Co-reporter:Peng Wang, Hong-Guang Xu, Guo-Jin Cao, Wen-Jing Zhang, Xi-Ling Xu, and Wei-Jun Zheng
The Journal of Physical Chemistry A November 22, 2017 Volume 121(Issue 46) pp:8973-8973
Publication Date(Web):October 31, 2017
DOI:10.1021/acs.jpca.7b09428
We conducted combined gas-phase anion photoelectron spectroscopy and density functional theory studies on nucleobase-silver complexes. The most probable structures of the nucleobase-Ag– complexes were determined by comparing the theoretical calculations with the experimental measurements. The vertical detachment energies (VDEs) of uracil-Ag–, thymine-Ag–, cytosine-Ag–, and guanine-Ag– were estimated to be 2.18 ± 0.08, 2.11 ± 0.08, 2.04 ± 0.08, and 2.20 ± 0.08 eV, respectively, based on their photoelectron spectra. Adenine-Ag– has two isomers coexisting in the experiment; the experimental VDEs of the two isomers are 2.18 and 2.53 eV, respectively. In the most probable isomers of nucleobases-Ag–, uracil, thymine, and cytosine interact with Ag– anion via N–H···Ag and C–H···Ag hydrogen bonds, while adenine and guanine interact with Ag– anion through two N–H···Ag hydrogen bonds. The N–H···Ag hydrogen bonds can be characterized as medium or strong hydrogen bonds. It is found that binding sites of the Ag anion to the nucleobases are affected by the deprotonation energies and the steric effects of two adjacent X–H groups.
Co-reporter:Gang Feng;Cheng-Wen Liu;Zhen Zeng;Gao-Lei Hou;Hong-Guang Xu
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 23) pp:15562-15569
Publication Date(Web):2017/06/14
DOI:10.1039/C7CP02965A
To understand the initial hydration processes of MgCl2, we measured photoelectron spectra of MgCl2(H2O)n− (n = 0–6) and conducted ab initio calculations on MgCl2(H2O)n− and their neutral counterparts up to n = 7. A dramatic drop in the vertical detachment energy (VDE) was observed upon addition of the first water molecule to bare MgCl2−. This large variation in VDE can be associated with the charge-transfer-to-solvent (CTTS) effect occurring in the MgCl2(H2O)n− clusters, as hydration induces transfer of the excess electron of MgCl2− to the water molecules. Investigation of the separation of Cl−–Mg2+ ion pair shows that, in MgCl2(H2O)n− anions, breaking of the first Mg–Cl bond occurs at n = 4, while breaking of the second Mg–Cl bond takes place at n = 6. For neutral MgCl2(H2O)n clusters, breaking of the first Mg–Cl bond starts at n = 7.
Co-reporter:Gao-Lei Hou, Cheng-Wen Liu, Ren-Zhong Li, Hong-Guang Xu, Yi Qin Gao, and Wei-Jun Zheng
The Journal of Physical Chemistry Letters 2017 Volume 8(Issue 1) pp:13-20
Publication Date(Web):December 7, 2016
DOI:10.1021/acs.jpclett.6b02670
Solvation of salts in water is a fundamental physical chemical process, but the underlying mechanism remains unclear. We investigated the contact ion pair (CIP) to solvent-separated ion pair (SSIP) transition in NaCl(H2O)n clusters with anion photoelectron spectroscopy and ab initio calculations. It is found that the SSIP type of structures show up at n = 2 for NaCl–(H2O)n anions. For neutral NaCl(H2O)n, the CIP structures are dominant at n < 9. At n = 9–12, the CIP structures and SSIP structures of NaCl(H2O)n are nearly degenerate in energy, coincident to the H2O:NaCl molar ratio of NaCl saturated solution and implying that the CIP and SSIP structures can coexist in concentrated solutions. These results are useful for understanding the solvation of salts at the molecular level.
Co-reporter:Li-Juan Zhao;Xi-Ling Xu;Hong-Guang Xu;Gang Feng
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 31) pp:21112-21118
Publication Date(Web):2017/08/09
DOI:10.1039/C7CP03870D
The interactions of FeO− with water molecules were studied by using photoelectron spectroscopy and density functional theoretical calculations. It is found that a dihydroxyl species, Fe(OH)2−/0, can be formed when FeO−/0 interacts with the first water molecule. The complexes formed via the interactions between FeO−/0 and n water molecules can be viewed as Fe(OH)2(H2O)n−1−/0, in which (n − 1)H2O molecules interact with a Fe(OH)2 core. For Fe(OH)2−/0 and Fe(OH)2(H2O)−, the Fe(OH)2 unit has two conformers with the two OH groups oriented differently. The vertical detachment energies (VDEs) of FeO2H2(H2O)n−1− (n = 1–4) are measured to be 1.25 ± 0.04, 1.66 ± 0.04, 2.06 ± 0.04, and 2.37 ± 0.04 eV, respectively, by experiment. It is also worth mentioning that in the FeO2H2(H2O)− anion the water molecule interacts with the Fe(OH)2 core by forming a hydrogen bond with one of the OH groups, while in neutral FeO2H2(H2O), the water molecule interacts with the Fe atom of the Fe(OH)2 core via its O atom.
Co-reporter:Sheng-Jie Lu, Lian-Rui Hu, Xi-Ling Xu, Hong-Guang Xu, Hui Chen and Wei-Jun Zheng
Physical Chemistry Chemical Physics 2016 vol. 18(Issue 30) pp:20321-20329
Publication Date(Web):24 Mar 2016
DOI:10.1039/C6CP00373G
Gold-doped germanium clusters, AuGen− (n = 2–12), were investigated by using anion photoelectron spectroscopy in combination with ab initio calculations. Their geometric structures were determined by comparison of the theoretical calculations with the experimental results. The results show that the most stable isomers of AuGen− with n = 2–10 are all exohedral structures with the Au atom capping the vertex, edge or face of Gen clusters, while AuGe11− is found to be the critical size of the endohedral structure. Interestingly, AuGe12− has an Ih symmetric icosahedral structure with the Au atom located at the center. The molecular orbital analysis of the AuGe12− cluster suggests that the interactions between the 5d orbitals of the Au atom and the 4s4p hybridized orbitals of the Ge atoms may stabilize the Ih symmetric icosahedral cage and promote the Au atom to be encapsulated in the cage of Ge12. The NICS(0) and NICS(1) values are calculated to be −143.7 ppm and −36.3 ppm, respectively, indicating that the icosahedral AuGe12− cluster is significantly aromatic.
Co-reporter:Ren-Zhong Li, Gao-Lei Hou, Cheng-Wen Liu, Hong-Guang Xu, Xiang Zhao, Yi Qin Gao and Wei-Jun Zheng
Physical Chemistry Chemical Physics 2016 vol. 18(Issue 1) pp:557-565
Publication Date(Web):18 Nov 2015
DOI:10.1039/C5CP05550D
We investigated (NaI)2−(H2O)n (n = 0–6) clusters to examine the initial solvation process of (NaI)2 in water, using negative ion photoelectron spectroscopy and theoretical calculations. The structures of these clusters and their neutrals were determined by comparing ab initio calculations with experimental results. It is found that bare (NaI)2− is a L-shaped structure and the corresponding neutral is a rhombus. In (NaI)2−(H2O), the water molecule prefers to interact with the middle Na atom of the L-shaped (NaI)2−. For (NaI)2−(H2O)n clusters with n = 2–3, two types of structures are nearly degenerate in energy: one is L-shaped and the other is pyramid-shaped. As for (NaI)2−(H2O)n with n = 4–6, the dominant structures are pyramid-shaped. For the anionic clusters, one of the Na–I distances increases abruptly when n = 2; for the neutral clusters, rapid lengthening of the Na–I distances occurs when n = 4. Additionally, analyses of the reduced density gradient were carried out, and the results reveal that Na+–water interactions dominate in (NaI)2−(H2O)n for n ≤ 4, whereas I−–water and water–water interactions are significantly enhanced when n increases to 5.
Co-reporter:Li-Juan Zhao, Hong-Guang Xu, Gang Feng, Peng Wang, Xi-Ling Xu and Wei-Jun Zheng
Physical Chemistry Chemical Physics 2016 vol. 18(Issue 8) pp:6175-6181
Publication Date(Web):22 Jan 2016
DOI:10.1039/C5CP07673K
We investigate BS2− and BSO− clusters using photoelectron spectroscopy and theoretical calculations. The electron affinities of BS2 and BSO are measured to be 3.80 ± 0.03 and 3.88 ± 0.03 eV, respectively, higher than those of halogen atoms. Thus, BS2 and BSO can be considered as superhalogens. The comparison of experimental and theoretical results confirmed that the ground state structures of BS2−, BSO−, and their neutrals are all linear. Analyses of natural bond orbitals suggest that both BS2− and BSO− have dual 3c-4e π hyperbonds.
