinum(II) complexes, of which two products were from acid hydrolysis of the vinyl ether. Benzoylation of the hemiacetals gave only one product. The absolute configurations and the coordination properties of the P-chiral cycloadducts have been established by single-crystal X-ray analysis.
Co-reporter:Yi Ding;Yongxin Li;Yi Zhang;Sumod A. Pullarkat
European Journal of Inorganic Chemistry 2008 Volume 2008( Issue 11) pp:1880-1891
Publication Date(Web):
DOI:10.1002/ejic.200701234
Abstract
A novel chiral palladacycle containing ortho-metallated (±)-1-(3,6-dimethylnaphthalen-2-yl)-N,N-dimethylethylaminewas designed and synthesized by a multi-step synthesis by using 2,7-dimethylnaphthalene as the starting material. The structure and palladacycle ring conformation of the triphenylphosphane derivative was investigated by X-ray structural analysis in the solid state and by 2D ROESY NMR spectroscopy in solution. Through a designed intramolecular steric interaction, the five-membered naphthylamine chelate can be locked into a fixed conformation, both in the solid state and in solution. The racemic cyclopalladated complex could be efficiently resolved through the formation of its (S)-prolinato and (S)-alaninato derivatives. The structure and absolute configuration of the two optically resolved palladium complexes were determined by X-ray diffraction. Both the (R,R) and (S,S)-di-μ-chlorido dimeric palladium complexes containing the resolved amine ligand was obtained chemoselectively by treating the corresponding prolinato and alaninato derivatives with 1 M hydrochloric acid. Despite the severe interchelate steric constraint within these new organopalladium complexes, the bulky monodentate ligand 3,4-dimethyl-1-phenylphosphole (dmpp) was able to split the chlorido bridges regiospecifically in the position trans to the NMe2 group. With the new palladacycle used as the chiral template, the stereoselectivity of Diels–Alder cycloaddition between dmpp and ethyl vinyl ketone significantly improves relative to that of other analogue complexes.(© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2008)
Co-reporter:Joseph K.P. Ng, Shuli Chen, Geok K. Tan, Pak-H. Leung
Tetrahedron: Asymmetry 2007 Volume 18(Issue 10) pp:1163-1169
Publication Date(Web):11 June 2007
DOI:10.1016/j.tetasy.2007.05.018
The conformational behaviors of the five-membered palladacycles derived from the ligands, 1-(1′-naphthyl)ethyldiphenylphosphine and 1-(1′-naphthyl)ethyldiphenylarsine were determined from their O,O′-acetylacetonate complexes. Despite the possibilities of both palladacycles to exist in both δ and λ conformations, these palladacycles were noted to be conformationally rigid and only one of these was adopted in both the solid state and in solution (CDCl3), as supported from their X-ray molecular structures and 2-D 1H–1H ROESY NMR studies, respectively.(Acetylacetonato-O,O′){(S)-1-[1-(diphenylphosphino)ethyl]naphthyl-C2,P}palladium(II)C29H27O2PPd[α]D = +594 (c 0.5, CH2Cl2)Absolute configuration: (S)(Acetylacetonato-O,O′){(S)-1-[1-(diphenylarsino)ethyl]naphthyl-C2,As}palladium(II)C29H27O2AsPd[α]D = +391 (c 0.4, CH2Cl2)Absolute configuration: (S)
Co-reporter:Wee-Chuan Yeo;Geok-Kheng Tan;Lip Lin Koh
European Journal of Inorganic Chemistry 2005 Volume 2005(Issue 23) pp:
Publication Date(Web):25 OCT 2005
DOI:10.1002/ejic.200500549
An organoplatinum complex containing the (S)-form of orthometalated [1-(dimethylamino)ethyl]naphthalene as the chiral auxiliary promotes the asymmetric [2+2] dimerization of (E)-2-(diphenylphosphanyl)styrene to generate two isomeric chelating diphosphanyl-substituted cyclobutane platinum template complexes in the ratio of 6:1. The four substituents on the cyclobutane rings adopt the pseudo-equatorial positions with an all-trans arrangement. The naphthylamine auxiliary can be removed chemoselectively from the template products by treatment with concentrated hydrochloric acid to form the corresponding dichloroplatinum(II) complexes, which, upon subsequent ligand displacement with aqueous cyanide, liberate the enantiomerically enriched free diphosphane ligand in high yields. Recomplexation of the liberated diphosphane to the (R)-form of the platinum template, followed by fractional crystallization, allows separation of the optically pure platinum template complex containing the diphosphane ligand (1R,2R,3R,4R)-1,2-bis(diphenylphosphanyl)-3,4-diphenylcyclobutane, from which the free diphosphane can be subsequently liberated efficiently. (© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2005)
