Co-reporter:Yun An, Yali Zhu, Yuan Yao and Junjun Liu
Physical Chemistry Chemical Physics 2016 vol. 18(Issue 14) pp:9838-9846
Publication Date(Web):22 Mar 2016
DOI:10.1039/C5CP07991H
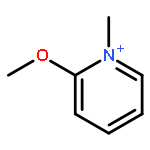
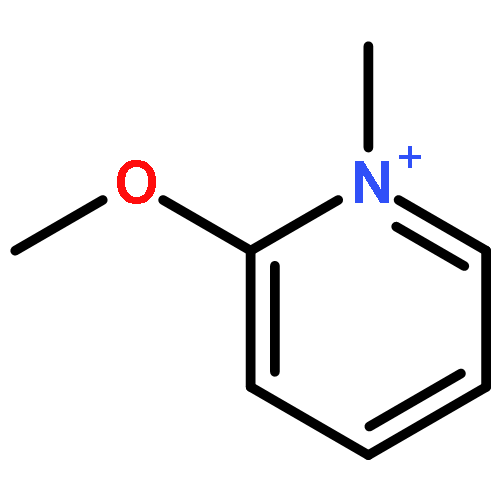
The main treatment for organophosphorus (OP) compound poisoning in clinics is to restore the activity of acetylcholinesterase (AChE) through oxime-induced reactivation of the phosphorylated OP–AChE adduct. It suffers from a competitive and irreversible aging reaction of the phosphorylated OP–AChE adduct, resulting in permanent inactivity of AChE. However, it was recently reported that N-methyl-2-methoxypyridinium species can act as methylating agents to methylate the methyl methane-phosphonate monoanion, in which the reaction mimics the reverse of the aging reaction of the phosphorylated OP–AChE adduct. If the aging reaction could be really reversed, the efficiency for the OP detoxification should be significantly improved, bringing up the possibility to develop an agent to reverse the aging process of the phosphorylated OP–AChE adduct. However, such a reaction with the N-methyl-2-methoxypyridinium species in the enzyme is still not reported so far. It is of great interest to know whether or not this reaction is observable in the enzyme, and more importantly, if it turns out to be not observable in the enzyme, why such a reaction proceeds quickly in aqueous solution but not in the enzyme. In the present study, we performed DFT calculations and quantum mechanical/molecular mechanical (QM/MM) calculations to reveal the fundamental mechanism for the methylation of both the methyl methane-phosphonate monoanion and the aged sarin–AChE adduct by N-methyl-2-methoxypyridinium species, respectively. The obtained results support the SN2 reaction mechanism, not the stepwise mechanism, for the methylation of the methyl methane-phosphonate monoanion by 9 reported N-methyl-2-methoxypyridinium compounds. The calculated free energy barriers are in good agreement with the experimental data. The methylation of the aged sarin–AChE adduct by one N-methyl-2-methoxypyridinium compound (labeled as compound 2) also employs the SN2 reaction mechanism with an extremely high free energy barrier of 30.4 ± 3.5 (or 26.6) kcal mol−1, implying that this reaction in the enzyme hardly occurs. Our results clearly show that compound 2 forms a strong π–π stacking interaction with the aromatic ring of the W86 residue of AChE, making itself unable to approach sarin for the reverse of the aging process. On the basis of the structure and mechanism, several possible strategies have been suggested for designing methylating agents with higher activity against the aged sarin–AChE adduct.
Co-reporter:Zhi-Bo Zhao, Yang Liu, Yuan Yao
Journal of Molecular Graphics and Modelling 2014 Volume 51() pp:168-172
Publication Date(Web):June 2014
DOI:10.1016/j.jmgm.2014.05.009
•The detailed binding of G6PD with the steroid DHEA derivatives has been studied by performing molecular docking, MD simulations, and binding free energy calculations.•The calculated binding free energies of the enzyme with the inhibitors using MM/PBSA method are qualitatively consistent with the experimental activity data.•The important, favorable interactions between the enzyme and the inhibitors have been revealed and main factors affecting the binding have been discussed.Glucose 6-phosphate dehydrogenase (G6PD), the first and the rate-limiting enzyme in the pentose phosphate pathway (PPP), catalyzes the oxidation of G6P to 6-phosphogluconolactone and the reduction of NADP+ to NADPH. Its key role in cancer promotes the development of a potent and selective inhibitor that might increase cancer cell death when combined with radiotherapy. In the present study, we investigated the detailed binding modes and binding free energies for G6PD interacting with a promising series of recently developed inhibitors, i.e., the steroid derivatives, by performing molecular docking, molecular dynamics (MD) simulations, and binding free energy calculations. The docking indicates that the inhibitors occupy the binding sites of both G6P and NADP+. The calculated binding free energies on the basis of the MD-simulated enzyme–inhibitor complexes are in good agreement with the experimental activity data for all of the examined inhibitors. The valuable insights into the detailed enzyme–inhibitor binding including the important intermolecular interactions, e.g., the hydrogen bond interaction and the hydrophobic interaction, have been provided. The computational results provide new insights into future rational design of more potent inhibitors of G6PD as a treatment for cancer.