Co-reporter:Guo-Jin Cao, Hong-Guang Xu, Xi-Ling Xu, Peng Wang, Wei-Jun Zheng
International Journal of Mass Spectrometry 2016 Volume 407() pp:118-125
Publication Date(Web):20 August 2016
DOI:10.1016/j.ijms.2016.07.008
•All of A2M+ and G2M+ cations are planar structures in nucleobase-M+-nucleobase style, containing a nearly linear N–M+–N bond.•The dissociation channels depend on the types of nucleobases and metal ions.•The loss of one nucleobase (adenine or guanine) molecule are the predominant channels for M = Cu and Ag.•The loss of AAu or GAu are the major channels for M = Au.To understand the interactions between nucleobases and coinage metal cations, we conducted combined photodissociation and density functional theory studies on A2M+ and G2M+ (A = adenine, G = guanine, M = Cu, Ag, and Au) cations. The nucleobase-metal complexes were produced by laser ablation and detected by a reflectron time-of-flight mass spectrometer. The mass peaks of A2M+ and G2M+ cations have high intensities in the mass spectra of AnM+ and GnM+ complexes, indicating that these cations have relatively high stabilities. They were mass-selected and then photodissociated by 266 nm photons. Their photodissociation spectra clearly show that the loss of adenine or guanine is the predominant channel for these complexes. The density functional theory calculations show that A2M+ and G2M+ complexes prefer planar structures with the metal cations interacting with the N atoms in the carbon-nitrogen rings of adenine and guanine. The calculated bond dissociation energies of different dissociation channels are in good agreement with the experimental observed fragment ions.The photodissociation experiments of A2M+A2M+and G2M+G2M+ complexes at 266 nm show that the loss of one nucleobase is the predominant channel for M = Cu and Ag, while the loss of AAu or GAu is the major channel for M = Au.
Co-reporter:Jinyun Yuan, Peng Wang, Gao-Lei Hou, Gang Feng, Wen-Jing Zhang, Xi-Ling Xu, Hong-Guang Xu, Jinlong Yang, and Wei-Jun Zheng
The Journal of Physical Chemistry A 2016 Volume 120(Issue 9) pp:1520-1528
Publication Date(Web):February 12, 2016
DOI:10.1021/acs.jpca.6b00241
The structural evolution and electronic properties of VnC2–/0 and VnC4–/0 (n = 1–6) clusters were investigated using photoelectron spectroscopy and density functional theory calculations. The adiabatic and vertical detachment energies of VnC2– and VnC4– (n = 1–6) clusters were obtained from their photoelectron spectra. The most stable structures were identified by comparing the results of our calculations with the experimental data. We found that the carbon atoms of VnC2–/0 and VnC4–/0 (n = 1–6) clusters were separated gradually with increasing number of vanadium atoms. For VnC2–/0 (n = 3–6) and VnC4–/0 (n = 4–6) clusters, the carbon atoms are separated by the vanadium atoms. The geometry of V4C4 is a cubic structure and the geometries of V5C4 and V6C4 are formed by one and two vanadium atoms capping the cubic V4C4 structure, respectively.
Co-reporter:Sheng-Jie Lu, Xi-Ling Xu, Gang Feng, Hong-Guang Xu, and Wei-Jun Zheng
The Journal of Physical Chemistry C 2016 Volume 120(Issue 44) pp:25628-25637
Publication Date(Web):October 18, 2016
DOI:10.1021/acs.jpcc.6b08598
AuSin– (n = 4–12) clusters were produced with a laser vaporization source and investigated by photoelectron spectroscopy. The swarm-intelligence-based CALYPSO structure search method and ab initio calculations were employed to determine their ground-state structures. The results revealed that the most stable isomers of AuSin– (n = 4–12) cluster anions are all exohedral structures, in which the Au atom caps the vertex, edge, or surface of the bare Sin clusters. The endohedral and exohedral structures of neutral AuSi11 are nearly degenerate in energy. The most stable structure of neutral AuSi12 is endohedral. The growth mechanism of AuSin– cluster anions is compared with those of AuGen–, AgSin–, and CuSin– clusters. It implies that the bond strengths of Au–Si and Au–Ge play important roles in the formation of cage structures for AuSi12– and AuGe12–, while the different atomic radii of coinage metals, different bond strengths, and the strong relativistic effect in Au atom are responsible for the different growth mechanisms of Si clusters doped with different coinage metals.
Co-reporter:Ke-Wei Ding, Xiao-Wei Li, Hong-Guang Xu, Tao-Qi Li, Zhong-Xue Ge, Qian Wang and Wei-Jun Zheng
Chemical Science 2015 vol. 6(Issue 8) pp:4723-4729
Publication Date(Web):11 May 2015
DOI:10.1039/C5SC01103E
TiNn+ clusters were generated by laser ablation and analyzed experimentally by mass spectrometry. The results showed that the mass peak of the TiN12+ cluster is dominant in the spectrum. The TiN12+ cluster was further investigated by photodissociation experiments with 266, 532 and 1064 nm photons. Density functional calculations were conducted to investigate stable structures of TiN12+ and the corresponding neutral cluster, TiN12. The theoretical calculations found that the most stable structure of TiN12+ is Ti(N2)6+ with Oh symmetry. The calculated binding energy is in good agreement with that obtained from the photodissociation experiments. The most stable structure of neutral TiN12 is Ti(N2)6 with D3d symmetry. The Ti–N bond strengths are greater than 0.94 eV in both Ti(N2)6+ and its neutral counterpart. The interaction between Ti and N2 weakens the N–N bond significantly. For neutral TiN12, the Ti(N3)4 azide, the N5TiN7 sandwich structure and the N6TiN6 structure are much higher in energy than the Ti(N2)6 complex. The DFT calculations predicted that the decomposition of Ti(N3)4, N5TiN7, and N6TiN6 into a Ti atom and six N2 molecules can release energies of about 139, 857, and 978 kJ mol−1 respectively.
Co-reporter:Xi-Ling Xu, Xiao-Jiao Deng, Hong-Guang Xu and Wei-Jun Zheng
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 46) pp:31011-31022
Publication Date(Web):27 Oct 2015
DOI:10.1039/C5CP04482K
CnSm− (n = 2–7; m = 1, 2) clusters were investigated by using photoelectron spectroscopy combined with density functional theory calculations. We found that the vertical detachment energies of both CnS− and CnS2− (n = 2–7) clusters exhibit a strong odd–even alternation with an increasing number of carbon atoms: the VDEs of even-n clusters are higher than those of adjacent odd-n clusters. The most stable structures of the anionic and neutral CnS (n = 2–7) clusters are linear with the S atom locating at one end of the carbon chain except that the structure of C3S− is slightly bent. The ground state isomers of the anionic and neutral CnS2 (n = 2–7) clusters are all linear structures with two S atoms locating at two ends of the carbon chain. The electron affinities of the neutral CnS (n = 2, 4–7) and CnS2 (n = 2–7) clusters are determined based on the experimental adiabatic detachment energies of the corresponding anion species, because the most stable structures of the neutral clusters are similar to those of the corresponding anions.
Co-reporter:Gang Feng, Gao-Lei Hou, Hong-Guang Xu, Zhen Zeng and Wei-Jun Zheng
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 8) pp:5624-5631
Publication Date(Web):15 Jan 2015
DOI:10.1039/C4CP05698A
The initial dissolution steps of lithium sulfate (Li2SO4) in water were investigated by performing anion photoelectron spectroscopy and density functional theory calculations on the Li2SO4(H2O)n− (n = 0–5) clusters. The plausible structures of these clusters and the corresponding neutral clusters were obtained using LC-ωPBE/6-311++G(d,p) calculations by comparing the experimental and theoretical vertical electron detachment energies. Two types of structures for bare Li2SO4−/0 were found: a turtle-shaped structure and a propeller-shaped structure. For Li2SO4(H2O)n− cluster anions with n = 1–3, two kinds of isomers derived from the turtle-shaped and propeller-shaped structures of bare Li2SO4− were identified. For n = 4–5, these two kinds of isomers present similar structural and energetic features and thus are not distinguishable. For the anionic clusters the water molecules prefer to firstly interact with one Li atom until fully coordinating it. While for the neutral clusters, the water molecules interact with the two Li atoms alternately, therefore, showing a pairwise solvation behavior. The Li–S distance increases smoothly upon addition of water molecules one by one. Addition of five water molecules to Li2SO4 cannot induce the dissociation of one Li+ ion because the water molecules are shared by two Li+ ions.