Co-reporter:Soh-Keang Loh, Geok-Kheng Tan, Lip Lin Koh, S. Selvaratnam, Pak-Hing Leung
Journal of Organometallic Chemistry 2005 Volume 690(21–22) pp:4933-4938
Publication Date(Web):1 November 2005
DOI:10.1016/j.jorganchem.2005.08.009
The organopalladium(II) complex containing the (S)-form of ortho-palladated (1-(dimethylamino)ethyl)naphthylalene has been successfully utilised as a chiral template to promote the asymmetric endo-cycloaddition reaction between coordinated 3,4-dimethyl-1-phenyl-phosphole and acrolein. The rate of this chiral template promoted reaction is dramatically affected by the solvent and temperature. In dichloromethane, the intermolecular cycloaddition reaction at room temperature gave a 2:1 mixture of the diastereomeric endo-substituted formyl-phosphanorbornene template complexes in 35 days. The major diastereomer could be isolated by fractional crystallization. The absolute configurations and the coordination properties of the endo-formylphosphines in the isolated template complex have been established by X-ray crystallography. The enantiomerically pure endo-cycloadduct (−)-5-(formyl)-2,3-dimethyl-7-phenyl-7-phosphabicyclo[2.2.1]hept-2-ene was obtained by the treatment of the major product with dppe. When the endo-cycloaddition reaction was conducted in the ionic liquid 1-hexyl-3-methylimidazolium tetrafluoroborate ([hmim][BF4]), the same 2:1 diastereomeric mixture was obtained in two days. When the reaction temperature was raised to 85 °C, the reaction generated the two diastereomeric endo-cycloadducts as a 1:1 mixture in 2 h. Similarly, a 1:1 mixture was obtained when the reaction was heated at 85 °C in 1,2-dichloroethane for 2 days.The organopalladium complex bearing the (S)-ortho-palladated (1-(dimethylamino)-ethyl)-naphthylalene successfully promoted the asymmetric endo-cycloaddition reaction between coordinated 3,4-dimethyl-1-phenyl-phosphole and acrolein to give complex 2 in both ionic liquid and organic solvent.
Co-reporter:Yu-Xiang Jia, Xiang-Yuan Yang, Wee Shan Tay, Yongxin Li, Sumod A. Pullarkat, Kai Xu, Hajime Hirao and Pak-Hing Leung
Dalton Transactions 2016 - vol. 45(Issue 5) pp:NaN2101-2101
Publication Date(Web):2015/10/07
DOI:10.1039/C5DT02049B
A 13C{1H} NMR based investigation was conducted to examine the electronic properties of C(aryl)–M bonds and their trans influence in P–C(aryl)–P pincer complexes. A series of structurally related platinum pincer complexes were rationally designed and their corresponding 13C–195Pt coupling constants were systematically examined. By methodical substitution of the ligand trans to the organometallic C(aryl)–Pt bond, this study revealed the significant influence of the ligands on the nature of the C(aryl)–M bonds. The single crystal X-ray analysis of the complexes and computational studies further confirmed the observations that the C–M bond exhibits significant π-character.
Co-reporter:Wee Shan Tay, Xiang-Yuan Yang, Yongxin Li, Sumod A. Pullarkat and Pak-Hing Leung
Chemical Communications 2017 - vol. 53(Issue 47) pp:NaN6310-6310
Publication Date(Web):2017/05/03
DOI:10.1039/C7CC02044A
A catalytic asymmetric hydroarsination reaction of an activated alkene viz. (E)-nitrostyrene was developed using chiral PCP Pt-, Pd- and Ni-pincer complexes as catalysts. The corresponding chiral tertiary arsine adduct was obtained in ees of up to 80% under mild reaction conditions using the PCP Ni–Cl pincer catalyst. The arsine adduct was furnished with catalyst loadings of 1–5 mol% and the reaction duration ranging from <5 min to 180 min. The subsequent coordination of the hydroarsination product to gold(I) chloride allowed for the confirmation of the stereochemistry of the arsine adduct via crystallographic analysis.
Co-reporter:Xiang-Yuan Yang, Wee Shan Tay, Yongxin Li, Sumod A. Pullarkat and Pak-Hing Leung
Chemical Communications 2016 - vol. 52(Issue 22) pp:NaN4214-4214
Publication Date(Web):2016/02/17
DOI:10.1039/C6CC00763E