Co-reporter:Yuan Yao and Ze-Sheng Li
Organic & Biomolecular Chemistry 2012 vol. 10(Issue 35) pp:7037-7044
Publication Date(Web):04 Jul 2012
DOI:10.1039/C2OB25605C
The reaction pathway of Schiff base hydrolysis catalyzed by type I dehydroquinate dehydratase (DHQD) from S. enterica has been studied by performing molecular dynamics (MD) simulations and density functional theory (DFT) calculations and the corresponding potential energy profile has also been identified. On the basis of the results, the catalytic hydrolysis process for the wild-type enzyme consists of three major reaction steps, including nucleophilic attack on the carbon atom involved in the carbon–nitrogen double bond of the Schiff base intermediate by a water molecule, deprotonation of the His143 residue, and dissociation between the product and the Lys170 residue of the enzyme. The remarkable difference between this and the previously proposed reaction mechanism is that the second step here, absent in the previously proposed reaction mechanism, plays an important role in facilitating the reaction through a key proton transfer by the His143 residue, resulting in a lower energy barrier. Comparison with our recently reported results on the Schiff base formation and dehydration processes clearly shows that the Schiff base hydrolysis is rate-determining in the overall reaction catalyzed by type I DHQD, consistent with the experimental prediction, and the calculated energy barrier of ∼16.0 kcal mol−1 is in good agreement with the experimentally derived activation free energy of ∼14.3 kcal mol−1. When the imidazole group of His143 residue is missing, the Schiff base hydrolysis is initiated by a hydroxide ion in the solution, rather than a water molecule, and both the reaction mechanism and the kinetics of Schiff base hydrolysis have been remarkably changed, clearly elucidating the catalytic role of the His143 residue in the reaction. The new mechanistic insights obtained here will be valuable for the rational design of high-activity inhibitors of type I DHQD as non-toxic antimicrobials, anti-fungals, and herbicides.
Co-reporter:Qi Pan, Yuan Yao, Ze-Sheng Li
Computational and Theoretical Chemistry 2012 Volume 1001() pp:60-66
Publication Date(Web):1 December 2012
DOI:10.1016/j.comptc.2012.10.009
Type II dehydroquinate dehydratase (DHQD), catalyzing the dehydration of dehydroquinate to dehydroshikimate, is considered as an attractive target for developing non-toxic antimicrobials, anti-fungals, and herbicides. The enzymes from different sources show distinguishable kinetic isotope effects, suggesting that they probably employ different reaction mechanisms. In the present study, the catalytic mechanism of type II DHQD from Mycobacterium tuberculosis has been reported by performing molecular dynamics simulations and quantum chemical calculations. The results revealed that this enzyme undergoes a two-step E1cB trans-elimination reaction mechanism and the calculated overall energy barrier of ∼17.7 kcal/mol is in excellent agreement with the experimental value. The developed enolate intermediate does not convert to enol intermediate by abstracting a solvent-derived proton and is therefore stabilized by Asn12 residue through strong hydrogen bonding interaction, reasonably explaining the observed kinetic isotope effect. Without the catalytic role of Asn12 residue, the overall energy barrier raises ∼4.5 kcal/mol.Graphical abstractHighlights► The reaction mechanism of the Mycobacterium tuberculosis type II DHQD was studied. ► It undergoes a two-step E1cB trans-elimination with an enolate intermediate. ► This mechanism can reasonably explain the experimental phenomena.