Co-reporter:Zhen Zeng, Gao-Lei Hou, Jian Song, Gang Feng, Hong-Guang Xu and Wei-Jun Zheng
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 14) pp:9135-9147
Publication Date(Web):25 Feb 2015
DOI:10.1039/C5CP00020C
The microsolvation of LiBO2 in water was investigated by conducting anion photoelectron spectroscopy and ab initio studies on the LiBO2(H2O)n− (n = 0–5) clusters. By comparing calculations with experiments, the structures of these clusters and their corresponding neutrals were assigned, and their structural evolutions were revealed. During the anionic structural evolution with n increasing to 5, hydroxyborate and metaborate channels were identified and the metaborate channel is more favorable. For the hydroxyborate structures, the anionic Li+–BO2− ion pair reacts with a water molecule to produce the LiBO(OH)2− moiety and three water molecules tend to dissolve this moiety. In the metaborate channel, two types of solvent-separated ion pair (SSIP) geometries were determined as the ring-type and linear-type. The transition from the contact ion pair (CIP) to the ring-type of SSIP starts at n = 3, while that to the linear-type of SSIP occurs at n = 4. In neutral LiBO2(H2O)n clusters, the first water molecule prefers to react with the Li+–BO2− ion pair to generate the LiBO(OH)2 moiety, analogous to the bulk crystal phase of α-LiBO2 with two O atoms substituted by two OH groups. The Li–O distance in the LiBO(OH)2 moiety increases with the increasing number of water molecules and elongates abruptly at n = 4. Our studies provide new insight into the initial dissolution of LiBO2 salt in water at the molecular level and may be correlated to the bulk state.
Co-reporter:Xiao-Jiao Deng
The Journal of Physical Chemistry C 2015 Volume 119(Issue 20) pp:11048-11055
Publication Date(Web):December 30, 2014
DOI:10.1021/jp511694c
The structural, electronic and magnetic properties of VGen–/0 (n = 3–12) clusters were investigated using anion photoelectron spectroscopy in combination with density functional theory calculations. We found that the dominant geometries are exohedral for the VGen–/0 clusters with n ≤ 7. The VGe8–/0 clusters have half-encapsulated boat-shaped structures, and the opening of the boat-shaped structure is gradually covered by the additional Ge atoms to form Gen cage from n = 9–11. At n = 12, a D3d distorted hexagonal prism cage structure is formed. According to the natural population analysis, for both anionic and neutral VGen clusters of n = 8–12, there is electron transfer from the Gen framework to the V atom and the total magnetic moments decrease to the minima. The electron transfer pattern and the minimization of the magnetic moments for these clusters are related to their structural evolution.
Co-reporter:Gao-Lei Hou, Gang Feng, Li-Juan Zhao, Hong-Guang Xu, and Wei-Jun Zheng
The Journal of Physical Chemistry A 2015 Volume 119(Issue 45) pp:11154-11161
Publication Date(Web):October 16, 2015
DOI:10.1021/acs.jpca.5b09205
The (KI)n– (n = 1–4) and K(KI)n– (n = 1–3) clusters were studied by negative ion photoelectron spectroscopy and ab initio calculations. Comparison between the theoretical vertical detachment energies and the experimental values revealed that multiple isomers may coexist in the experiments. The existence of two isomers for K(KI)− and K(KI)2– were confirmed directly by isomer-depletion experiments, in which the low adiabatic detachment energy isomers were depleted by a 1064 nm laser beam before the anions were photodetached by a 532 nm laser beam. Our results show that the most stable structures of the K(KI)−, (KI)2–, and K(KI)2– anions are chain structures, while those of their neutral counterparts are planar. Three-dimensional structures start to appear at n = 3 for (KI)n–/0 and K(KI)n–/0. In the K(KI)n– cluster anions, the excess electron is localized on the extra K atom and forms an electron pair with the existing s electron of the K atom; the resulting negatively charged K prefers to interact with the other positively charged K atoms rather than with the I atoms. Both the anionic and neutral (KI)4 clusters have cuboid structures, which may be regarded as the smallest structural motif of KI crystal.
Co-reporter:Zhen Zeng, Cheng-Wen Liu, Gao-Lei Hou, Gang Feng, Hong-Guang Xu, Yi Qin Gao, and Wei-Jun Zheng
The Journal of Physical Chemistry A 2015 Volume 119(Issue 12) pp:2845-2856
Publication Date(Web):February 27, 2015
DOI:10.1021/jp512177j
The Li(H2O)n– and Cs(H2O)n– (n = 0–6) clusters were studied using anion photoelectron spectroscopy combined with ab initio calculations. It was found that Li tends to be surrounded by water molecules with no water–water H-bonds being formed in the first hydration shell; while Cs sticks on the surface of water–water H-bonds network. The Li atom in its anionic or neutral state is surrounded by four water molecules through Li–O interactions within the first hydration shell; while the case of Cs is different. For the anionic Cs(H2O)n– clusters, two types of structures, namely H-end and O-end structures, were identified, with nearly degenerate energies. For the neutral Cs(H2O)n clusters, only O-end structures exist and the first hydration shell of the Cs atom has four water molecules. The different hydration nature of Li and Cs atoms can be ascribed to the delicate balance between the alkali metal–water interactions and the water–water interactions as well as the effect of excess electron.
Co-reporter:Xiaoming Huang, Hong-Guang Xu, Shengjie Lu, Yan Su, R. B. King, Jijun Zhao and Weijun Zheng
Nanoscale 2014 vol. 6(Issue 24) pp:14617-14621
Publication Date(Web):11 Sep 2014
DOI:10.1039/C4NR03130J
Our studies show that VSi12− adopts a V-centered hexagonal prism with a singlet spin state. The addition of the second V atom leads to a capped hexagonal antiprism for V2Si12− in a doublet spin state. Most interestingly, V3Si12− exhibits a ferrimagnetic, bicapped hexagonal antiprism wheel-like structure with a total spin of 4μB.
Co-reporter:Xiang-Yu Kong, Hong-Guang Xu, Pratik Koirala, Wei-Jun Zheng, Anil K. Kandalam and Puru Jena
Physical Chemistry Chemical Physics 2014 vol. 16(Issue 47) pp:26067-26074
Publication Date(Web):24 Oct 2014
DOI:10.1039/C4CP04299A
The electronic and structural properties of neutral and anionic Agn(BO2)m (n = 1–3, m = 1–2) clusters are investigated by using mass-selected anion photoelectron spectroscopy and density functional theory calculations. Agreement between the measured and calculated vertical detachment energies (VDEs) allows us to validate the equilibrium geometries of [Agn(BO2)m]− clusters obtained from theory. The ground state structures of anionic Ag2(BO2) and Agn(BO2)2 (n = 1–3) clusters are found to be very different from those of their neutral counterparts. The structures of anionic clusters are chain-like while those of the neutral clusters are closed-rings. The presence of multiple isomers for [Ag2(BO2)2]− and [Ag3(BO2)2]− in the cluster beam has also been confirmed. Several of these clusters are found to be hyperhalogens.
Co-reporter:Jin-Yun Yuan, Hong-Guang Xu and Wei-Jun Zheng
Physical Chemistry Chemical Physics 2014 vol. 16(Issue 11) pp:5434-5439
Publication Date(Web):2013/12/23
DOI:10.1039/C3CP54758B
ConC2− (n = 1–5) cluster anions were investigated using anion photoelectron spectroscopy. The adiabatic detachment energies (ADEs) and the vertical detachment energies (VDEs) of the ConC2− (n = 1–5) cluster anions were determined from their photoelectron spectra. Density functional calculations were performed for the ConC2 (n = 1–5) cluster anions and neutrals. Our studies show that the structures of ConC2− (n = 1–5) can be described as attaching C2 to the top sites, bridge sites, or hollow sites of the Con clusters. The C2 retains an integral structure unit in the ConC2 (n = 1–5) cluster anions and neutrals, rather than being separated by the Con clusters. The C2 unit in the ConC2 (n = 1–5) cluster anions and neutrals has the characteristics of a double-bond.