Cyclopalladation of the pyridyl-substituted chiral phosphine sulfide (N–PS) and oxide (N–PO) compounds afforded the asymmetric N–C(sp3)*–S and N–C(sp3)*–O pincer complexes. When applied as catalysts in asymmetric hydrophosphination, the newly developed aliphatic pincer catalyst could be recycled over three runs and obtained in large quantities via a one-pot “self-breeding” catalytic protocol.
Co-reporter:Jeanette See Leng Yap, Bin Bin Li, Jonathan Wong, Yongxin Li, Sumod A. Pullarkat and Pak-Hing Leung
Dalton Transactions 2014 - vol. 43(Issue 15) pp:NaN5784-5784
Publication Date(Web):2014/01/29
DOI:10.1039/C4DT00062E
A novel amine ligand, 1-(2,5-dichlorophenyl)-N,N-dimethylethanamine, was synthesized from 1-(2,5-dichlorophenyl)ethanone via a three step synthetic route. Direct ortho-palladation of the amine ligand with Pd(OAc)2 gave the racemic dimeric complex in high yield. This racemic palladacycle was efficiently resolved through the formation of its (S)-prolinato derivatives. The resulting diastereomeric complexes were separated efficiently by column chromatography. In the solid state, the structure and absolute configuration of the two optically resolved palladium complexes were determined by single crystal X-ray crystallography. In solution, their absolute conformations were also investigated by the 2D 1H–1H rotating frame nuclear Overhauser enhancement (ROESY) NMR spectroscopy. Both (R,R) and (S,S)-di-μ-chloro dimeric palladium complexes could be obtained chemoselectively by treating the corresponding prolinato derivatives with dilute hydrochloric acid. The amine auxiliary could be subsequently removed from the palladium center by treatment with concentrated hydrochloric acid. The enantiomerically pure palladacycle was used to promote the asymmetric hydrophosphination reaction between diphenylphosphine and dimethyl acetylenedicarboxylate. The 31P{1H} NMR spectroscopy indicated that only one stereo-isomeric product was formed.
Co-reporter:Ke Chen, Sumod A. Pullarkat, Mengtao Ma, Yongxin Li and Pak-Hing Leung
Dalton Transactions 2012 - vol. 41(Issue 17) pp:NaN5400-5400
Publication Date(Web):2012/02/09
DOI:10.1039/C2DT12379G
A series of enantiomerically pure 1,2-diester substituted P,N-ligands incorporating two chiral carbons in the backbone were generated in high yields and high stereoselectivity from acetylenedicarboxylate via initial hydrophosphination using diphenylphosphine followed by hydroamination with various primary and secondary amines. The reactions were activated and stereochemically controlled by the organopalladium complex containing ortho-palladated (S)-(1-(dimethylamino)ethyl)naphthalene under mild conditions. The absolute stereochemistry and the coordination chemistry of P,N-products were determined by the single crystal X-ray diffraction analysis. All the chiral P,N-ligands could be liberated from the palladium template without loss of optical purity. Subsequent recomplexation to selected chiral palladium centers confirmed the optical purity of the new functionalized chiral P,N-ligands.
Co-reporter:Xiang-Yuan Yang, Jun Hao Gan, Yongxin Li, Sumod A. Pullarkat and Pak-Hing Leung
Dalton Transactions 2015 - vol. 44(Issue 3) pp:NaN1263-1263
Publication Date(Web):2014/11/12
DOI:10.1039/C4DT02673J
Both PC-cyclometalated and PCP-pincer type palladium catalysts have recently been found to be robust and efficacious catalysts for the asymmetric P–H addition reaction involving activated olefins. Our studies on the asymmetric P–H addition of diphenylphosphine to malonate ester and α,β,γ,δ-alkylidenemalonate ester revealed for the first time that the catalyst choice can have a dramatic impact in terms of reactivity as well as regio- and stereo-control for this asymmetric hydrofunctionalization reaction. Besides showing significantly contrasting reactivity and stereoselectivity in the hydrophosphination reaction involving malonate ester, in the case of α,β,γ,δ-alkylidenemalonate ester, a novel regiodivergent method was developed with the 1,4-adduct being obtained exclusively with the PC-catalyst while the pincer catalyst produced only the 1,6-adduct.
Co-reporter:Renta Jonathan Chew, Kai Yuan Teo, Yinhua Huang, Bin-Bin Li, Yongxin Li, Sumod A. Pullarkat and Pak-Hing Leung
Chemical Communications 2014 - vol. 50(Issue 63) pp:NaN8770-8770
Publication Date(Web):2014/06/26
DOI:10.1039/C4CC01610F