Co-reporter:Yuan Yao and Ze-Sheng Li
Organic & Biomolecular Chemistry 2012 - vol. 10(Issue 35) pp:NaN7044-7044
Publication Date(Web):2012/07/04
DOI:10.1039/C2OB25605C
The reaction pathway of Schiff base hydrolysis catalyzed by type I dehydroquinate dehydratase (DHQD) from S. enterica has been studied by performing molecular dynamics (MD) simulations and density functional theory (DFT) calculations and the corresponding potential energy profile has also been identified. On the basis of the results, the catalytic hydrolysis process for the wild-type enzyme consists of three major reaction steps, including nucleophilic attack on the carbon atom involved in the carbon–nitrogen double bond of the Schiff base intermediate by a water molecule, deprotonation of the His143 residue, and dissociation between the product and the Lys170 residue of the enzyme. The remarkable difference between this and the previously proposed reaction mechanism is that the second step here, absent in the previously proposed reaction mechanism, plays an important role in facilitating the reaction through a key proton transfer by the His143 residue, resulting in a lower energy barrier. Comparison with our recently reported results on the Schiff base formation and dehydration processes clearly shows that the Schiff base hydrolysis is rate-determining in the overall reaction catalyzed by type I DHQD, consistent with the experimental prediction, and the calculated energy barrier of ∼16.0 kcal mol−1 is in good agreement with the experimentally derived activation free energy of ∼14.3 kcal mol−1. When the imidazole group of His143 residue is missing, the Schiff base hydrolysis is initiated by a hydroxide ion in the solution, rather than a water molecule, and both the reaction mechanism and the kinetics of Schiff base hydrolysis have been remarkably changed, clearly elucidating the catalytic role of the His143 residue in the reaction. The new mechanistic insights obtained here will be valuable for the rational design of high-activity inhibitors of type I DHQD as non-toxic antimicrobials, anti-fungals, and herbicides.
Co-reporter:Yun An, Yali Zhu, Yuan Yao and Junjun Liu
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 14) pp:NaN9846-9846
Publication Date(Web):2016/03/22
DOI:10.1039/C5CP07991H
The main treatment for organophosphorus (OP) compound poisoning in clinics is to restore the activity of acetylcholinesterase (AChE) through oxime-induced reactivation of the phosphorylated OP–AChE adduct. It suffers from a competitive and irreversible aging reaction of the phosphorylated OP–AChE adduct, resulting in permanent inactivity of AChE. However, it was recently reported that N-methyl-2-methoxypyridinium species can act as methylating agents to methylate the methyl methane-phosphonate monoanion, in which the reaction mimics the reverse of the aging reaction of the phosphorylated OP–AChE adduct. If the aging reaction could be really reversed, the efficiency for the OP detoxification should be significantly improved, bringing up the possibility to develop an agent to reverse the aging process of the phosphorylated OP–AChE adduct. However, such a reaction with the N-methyl-2-methoxypyridinium species in the enzyme is still not reported so far. It is of great interest to know whether or not this reaction is observable in the enzyme, and more importantly, if it turns out to be not observable in the enzyme, why such a reaction proceeds quickly in aqueous solution but not in the enzyme. In the present study, we performed DFT calculations and quantum mechanical/molecular mechanical (QM/MM) calculations to reveal the fundamental mechanism for the methylation of both the methyl methane-phosphonate monoanion and the aged sarin–AChE adduct by N-methyl-2-methoxypyridinium species, respectively. The obtained results support the SN2 reaction mechanism, not the stepwise mechanism, for the methylation of the methyl methane-phosphonate monoanion by 9 reported N-methyl-2-methoxypyridinium compounds. The calculated free energy barriers are in good agreement with the experimental data. The methylation of the aged sarin–AChE adduct by one N-methyl-2-methoxypyridinium compound (labeled as compound 2) also employs the SN2 reaction mechanism with an extremely high free energy barrier of 30.4 ± 3.5 (or 26.6) kcal mol−1, implying that this reaction in the enzyme hardly occurs. Our results clearly show that compound 2 forms a strong π–π stacking interaction with the aromatic ring of the W86 residue of AChE, making itself unable to approach sarin for the reverse of the aging process. On the basis of the structure and mechanism, several possible strategies have been suggested for designing methylating agents with higher activity against the aged sarin–AChE adduct.