Co-reporter:Wen-Juan Tian, Hong-Guang Xu, Xiang-Yu Kong, Qiang Chen, Wei-Jun Zheng, Hua-Jin Zhai and Si-Dian Li
Physical Chemistry Chemical Physics 2014 vol. 16(Issue 11) pp:5129-5136
Publication Date(Web):16 Jan 2014
DOI:10.1039/C3CP55362K
We report on the structural and electronic properties and chemical bonding in a series of lithium and gold alloyed boron oxide clusters: B2O3−, LiB2O3−, AuB2O3−, and LiAuB2O3−. The clusters have been produced by laser vaporization and characterized using photoelectron spectroscopy, in combination with the Coalescence Kick and Basin Hopping global-minimum searches and density-functional theory and molecular orbital theory calculations. Electron affinities of B2O3, LiB2O3, AuB2O3, and LiAuB2O3 neutral clusters are measured to be 1.45 ± 0.08, 4.25 ± 0.08, 6.05 ± 0.08, and 2.40 ± 0.08 eV, respectively. The experimental and computational data allow the cluster structures to be established for the anions as well as their neutrals. While B2O3− (C2v) is bent, the three alloy clusters, LiB2O3− (C∞v), AuB2O3− (Cs), and LiAuB2O3− (C∞v), adopt linear or quasi-linear geometries with a metal center inserted between BO and OBO subunits, featuring charge transfer complexes, covalent gold, hyperhalogen, and dual three-center four-electron (3c-4e) π hyperbonds. The current results suggest the possibility of altering and fine-tuning the properties of boron oxides via alloying, which may lead to markedly different electronic structures and chemical reactivities. The LiB2O3 cluster belongs to the class of oxidizing agents called superhalogens, whereas AuB2O3 is a hyperhalogen species. Dual 3c-4e π hyperbonds represent a critical bonding element in boron oxides and are considered to be the root of delocalized bonding and aromaticity therein.
Co-reporter:Guo-Jin Cao, Hong-Guang Xu, Wei-Jun Zheng and Jun Li
Physical Chemistry Chemical Physics 2014 vol. 16(Issue 7) pp:2928-2935
Publication Date(Web):02 Dec 2013
DOI:10.1039/C3CP54478H
Combined anion photoelectron spectroscopy and relativistic quantum chemical studies are conducted on nucleobase–Au2− cluster anions. The vertical detachment energies of uracil–Au2− (UAu2−), thymine–Au2− (TAu2−), cytosine–Au2− (CAu2−), adenine–Au2− (AAu2−), guanine–Au2− (GAu2−) are determined to be 2.71 ± 0.08 eV, 2.74 ± 0.08 eV, 2.67 ± 0.08 eV, 2.65 ± 0.08 eV and 2.73 ± 0.08 eV, respectively, based on the measured photoelectron spectra. Through computational geometry optimizations we have identified the lowest-energy structures of these nucleobase–Au2− cluster anions. The structures are further confirmed by comparison of theoretically calculated vertical and adiabatic electron detachment energies with experimental measurements. The results reveal that the Au2− anion remains as an intact unit and interacts with the nucleobases through N–H⋯Au or C–H⋯Au nonconventional hydrogen bonds. The nucleobase–Au2− cluster anions have relatively weak N–H⋯Au hydrogen bonds and strong C–H⋯Au hydrogen bonds compared to those of nucleobase–Au− anions.
Co-reporter:Ren-Zhong Li, Hong-Guang Xu, Xi-Ling Xu, Wei-Jun Zheng
Chemical Physics Letters 2014 Volume 607() pp:105-109
Publication Date(Web):27 June 2014
DOI:10.1016/j.cplett.2014.05.051
•The structures of ConOH− (n = 1–3) were determined.•The excess electron of ConOH− (n = 1–3) cluster anions is mainly localized on the Co atoms.•The Co–O bond of CoOH−/0 has covalent characters.We investigated ConOH− (n = 1–3) clusters with photoelectron spectroscopy and density functional theory calculations. The vertical detachment energies of ConOH− (n = 1–3) were measured to be 1.41 ± 0.04, 1.22 ± 0.08, 1.62 ± 0.08 eV, respectively. The electron affinity and term energy of CoOH neutral were determined to be 1.33 ± 0.04 and 0.25 ± 0.04 eV, respectively. The most probable geometries of CoOH− and Co2OH− were determined to be L-shaped structures, and that of Co3OH− can be considered as one of the Co atoms of Co3 triangle bonded to the oxygen atom of OH group. The excess electron of the ConOH− anion is mainly localized on the Con site.Graphical abstract
Co-reporter:Jinyun Yuan, Gao-Lei Hou, Baocheng Yang, Hong-Guang Xu, and Wei-Jun Zheng
The Journal of Physical Chemistry A 2014 Volume 118(Issue 34) pp:6757-6762
Publication Date(Web):July 30, 2014
DOI:10.1021/jp504933a
The anionic and neutral ConC2H2 (n = 1–3) clusters were investigated using anion photoelectron spectroscopy and density functional calculations. The adiabatic detachment energies and vertical detachment energies of ConC2H2– (n = 1–3) were determined. Our results show that the most stable geometries of anionic ConC2H2– (n = 1–2) and neutral ConC2H2 (n = 1–3) are composed of ConC2H clusters adsorbing a hydrogen atom on the top or bridge sites of Con, whereas Co3C2H2– consists of a five-member ring of Co3C2 carbide adsorbing two hydrogen atoms on two bridge sites of Co3. The reaction mechanisms show that the inserted isomer HCoC2H can convert into the vinylidene complex Co═C═CH2 via a side-on isomer M-η2-(C2H2).
Co-reporter:Cheng-Wen Liu, Feng Wang, Lijiang Yang, Xin-Zheng Li, Wei-Jun Zheng, and Yi Qin Gao
The Journal of Physical Chemistry B 2014 Volume 118(Issue 3) pp:743-751
Publication Date(Web):January 3, 2014
DOI:10.1021/jp408439j
How salts affect water structure is an important topic in many research fields. Salt–water clusters can be used as model systems to extract interaction information that is difficult to obtain directly from bulk solutions. In the present study, integrated tempering sampling1,2 molecular dynamics (MD) are combined with quantum mechanics (QM) calculations to overcome the sampling problem in cluster structure searches. We used LiI(H2O)n and CsI(H2O)n as representatives to investigate the microsolvation of ion pairs. It was found that Li+–I– and Cs+–I– ion pairs interact with water molecules in very different ways, and the corresponding salt–water clusters have distinctly different structures. LiI strongly affects water–water interactions, and the LiI(H2O)n (n ≥ 5) clusters build around a Li+(H2O)4 motif. CsI only slightly perturbs the water cluster structure, and CsI(H2O)n favors the clathrate-like structure when n = 18 or 20. Consistent with the law of “matching water affinities”, Li+ and I– are more easily separated by solvent molecules than the Cs+–I– ion pair.
Co-reporter:Xiao-Jiao Deng;Dr. Xiang-Yu Kong;Dr. Xi-Ling Xu;Dr. Hong-Guang Xu; Dr. Wei-Jun Zheng
ChemPhysChem 2014 Volume 15( Issue 18) pp:3987-3993
Publication Date(Web):
DOI:10.1002/cphc.201402615
Abstract
A series of cobalt-doped germanium clusters, CoGen−/0 (n=2–11), are investigated by using anion photoelectron spectroscopy combined with density functional theory calculations. For both anionic and neutral CoGen (n=2–11) clusters, the critical size of the transition from exo- to endohedral structures is n=9. Natural population analysis shows that there is electron transfer from the Gen framework to the Co atom at n=7–11 for both anionic and neutral CoGen clusters. The magnetic moments of the anionic and neutral CoGen clusters decrease to the lowest values at n=10 and 11. The transfer of electrons from the Gen framework to the Co atom and the minimization of the magnetic moments are related to the evolution of CoGen structures from exo- to endohedral.
Co-reporter:Jing-Heng Meng;Xiao-Jiao Deng;Zi-Yu Li;Dr. Sheng-Gui He;Dr. Wei-Jun Zheng
Chemistry - A European Journal 2014 Volume 20( Issue 19) pp:5580-5583
Publication Date(Web):
DOI:10.1002/chem.201400218
Abstract
The first example of a metal oxide cluster anion, La6O10− that can activate methane under ambient conditions is reported. This reaction is facilitated by the oxygen-centered radical (O−⋅) and follows the hydrogen atom transfer mechanism. The La6O10− has a high vertical electron detachment energy (VDE=4.06 eV) and a high symmetry (C4v).