An enantioselective hydrophosphination of β,γ-unsaturated α-ketoesters and amides has been developed using a chiral palladacycle catalyst. Adducts can be obtained in excellent yields and enantioselectivities, providing direct access to chiral tertiary phosphines which are synthetically useful intermediates in the preparation of bidentate ligands.
Co-reporter:Yinhua Huang, Sumod A. Pullarkat, Yongxin Li and Pak-Hing Leung
Chemical Communications 2010 - vol. 46(Issue 37) pp:NaN6952-6952
Publication Date(Web):2010/08/23
DOI:10.1039/C0CC00925C
Chiral tertiary phosphines were synthesized by asymmetric hydrophosphination of aromatic enones catalyzed by an organopalladium complex with high yields and stereoselectivity. The procedure offers practical access to chiral tertiary phosphines.
Co-reporter:Mengtao Ma, Ruifeng Lu, Sumod A. Pullarkat, Weiqiao Deng and Pak-Hing Leung
Dalton Transactions 2010 - vol. 39(Issue 23) pp:NaN5461-5461
Publication Date(Web):2010/05/17
DOI:10.1039/B924613D
The asymmetric cycloaddition reactions of 3,4-dimethyl-1-phenylarsole and (Z/E)-diphenyl-1-propenylphosphine/diphenyl-1-styrylphosphine promoted by a chiral organopalladium(II) complex derived from (S)-[1-(dimethylamino)ethyl]naphthalene proceeded stereoselectively to generate different exo/endo-products. The reactions involving the (Z/E)-methyl substituted phosphines gave the individual optically pure exo- and endo-cycloadducts in very high stereoselectivity (33:1). However, when the methyl group was replaced by a Ph moiety both (Z/E)-phenyl substituted phosphines produced the same endo-cycloadduct in a stereoselectivity of 15:1. Every reaction produced five new chiral centers (four of them are sterically independent) in a single step and all three optically pure As–P heterobidentate ligands were obtained in high yields. The mechanism involved in the conversion of exo- to endo-product was investigated via density functional theory calculations. Computational results were consistent with the experimentally observed endo/exo-selectivity.
Co-reporter:Mingjun Yuan, Sumod A. Pullarkat, Crystal Huixian Yeong, Yongxin Li, Deepa Krishnan and Pak-Hing Leung
Dalton Transactions 2009(Issue 19) pp:NaN3670-3670
Publication Date(Web):2009/03/26
DOI:10.1039/B904090K
Optically pure P-chiral 1,2- and 1,3-diphosphines were synthesized chemoselectively and stereoselectively from the chiral organopalladium complex promoted asymmetric Diels–Alder reaction between 3,4-dimethyl-1-phenylphosphole (DMPP) and cis- and trans-ester substituted allylic phosphines.
Co-reporter:Xiang-Yuan Yang, Yu-Xiang Jia, Wee Shan Tay, Yongxin Li, Sumod A. Pullarkat and Pak-Hing Leung
Dalton Transactions 2016 - vol. 45(Issue 34) pp:NaN13455-13455
Publication Date(Web):2016/07/22
DOI:10.1039/C6DT02588A
The impact of the structural attributes of chiral PC- and PCP-palladium catalysts was investigated in the asymmetric hydrophosphination of various heterocycle-functionalized enone substrates. Due to the architecture of the catalysts, they are confronted with potential catalyst deactivation arising from the coordination of the electron-rich heteroatoms (P, O, N and S) to the metal center. A systematic variation of the location and identity of the heteroatoms demonstrated the impact of structural modifications on the substrates, which have a significant influence on both yields (16–99%) and enantioselectivities (0–99%). A detailed discussion on the distinct catalytic mechanisms (intra- vs. inter-molecular addition) provides important information to explain the results obtained.
Co-reporter:Xi-Rui Li, Xiang-Yuan Yang, Yongxin Li, Sumod A. Pullarkat and Pak-Hing Leung
Dalton Transactions 2017 - vol. 46(Issue 4) pp:NaN1316-1316
Publication Date(Web):2016/12/27
DOI:10.1039/C6DT04447F
A chiral phosphine auxiliary was generated with excellent ee via catalytic asymmetric hydrophosphination of 3-(naphthalen-1-ylmethylene)pentane-2,4-dione. The subsequent metal complexation of the monophosphine yielded two different coordination complexes depending on the reaction conditions. The ortho-palladation of both coordination complexes resulted in the formation of a single dimeric phosphapalladacycle complex that could be further converted to the monomeric bisacetonitrile derivative. Moreover, the palladium complex exhibits interesting oxophilicity as the stable bisaquo derivative could be isolated and characterized crystallographically. The catalytic potential of the phosphapalladacycle was also demonstrated.