![Bicyclo[2.2.1]heptane-2-thione, 1,7,7-trimethyl-, S-oxide, (1R)-](http://img.cochemist.com/ccimg/59200/59121-52-7.png)
![Bicyclo[2.2.1]heptane-2-thione, 1,7,7-trimethyl-, S-oxide, (1R)-](http://img.cochemist.com/ccimg/59200/59121-52-7_b.png)




![Dibenzo[b,e]thiepin-11(6H)-one, 5-oxide](http://img.cochemist.com/ccimg/34200/34129-26-5.png)
![Dibenzo[b,e]thiepin-11(6H)-one, 5-oxide](http://img.cochemist.com/ccimg/34200/34129-26-5_b.png)
![Ethanone, 1-[4-(methylsulfinyl)phenyl]-](http://img.cochemist.com/ccimg/32400/32361-73-2.png)
![Ethanone, 1-[4-(methylsulfinyl)phenyl]-](http://img.cochemist.com/ccimg/32400/32361-73-2_b.png)

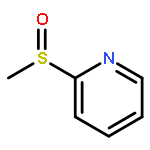
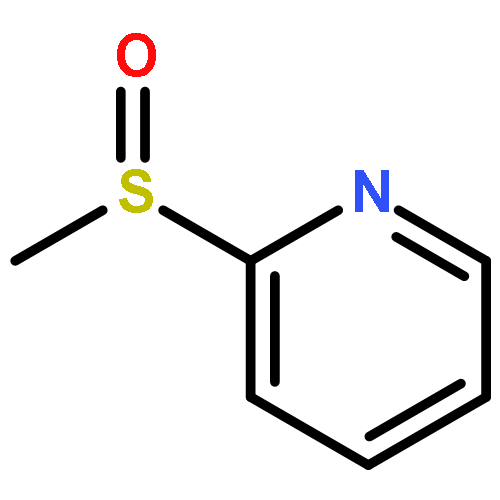
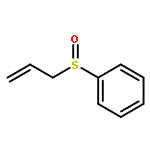
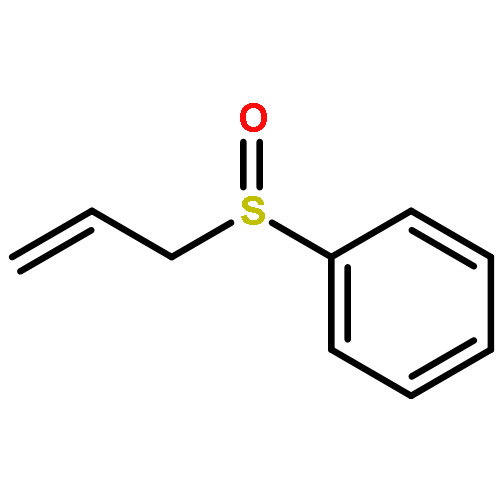
![2,2,4-TRIMETHYL-3-THIABICYCLO[2.2.2]OCTANE](http://img.cochemist.com/ccimg/5800/5718-74-1.png)
![2,2,4-TRIMETHYL-3-THIABICYCLO[2.2.2]OCTANE](http://img.cochemist.com/ccimg/5800/5718-74-1_b.png)



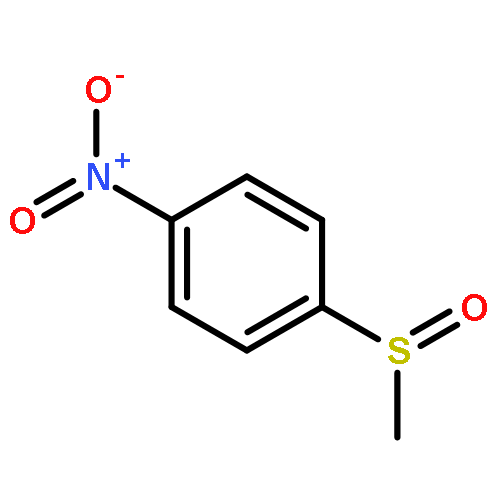


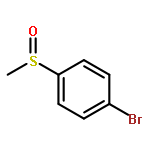
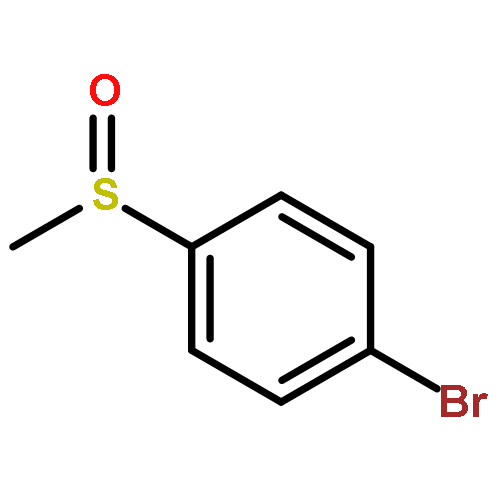
![Benzene, [(phenylmethyl)sulfinyl]-](/data/chemimg/1595300/833-82-9.png)
![Benzene, [(phenylmethyl)sulfinyl]-](/data/chemimg/1595300/833-82-9_b.png)
![Benzene,[(methylsulfinyl)methyl]-](http://img.cochemist.com/ccimg/900/824-86-2.png)
![Benzene,[(methylsulfinyl)methyl]-](http://img.cochemist.com/ccimg/900/824-86-2_b.png)