Co-reporter:Ren-Zhong Li ; Cheng-Wen Liu ; Yi Qin Gao ; Hong Jiang ; Hong-Guang Xu
Journal of the American Chemical Society 2013 Volume 135(Issue 13) pp:5190-5199
Publication Date(Web):February 23, 2013
DOI:10.1021/ja4006942
In order to understand the microsolvation of LiI and CsI in water and provide information about the dependence of solvation processes on different ions, we investigated the LiI(H2O)n– and CsI(H2O)n– (n = 0–6) clusters using photoelectron spectroscopy. The structures of these clusters and their corresponding neutrals were investigated with ab initio calculations and confirmed by comparing with the photoelectron spectroscopy experiments. Our studies show that the structural evolutions of LiI(H2O)n and CsI(H2O)n clusters are very different. The Li–I distance in LiI(H2O)n– increases abruptly at n = 3, whereas the abrupt elongation of the Li–I distance in neutral LiI(H2O)n occurs at n = 5. In contrast to the LiI(H2O)n– clusters, the Cs–I distance in CsI(H2O)n– increases significantly at n = 3, reaches a maximum at n = 4, and decreases again as n increases further. There is no abrupt change of the Cs–I distance in neutral CsI(H2O)n as n increases from 0 to 6. Water molecules interact strongly with the Li ion; consequently, water molecule(s) can insert within the Li+–I– ion pair. In contrast, five or six water molecules are not enough to induce obvious separation of the Cs+–I– ion pair since the Cs–water interaction is relatively weak compared to the Li–water interaction. Our work has shown that the structural variation and microsolvation in MI(H2O)n clusters are determined by the delicate balance between ion–ion, ion–water, and water–water interactions, which may have significant implications for the general understanding of salt effects in water solution.
Co-reporter:Hong-Guang Xu, Xiao-Na Li, Xiang-Yu Kong, Sheng-Gui He and Wei-Jun Zheng
Physical Chemistry Chemical Physics 2013 vol. 15(Issue 40) pp:17126-17133
Publication Date(Web):14 Aug 2013
DOI:10.1039/C3CP52823E
We investigated the interaction of TiO+ with water by conducting infrared photodissociation spectroscopy and density functional theory calculations on TiO(H2O)Ar+ and TiO(H2O)5–7+ clusters. The studies show that TiO(H2O)Ar+ has two isomers, Ti(OH)2Ar+ and (H2O)–TiOAr+, coexisting in our experiments. The structure of TiO(H2O)5+ is characterized by attaching four water molecules to a Ti(OH)2+ core with their O atoms interacting with the Ti atom directly. With the increasing number of water molecules, the additional water molecules start to form hydrogen bonds with the inner shell water molecules and the OH groups of Ti(OH)2+ instead of coordinating directly with the Ti atom. Therefore, the structures of TiO(H2O)6+ and TiO(H2O)7+ clusters are evolved from that of TiO(H2O)5+ by adding the sixth and seventh water molecules to the second solvent-shell. Our results demonstrate that a Ti(OH)2+ type of product is dominant when TiO+ interacts with water, especially when more water molecules are involved.
Co-reporter:Ren-Zhong Li, Jun Liang, Xi-Ling Xu, Hong-Guang Xu, Wei-Jun Zheng
Chemical Physics Letters 2013 Volume 575() pp:12-17
Publication Date(Web):21 June 2013
DOI:10.1016/j.cplett.2013.04.066
•ConO− (n = 1–3) were investigated with photoelectron spectroscopy and calculations.•The structures of ConO− (n = 1–3) were determined.•The HOMOs of ConO− (n = 1–3) cluster anions are mainly localized on the Co atoms.•The oxygen atom only had a minor influence on the electronic structures.ConO− (n = 1–3) clusters were investigated with photoelectron spectroscopy and density functional calculations. The vertical detachment energies (VDEs) of ConO− (n = 1–3) were measured to be 1.54 ± 0.04, 1.43 ± 0.08, and 1.42 ± 0.08 eV respectively from their photoelectron spectra. The electron affinity and term energy of CoO were determined to be 1.54 ± 0.04 eV and 0.31 ± 0.04 eV respectively based on the vibrationally resolved photoelectron spectrum of CoO− and theoretical calculations. The structures of ConO− (n = 1–3) were determined by comparison of photoelectron experiments and calculations. The analysis of molecular orbitals shows that the HOMOs of ConO− (n = 1–3) cluster anions are mainly localized on the Co atoms.
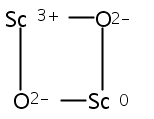
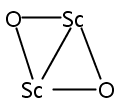
Co-reporter:Jinyun Yuan, Hong-Guang Xu, Xiangyu Kong, Weijun Zheng
Chemical Physics Letters 2013 Volume 564() pp:6-10
Publication Date(Web):28 March 2013
DOI:10.1016/j.cplett.2013.01.068
The ScmOn- (m = 2–5, n = 2–3) cluster anions were investigated using photoelectron spectroscopy and density functional theory (DFT) calculations. The adiabatic detachment energies (ADEs) and vertical detachment energies (VDEs) of these cluster anions were estimated from their photoelectron spectra. The most probable structures of ScmOnScmOn (m = 2–5, n = 2–3) cluster anions and neutrals were determined by combining DFT calculations with the photoelectron spectroscopy experiments. The structures of ScmOn- (m = 2–5, n = 2–3) can be characterized as attaching oxygen atoms to the top sites, bridge sites, or hollow sites of Scm clusters. There is no direct interaction between the oxygen atoms.Graphical abstractHighlights► ScmOn- (m = 2–5, n = 2–3) has the O atoms attached to the top, bridge, or hollow sites of Scm cluster. ► There is no direct interaction between the oxygen atoms. ► The addition of oxygen atoms weakens the Sc–Sc bonds of the Scm (m = 2–5) clusters.
Co-reporter: Dr. Hui Chen;Xiang-Yu Kong; Dr. Weijun Zheng; Dr. Jiannian Yao; Dr. Anil K. Kalam; Dr. Puru Jena
ChemPhysChem 2013 Volume 14( Issue 14) pp:3303-3308
Publication Date(Web):
DOI:10.1002/cphc.201300677
Abstract
Hyperhalogens were recently identified as a new class of highly electronagative species which are composed of metals and superhalogens. In this work, high-level theoretical calculations and photoelectron spectroscopy experiments are systematically conducted to investigate a series of coinage-metal-containing hyperhalogen anions, Cu(BO2)2−, Ag(BO2)2−, and Au(BO2)2−. The vertical electron detachment energy (VDE) of Ag(BO2)2− is anomalously higher than those of Au(BO2)2− and Cu(BO2)2−. In quantitative agreement with the experiment, high-level ab initio calculations reveal that spin–orbit coupling (SOC) lowers the VDE of Au(BO2)2− significantly. The sizable magnitude of about 0.5 eV of SOC effect on the VDE of Au(BO2)2− demonstrates that SOC plays an important role in the electronic structure of gold hyperhalogens. This study represents a new paradigm for relativistic electronic structure calculations for the one-electron-removal process of ionic AuIL2 complexes, which is characterized by a substantial SOC effect.
Co-reporter:Jinyun Yuan, Yuchao Zhao, Gaolei Hou, Zhen Gao, Weijun Zheng
International Journal of Mass Spectrometry 2012 Volume 309() pp:49-55
Publication Date(Web):1 January 2012
DOI:10.1016/j.ijms.2011.08.026
Al+(C2H4)n clusters were produced by reaction of laser ablated Al+ ions with ethene molecules seeded in a pulsed molecular beam, and detected with a reflectron time-of-flight mass spectrometer. The mass spectrum shows that an Al+ ion can combine with at least eight ethene molecules to form Al+(C2H4)n clusters. Based on the photodissociation experiments and ab initio calculations, we suggest that an Al+ ion can strongly interact with one or two C atoms to form Al–C σ-bonds and trigger addition reaction of ethene molecules to develop chain or ring structures. The interaction of Al+ ion with ethene is different from the interaction between V+ ion and ethene molecules reported previously (Int. J. Mass. Spectrom. 295 (2010) 36), wherein a V+ ion can only combine with no more than four ethene molecules at similar experimental conditions and causes little change in the structures of the ethene molecules.Graphical abstractHighlights► We propose that Al–C σ-bonds can be formed in Al+(C2H4)n (n > 1) clusters. ► The strong interaction between Al+ and ethene molecules weakens the CC bond significantly. ► Al+ can trigger addition reaction of ethene molecules to develop chain or ring structures.