![Piperidine, 1-[(2E)-3-phenyl-2-propenyl]-](http://img.cochemist.com/ccimg/5900/5882-82-6.png)
![Piperidine, 1-[(2E)-3-phenyl-2-propenyl]-](http://img.cochemist.com/ccimg/5900/5882-82-6_b.png)




![Isoxazole, 5-[(1E)-2-(4-methoxyphenyl)ethenyl]-3-methyl-4-nitro-](http://img.cochemist.com/ccimg/52000/51978-98-4.png)
![Isoxazole, 5-[(1E)-2-(4-methoxyphenyl)ethenyl]-3-methyl-4-nitro-](http://img.cochemist.com/ccimg/52000/51978-98-4_b.png)






![Ethanone, 1-[2,4-bis(1,1-dimethylethyl)phenyl]-](http://img.cochemist.com/ccimg/1200/1144-40-7.png)
![Ethanone, 1-[2,4-bis(1,1-dimethylethyl)phenyl]-](http://img.cochemist.com/ccimg/1200/1144-40-7_b.png)






![2,4-PENTADIEN-1-ONE, 1-[1,1'-BIPHENYL]-4-YL-5-PHENYL-, (E,E)-](http://img.cochemist.com/ccimg/90900/90812-15-0.png)
![2,4-PENTADIEN-1-ONE, 1-[1,1'-BIPHENYL]-4-YL-5-PHENYL-, (E,E)-](http://img.cochemist.com/ccimg/90900/90812-15-0_b.png)
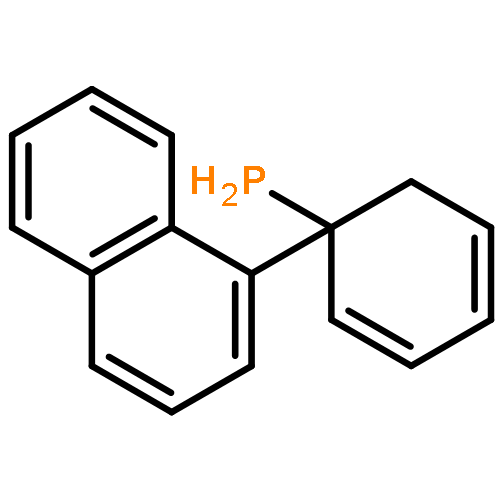






![MORPHOLINE, 4-[(2E)-3-PHENYL-2-PROPENYL]-](http://img.cochemist.com/ccimg/85700/85620-82-2.png)
![MORPHOLINE, 4-[(2E)-3-PHENYL-2-PROPENYL]-](http://img.cochemist.com/ccimg/85700/85620-82-2_b.png)






![1H-Imidazole, 1-[(1R)-1-phenylethyl]-](http://img.cochemist.com/ccimg/844700/844658-92-0.png)
![1H-Imidazole, 1-[(1R)-1-phenylethyl]-](http://img.cochemist.com/ccimg/844700/844658-92-0_b.png)




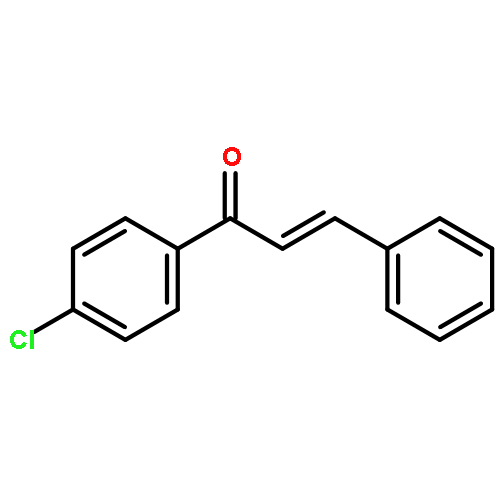









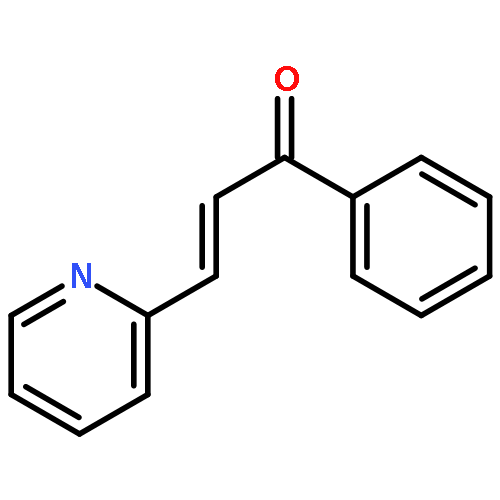
















![2-Propen-1-one, 1-phenyl-3-[4-(trifluoromethyl)phenyl]-](/data/chemimg/1516700/61637-11-4.png)
![2-Propen-1-one, 1-phenyl-3-[4-(trifluoromethyl)phenyl]-](/data/chemimg/1516700/61637-11-4_b.png)


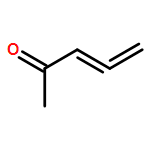
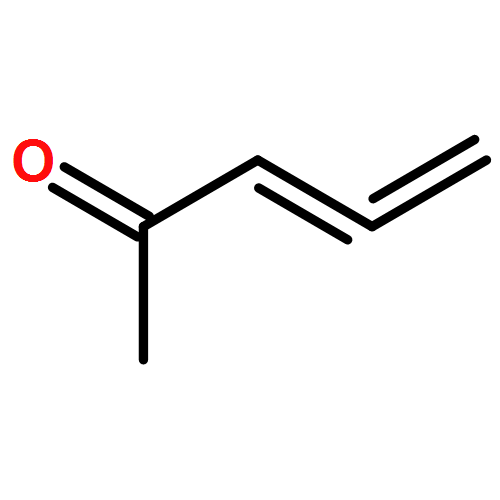











![Ethanone, 1-[1,1'-biphenyl]-4-yl-2-(triphenylphosphoranylidene)-](http://img.cochemist.com/ccimg/3000/2959-60-6.png)
![Ethanone, 1-[1,1'-biphenyl]-4-yl-2-(triphenylphosphoranylidene)-](http://img.cochemist.com/ccimg/3000/2959-60-6_b.png)












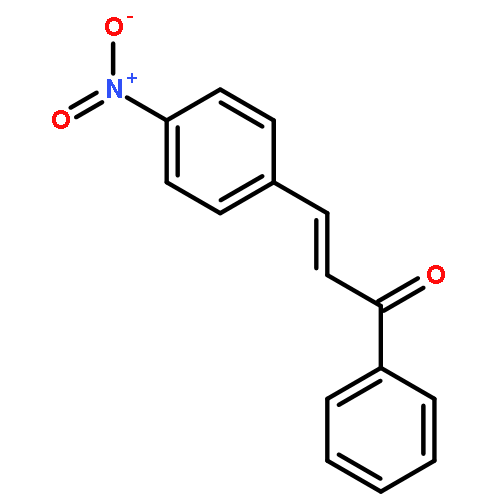








![1-Naphthalenemethanamine, N-[(2E)-3-phenyl-2-propenyl]-](http://img.cochemist.com/ccimg/92700/92610-10-1.png)
![1-Naphthalenemethanamine, N-[(2E)-3-phenyl-2-propenyl]-](http://img.cochemist.com/ccimg/92700/92610-10-1_b.png)