Co-reporter:Yuan Feng, Gao-Lei Hou, Hong-Guang Xu, Zeng-Guang Zhang, Wei-Jun Zheng
Chemical Physics Letters 2012 Volume 545() pp:21-25
Publication Date(Web):30 August 2012
DOI:10.1016/j.cplett.2012.07.030
Abstract
CunBO2(OH)− (n = 1, 2) clusters were studied by anion photoelectron spectroscopy and density functional calculations. From the experimental photoelectron spectra, the adiabatic detachment energy (ADE) and vertical detachment energy (VDE) of CuBO2(OH)− are determined to be 4.00 ± 0.08 and 4.26 ± 0.08 eV, and those of Cu2BO2(OH)− to be 2.30 ± 0.08 and 2.58 ± 0.08 eV. The structures of CunBO2(OH)− and their corresponding neutrals are assigned by comparison between theoretical calculations and experimental measurements. Both experiment and theory show that CuBO2(OH) can be viewed as a superhalogen, thus, confirmed that OH can behave like a halogen atom to participate in superhalogen formation.
Co-reporter:Weijun Zheng, Y. Seol Kim and Ralf I. Kaiser
Physical Chemistry Chemical Physics 2011 vol. 13(Issue 35) pp:15749-15754
Publication Date(Web):17 May 2011
DOI:10.1039/C1CP20528E
We investigated the irradiation of low temperature H218O/N2 ice mixtures with energetic electrons in an ultrahigh vacuum chamber. The newly formed species, such as nitric oxide (N18O), nitrous oxide (NN18O), hydrogen peroxide (H218O2) and hydrazine (N2H4), were identified in the experiments with infrared absorption spectroscopy and mass spectrometry. The results suggest that the unimolecular decomposition of water molecules within water ices at 10 K can lead to the formation of transient, suprathermal oxygen atoms. These oxygen atoms may play an important role in the formation of oxygen-containing biomolecules such as amino acids and sugar, as well as the decomposition of the biomolecules in the ices.
Co-reporter:Yu-Chao Zhao, Jinyun Yuan, Zeng-Guang Zhang, Hong-Guang Xu and Weijun Zheng
Dalton Transactions 2011 vol. 40(Issue 11) pp:2502-2508
Publication Date(Web):02 Feb 2011
DOI:10.1039/C0DT01179G
Manganese polysulfide cations, MnSx+ (x = 1–10), were studied with mass-selected photodissociation experiments and density functional calculations. We found that MnS+, MnS2+ and MnS3+ undergo dissociation at 355 nm by loss of S, S2 and S3, respectively. The dissociation of larger clusters is relatively complex because of the existence of multiple isomers and multiple dissociation channels. The geometric structures of the low-lying isomers found by theoretical calculations are consistent with the dissociation channels observed in the experiments. The dissociation of MnSx+ clusters occurs mainly by breaking of the Mn–S bonds since they are weaker than the S–S bonds.
Co-reporter:Yuan Feng, Min Cheng, Xiang-Yu Kong, Hong-Guang Xu and Wei-Jun Zheng
Physical Chemistry Chemical Physics 2011 vol. 13(Issue 35) pp:15865-15872
Publication Date(Web):03 Aug 2011
DOI:10.1039/C1CP20831D
We investigated the microscopic solvation of NaBO2 in water by conducting photoelectron spectroscopy and ab initio studies on NaBO2−(H2O)n (n = 0–4) clusters. The vertical detachment energy (VDE) of NaBO2− is estimated to be 1.00 ± 0.08 eV. The photoelectron spectra of NaBO2−(H2O)1 and NaBO2−(H2O)2 are similar to that of bare NaBO2−, except that their VDEs shift to higher electron binding energies (EBE). For the spectra of NaBO2−(H2O)3 and NaBO2−(H2O)4, a low EBE feature appears dramatically in addition to the features observed in the spectra of NaBO2−(H2O)0–2. Our study shows that the water molecules mainly interact with the BO2− unit in NaBO2−(H2O)1 and NaBO2−(H2O)2 clusters to form Na–BO2−(H2O)n type structures, while in NaBO2−(H2O)3 and NaBO2−(H2O)4 clusters, the water molecules can interact strongly with the Na atom, therefore, the Na–BO2−(H2O)n and Na(H2O)n⋯BO2− types of structures coexist. That can be seen as an initial step of the transition from a contact ion pair (CIP) structure to a solvent-separated ion pair (SSIP) structure for the dissolution of NaBO2.
Co-reporter:Jinyun Yuan, Hong-Guang Xu, Zeng-Guang Zhang, Yuan Feng, and Weijun Zheng
The Journal of Physical Chemistry A 2011 Volume 115(Issue 2) pp:182-186
Publication Date(Web):December 14, 2010
DOI:10.1021/jp108847f
We investigated the adsorption of C2H radical on small cobalt clusters by mass spectrometry and by measuring the photoelectron spectra of ConC2H− (n = 1−5) cluster anions. The most stable structures of ConC2H− (n = 1−5) and their neutrals were determined by comparing the experimental results with theoretical calculations. Our studies show that C2H radical still maintains its integrity as a structural unit in ConC2H− clusters, rather than being divided by Con clusters. The most stable isomers of Co1−2C2H− clusters are linear with the C2H interacting with only one Co atom, while those of Co3−5C2H− cluster anions are quasi-planar structures with the carbon−carbon bonds bending slightly toward the Co3−5 clusters. The carbon−carbon bond of C2H is lengthened more in Co3−5C2H− clusters than in Co1−2C2H−.
Co-reporter:Zeng-Guang Zhang, Hong-Guang Xu, Xiangyu Kong, and Weijun Zheng
The Journal of Physical Chemistry A 2011 Volume 115(Issue 1) pp:13-18
Publication Date(Web):December 9, 2010
DOI:10.1021/jp109221m
Small aluminum−vanadium oxide clusters, AlVOy− (y = 1−3) and AlxVO2− (x = 2, 3), were investigated with anion photoelectron spectroscopy and density functional calculations. The adiabatic detachment energies of AlVOy− were estimated to be 1.06 ± 0.05, 1.50 ± 0.08, and 2.83 ± 0.08 eV for y = 1, 2, and 3. Those of Al2VO2− and Al3VO2− were estimated to be 1.22 ± 0.08 and 1.25 ± 0.08 eV. Comparison of theoretical calculations with experimental measurement suggests that the most probable structure of AlVO− cluster is quasilinear with O atom in the middle. AlVO2− has an irregular chain structure of Al−O−V−O and a C2v cyclic structure very close in energy. The structure of AlVO3− cluster is evolved from the C2v cyclic AlVO2− structure by adding the third O atom to the V atom. Al2VO2− has a pair of nearly degenerate Al−O−V−O−Al chain structures that can be considered as cis and trans forms. Al3VO2− probably has two low-lying isomers each containing a four-membered ring. The structures of the corresponding neutral clusters are discussed.
Co-reporter:Jinyun Yuan, Zeng-Guang Zhang, Yuchao Zhao, Gao-Lei Hou, Hong-Guang Xu, Weijun Zheng
International Journal of Mass Spectrometry 2010 Volume 295(1–2) pp:36-42
Publication Date(Web):15 July 2010
DOI:10.1016/j.ijms.2010.06.018
V+(C2H4)n were produced in a supersonic molecular beam by laser vaporization and were mass-analyzed with a reflectron time-of-flight mass spectrometer. V+(C2H4)n (n = 1–3) are predominant mass peaks in the mass spectrum. These species were mass-selected and photodissociated with 1064, 532 and 355 nm photons. Dissociation occurs by elimination of neutral ethene molecules. The fragment ion yields were studied as a function of photon fluxes in order to give insight into the dissociation mechanisms. The geometric structures, bond dissociation energies and ground electronic state of V+(C2H4)n (n = 1–4) were investigated using density functional theory (DFT). It has been confirmed that the most stable structures of V+(C2H4)n are all in quintet states.V+(C2H4)n (n = 1–3) were photodissociated with 1064, 532 and 355 nm photons. The dissociation occurred by elimination of neutral ethene molecules.