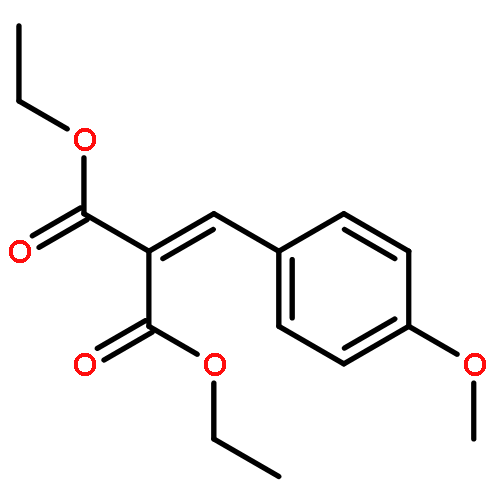








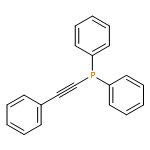
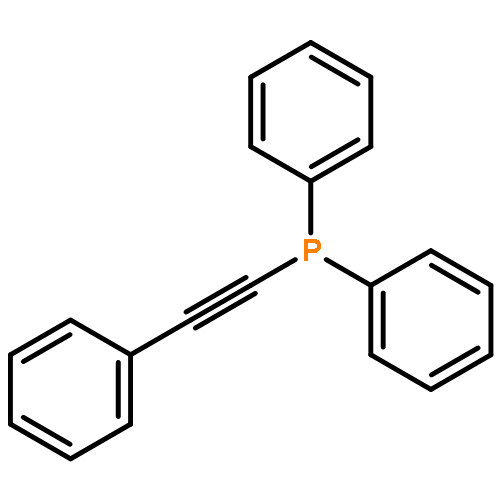












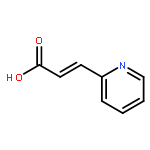
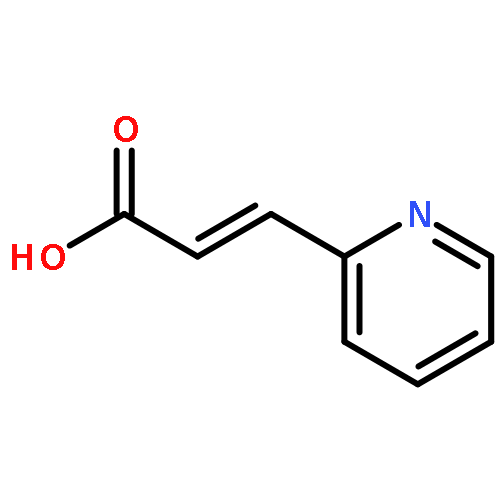




![Phosphine, diphenyl[(1E)-2-phenylethenyl]-](http://img.cochemist.com/ccimg/14100/14090-06-3.png)
![Phosphine, diphenyl[(1E)-2-phenylethenyl]-](http://img.cochemist.com/ccimg/14100/14090-06-3_b.png)


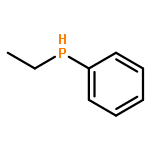
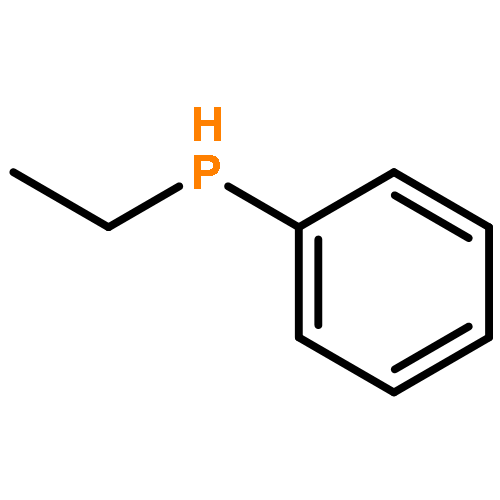