Co-reporter:Hong-Guang Xu, Zeng-Guang Zhang, Yuan Feng, Weijun Zheng
Chemical Physics Letters 2010 Volume 498(1–3) pp:22-26
Publication Date(Web):30 September 2010
DOI:10.1016/j.cplett.2010.08.027
We conducted a photoelectron spectroscopy and density-functional study on Sc2Sin− (n = 2–6) clusters. The adiabatic detachment energies of Sc2Si2–6− were estimated to be 1.42 ± 0.08, 1.37 ± 0.08, 1.33 ± 0.08, 1.9 ± 0.2, and 2.0 ± 0.2 eV respectively from their photoelectron spectra. Comparison of theoretical and experimental results indicates that each of these clusters has more than one isomer in the experiments. In the most stable structures of Sc2Si3–6− clusters, the silicon atoms form an n-membered silicon ring, and the two Sc atoms cap to the opposite sides of the ring. The Sc–Sc interaction in Sc2Sin− clusters is very weak comparing to the strong V–V interaction in V2Sin− clusters.In the most stable structures of Sc2Si3–6− clusters, the silicon atoms form an n-membered silicon ring, and the two Sc atoms cap to the opposite sides of the ring.
Co-reporter:Hong-Guang Xu, Zeng-Guang Zhang, Yuan Feng, Jinyun Yuan, Yuchao Zhao, Weijun Zheng
Chemical Physics Letters 2010 Volume 487(4–6) pp:204-208
Publication Date(Web):5 March 2010
DOI:10.1016/j.cplett.2010.01.050
Co-reporter:Weijun Zheng, David Jewitt and Ralf I. Kaiser
The Journal of Physical Chemistry A 2009 Volume 113(Issue 42) pp:11174-11181
Publication Date(Web):August 12, 2009
DOI:10.1021/jp903817y
The crystalline state of water ice in the Solar System depends on the temperature history of the ice and the influence of energetic particles to which it has been exposed. We measured the infrared absorption spectra of amorphous and crystalline water ice in the 10−50 K and 10−140 K temperature ranges, respectively, and conducted a systematic experimental study to investigate the amorphization of crystalline water ice via ionizing radiation irradiation at doses of up to 160 ± 30 eV per molecule. We found that crystalline water ice can be converted only partially to amorphous ice by electron irradiation. The experiments showed that a fraction of the 1.65 μm band, which is characteristic for crystalline water ice, survived the irradiation, to a degree that strongly depends on the temperature. Quantitative kinetic fits of the temporal evolution of the 1.65 μm band clearly demonstrate that there is a balance between thermal recrystallization and irradiation-induced amorphization, with thermal recrystallizaton dominant at higher temperatures. Our experiments show the amorphization at 40 K was incomplete, in contradiction to Mastrapa and Brown’s conclusion (Icarus 2006, 183, 207.). At 50 K, the recrystallization due to thermal effects is strong, and most of the crystalline ice survived. Temperatures of most icy objects in the Solar System, including Jovian satellites, Saturnian satellites (including Titan), and Kuiper Belt Objects, are equal to or above 50 K; this explains why water ice detected on those objects is mostly crystalline.
Co-reporter:Zhen Zeng, Gao-Lei Hou, Jian Song, Gang Feng, Hong-Guang Xu and Wei-Jun Zheng
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 14) pp:NaN9147-9147
Publication Date(Web):2015/02/25
DOI:10.1039/C5CP00020C
The microsolvation of LiBO2 in water was investigated by conducting anion photoelectron spectroscopy and ab initio studies on the LiBO2(H2O)n− (n = 0–5) clusters. By comparing calculations with experiments, the structures of these clusters and their corresponding neutrals were assigned, and their structural evolutions were revealed. During the anionic structural evolution with n increasing to 5, hydroxyborate and metaborate channels were identified and the metaborate channel is more favorable. For the hydroxyborate structures, the anionic Li+–BO2− ion pair reacts with a water molecule to produce the LiBO(OH)2− moiety and three water molecules tend to dissolve this moiety. In the metaborate channel, two types of solvent-separated ion pair (SSIP) geometries were determined as the ring-type and linear-type. The transition from the contact ion pair (CIP) to the ring-type of SSIP starts at n = 3, while that to the linear-type of SSIP occurs at n = 4. In neutral LiBO2(H2O)n clusters, the first water molecule prefers to react with the Li+–BO2− ion pair to generate the LiBO(OH)2 moiety, analogous to the bulk crystal phase of α-LiBO2 with two O atoms substituted by two OH groups. The Li–O distance in the LiBO(OH)2 moiety increases with the increasing number of water molecules and elongates abruptly at n = 4. Our studies provide new insight into the initial dissolution of LiBO2 salt in water at the molecular level and may be correlated to the bulk state.
Co-reporter:Yuan Feng, Min Cheng, Xiang-Yu Kong, Hong-Guang Xu and Wei-Jun Zheng
Physical Chemistry Chemical Physics 2011 - vol. 13(Issue 35) pp:NaN15872-15872
Publication Date(Web):2011/08/03
DOI:10.1039/C1CP20831D
We investigated the microscopic solvation of NaBO2 in water by conducting photoelectron spectroscopy and ab initio studies on NaBO2−(H2O)n (n = 0–4) clusters. The vertical detachment energy (VDE) of NaBO2− is estimated to be 1.00 ± 0.08 eV. The photoelectron spectra of NaBO2−(H2O)1 and NaBO2−(H2O)2 are similar to that of bare NaBO2−, except that their VDEs shift to higher electron binding energies (EBE). For the spectra of NaBO2−(H2O)3 and NaBO2−(H2O)4, a low EBE feature appears dramatically in addition to the features observed in the spectra of NaBO2−(H2O)0–2. Our study shows that the water molecules mainly interact with the BO2− unit in NaBO2−(H2O)1 and NaBO2−(H2O)2 clusters to form Na–BO2−(H2O)n type structures, while in NaBO2−(H2O)3 and NaBO2−(H2O)4 clusters, the water molecules can interact strongly with the Na atom, therefore, the Na–BO2−(H2O)n and Na(H2O)n⋯BO2− types of structures coexist. That can be seen as an initial step of the transition from a contact ion pair (CIP) structure to a solvent-separated ion pair (SSIP) structure for the dissolution of NaBO2.
Co-reporter:Weijun Zheng, Y. Seol Kim and Ralf I. Kaiser
Physical Chemistry Chemical Physics 2011 - vol. 13(Issue 35) pp:NaN15754-15754
Publication Date(Web):2011/05/17
DOI:10.1039/C1CP20528E
We investigated the irradiation of low temperature H218O/N2 ice mixtures with energetic electrons in an ultrahigh vacuum chamber. The newly formed species, such as nitric oxide (N18O), nitrous oxide (NN18O), hydrogen peroxide (H218O2) and hydrazine (N2H4), were identified in the experiments with infrared absorption spectroscopy and mass spectrometry. The results suggest that the unimolecular decomposition of water molecules within water ices at 10 K can lead to the formation of transient, suprathermal oxygen atoms. These oxygen atoms may play an important role in the formation of oxygen-containing biomolecules such as amino acids and sugar, as well as the decomposition of the biomolecules in the ices.
Co-reporter:Xiang-Yu Kong, Hong-Guang Xu, Pratik Koirala, Wei-Jun Zheng, Anil K. Kandalam and Puru Jena
Physical Chemistry Chemical Physics 2014 - vol. 16(Issue 47) pp:NaN26074-26074
Publication Date(Web):2014/10/24
DOI:10.1039/C4CP04299A
The electronic and structural properties of neutral and anionic Agn(BO2)m (n = 1–3, m = 1–2) clusters are investigated by using mass-selected anion photoelectron spectroscopy and density functional theory calculations. Agreement between the measured and calculated vertical detachment energies (VDEs) allows us to validate the equilibrium geometries of [Agn(BO2)m]− clusters obtained from theory. The ground state structures of anionic Ag2(BO2) and Agn(BO2)2 (n = 1–3) clusters are found to be very different from those of their neutral counterparts. The structures of anionic clusters are chain-like while those of the neutral clusters are closed-rings. The presence of multiple isomers for [Ag2(BO2)2]− and [Ag3(BO2)2]− in the cluster beam has also been confirmed. Several of these clusters are found to be hyperhalogens.