![Morpholine, 4-[(2E)-1,3-diphenyl-2-propenyl]-](http://img.cochemist.com/ccimg/201900/201852-22-4.png)
![Morpholine, 4-[(2E)-1,3-diphenyl-2-propenyl]-](http://img.cochemist.com/ccimg/201900/201852-22-4_b.png)




![Benzenesulfonamide, N-[3-(4-chlorophenyl)-1-phenyl-2-propen-1-ylidene]-4-methyl-](/data/chemimg/144700/1116669-73-8.png)
![Benzenesulfonamide, N-[3-(4-chlorophenyl)-1-phenyl-2-propen-1-ylidene]-4-methyl-](/data/chemimg/144700/1116669-73-8_b.png)

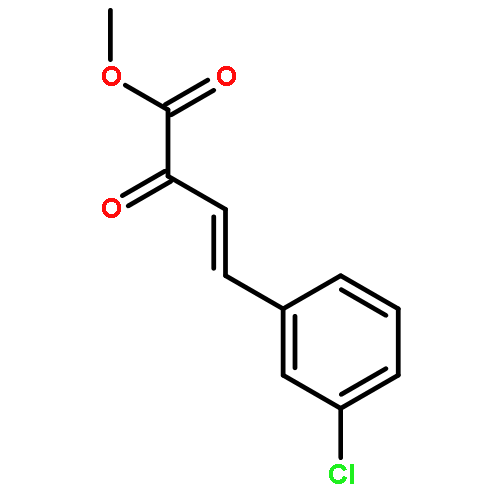






![Benzenemethanamine,N-[(2E)-3-phenyl-2-propen-1-yl]-](http://img.cochemist.com/ccimg/40100/40032-55-1.png)
![Benzenemethanamine,N-[(2E)-3-phenyl-2-propen-1-yl]-](http://img.cochemist.com/ccimg/40100/40032-55-1_b.png)






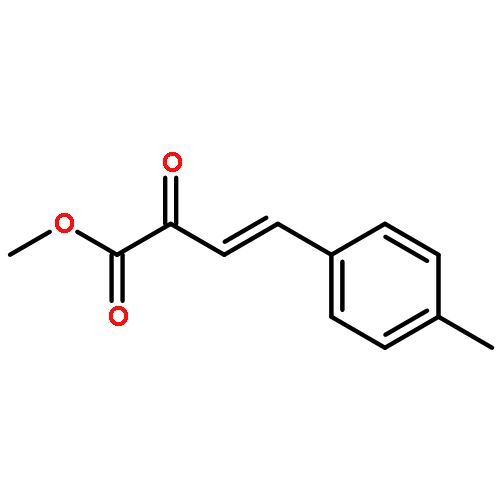
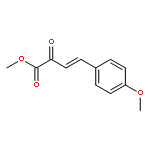

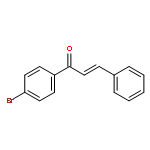
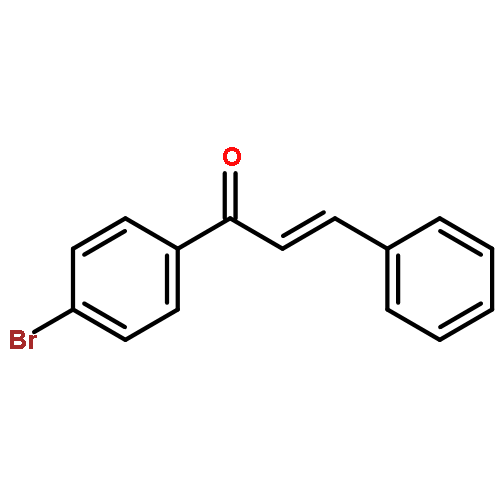


![Benzenemethanamine, N-(phenylmethyl)-N-[(2E)-3-phenyl-2-propen-1-yl]-](/data/chemimg/109900/147452-43-5.png)
![Benzenemethanamine, N-(phenylmethyl)-N-[(2E)-3-phenyl-2-propen-1-yl]-](/data/chemimg/109900/147452-43-5_b.png)