Co-reporter:Wen-Juan Tian, Hong-Guang Xu, Xiang-Yu Kong, Qiang Chen, Wei-Jun Zheng, Hua-Jin Zhai and Si-Dian Li
Physical Chemistry Chemical Physics 2014 - vol. 16(Issue 11) pp:NaN5136-5136
Publication Date(Web):2014/01/16
DOI:10.1039/C3CP55362K
We report on the structural and electronic properties and chemical bonding in a series of lithium and gold alloyed boron oxide clusters: B2O3−, LiB2O3−, AuB2O3−, and LiAuB2O3−. The clusters have been produced by laser vaporization and characterized using photoelectron spectroscopy, in combination with the Coalescence Kick and Basin Hopping global-minimum searches and density-functional theory and molecular orbital theory calculations. Electron affinities of B2O3, LiB2O3, AuB2O3, and LiAuB2O3 neutral clusters are measured to be 1.45 ± 0.08, 4.25 ± 0.08, 6.05 ± 0.08, and 2.40 ± 0.08 eV, respectively. The experimental and computational data allow the cluster structures to be established for the anions as well as their neutrals. While B2O3− (C2v) is bent, the three alloy clusters, LiB2O3− (C∞v), AuB2O3− (Cs), and LiAuB2O3− (C∞v), adopt linear or quasi-linear geometries with a metal center inserted between BO and OBO subunits, featuring charge transfer complexes, covalent gold, hyperhalogen, and dual three-center four-electron (3c-4e) π hyperbonds. The current results suggest the possibility of altering and fine-tuning the properties of boron oxides via alloying, which may lead to markedly different electronic structures and chemical reactivities. The LiB2O3 cluster belongs to the class of oxidizing agents called superhalogens, whereas AuB2O3 is a hyperhalogen species. Dual 3c-4e π hyperbonds represent a critical bonding element in boron oxides and are considered to be the root of delocalized bonding and aromaticity therein.
Co-reporter:Xi-Ling Xu, Xiao-Jiao Deng, Hong-Guang Xu and Wei-Jun Zheng
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 46) pp:NaN31022-31022
Publication Date(Web):2015/10/27
DOI:10.1039/C5CP04482K
CnSm− (n = 2–7; m = 1, 2) clusters were investigated by using photoelectron spectroscopy combined with density functional theory calculations. We found that the vertical detachment energies of both CnS− and CnS2− (n = 2–7) clusters exhibit a strong odd–even alternation with an increasing number of carbon atoms: the VDEs of even-n clusters are higher than those of adjacent odd-n clusters. The most stable structures of the anionic and neutral CnS (n = 2–7) clusters are linear with the S atom locating at one end of the carbon chain except that the structure of C3S− is slightly bent. The ground state isomers of the anionic and neutral CnS2 (n = 2–7) clusters are all linear structures with two S atoms locating at two ends of the carbon chain. The electron affinities of the neutral CnS (n = 2, 4–7) and CnS2 (n = 2–7) clusters are determined based on the experimental adiabatic detachment energies of the corresponding anion species, because the most stable structures of the neutral clusters are similar to those of the corresponding anions.
Co-reporter:Gang Feng, Gao-Lei Hou, Hong-Guang Xu, Zhen Zeng and Wei-Jun Zheng
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 8) pp:NaN5631-5631
Publication Date(Web):2015/01/15
DOI:10.1039/C4CP05698A
The initial dissolution steps of lithium sulfate (Li2SO4) in water were investigated by performing anion photoelectron spectroscopy and density functional theory calculations on the Li2SO4(H2O)n− (n = 0–5) clusters. The plausible structures of these clusters and the corresponding neutral clusters were obtained using LC-ωPBE/6-311++G(d,p) calculations by comparing the experimental and theoretical vertical electron detachment energies. Two types of structures for bare Li2SO4−/0 were found: a turtle-shaped structure and a propeller-shaped structure. For Li2SO4(H2O)n− cluster anions with n = 1–3, two kinds of isomers derived from the turtle-shaped and propeller-shaped structures of bare Li2SO4− were identified. For n = 4–5, these two kinds of isomers present similar structural and energetic features and thus are not distinguishable. For the anionic clusters the water molecules prefer to firstly interact with one Li atom until fully coordinating it. While for the neutral clusters, the water molecules interact with the two Li atoms alternately, therefore, showing a pairwise solvation behavior. The Li–S distance increases smoothly upon addition of water molecules one by one. Addition of five water molecules to Li2SO4 cannot induce the dissociation of one Li+ ion because the water molecules are shared by two Li+ ions.
Co-reporter:Li-Juan Zhao, Hong-Guang Xu, Gang Feng, Peng Wang, Xi-Ling Xu and Wei-Jun Zheng
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 8) pp:NaN6181-6181
Publication Date(Web):2016/01/22
DOI:10.1039/C5CP07673K
We investigate BS2− and BSO− clusters using photoelectron spectroscopy and theoretical calculations. The electron affinities of BS2 and BSO are measured to be 3.80 ± 0.03 and 3.88 ± 0.03 eV, respectively, higher than those of halogen atoms. Thus, BS2 and BSO can be considered as superhalogens. The comparison of experimental and theoretical results confirmed that the ground state structures of BS2−, BSO−, and their neutrals are all linear. Analyses of natural bond orbitals suggest that both BS2− and BSO− have dual 3c-4e π hyperbonds.
Co-reporter:Ren-Zhong Li, Gao-Lei Hou, Cheng-Wen Liu, Hong-Guang Xu, Xiang Zhao, Yi Qin Gao and Wei-Jun Zheng
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 1) pp:NaN565-565
Publication Date(Web):2015/11/18
DOI:10.1039/C5CP05550D
We investigated (NaI)2−(H2O)n (n = 0–6) clusters to examine the initial solvation process of (NaI)2 in water, using negative ion photoelectron spectroscopy and theoretical calculations. The structures of these clusters and their neutrals were determined by comparing ab initio calculations with experimental results. It is found that bare (NaI)2− is a L-shaped structure and the corresponding neutral is a rhombus. In (NaI)2−(H2O), the water molecule prefers to interact with the middle Na atom of the L-shaped (NaI)2−. For (NaI)2−(H2O)n clusters with n = 2–3, two types of structures are nearly degenerate in energy: one is L-shaped and the other is pyramid-shaped. As for (NaI)2−(H2O)n with n = 4–6, the dominant structures are pyramid-shaped. For the anionic clusters, one of the Na–I distances increases abruptly when n = 2; for the neutral clusters, rapid lengthening of the Na–I distances occurs when n = 4. Additionally, analyses of the reduced density gradient were carried out, and the results reveal that Na+–water interactions dominate in (NaI)2−(H2O)n for n ≤ 4, whereas I−–water and water–water interactions are significantly enhanced when n increases to 5.
Co-reporter:Yu-Chao Zhao, Jinyun Yuan, Zeng-Guang Zhang, Hong-Guang Xu and Weijun Zheng
Dalton Transactions 2011 - vol. 40(Issue 11) pp:NaN2508-2508
Publication Date(Web):2011/02/02
DOI:10.1039/C0DT01179G
Manganese polysulfide cations, MnSx+ (x = 1–10), were studied with mass-selected photodissociation experiments and density functional calculations. We found that MnS+, MnS2+ and MnS3+ undergo dissociation at 355 nm by loss of S, S2 and S3, respectively. The dissociation of larger clusters is relatively complex because of the existence of multiple isomers and multiple dissociation channels. The geometric structures of the low-lying isomers found by theoretical calculations are consistent with the dissociation channels observed in the experiments. The dissociation of MnSx+ clusters occurs mainly by breaking of the Mn–S bonds since they are weaker than the S–S bonds.
Co-reporter:Ke-Wei Ding, Xiao-Wei Li, Hong-Guang Xu, Tao-Qi Li, Zhong-Xue Ge, Qian Wang and Wei-Jun Zheng
Chemical Science (2010-Present) 2015 - vol. 6(Issue 8) pp:NaN4729-4729
Publication Date(Web):2015/05/11
DOI:10.1039/C5SC01103E
TiNn+ clusters were generated by laser ablation and analyzed experimentally by mass spectrometry. The results showed that the mass peak of the TiN12+ cluster is dominant in the spectrum. The TiN12+ cluster was further investigated by photodissociation experiments with 266, 532 and 1064 nm photons. Density functional calculations were conducted to investigate stable structures of TiN12+ and the corresponding neutral cluster, TiN12. The theoretical calculations found that the most stable structure of TiN12+ is Ti(N2)6+ with Oh symmetry. The calculated binding energy is in good agreement with that obtained from the photodissociation experiments. The most stable structure of neutral TiN12 is Ti(N2)6 with D3d symmetry. The Ti–N bond strengths are greater than 0.94 eV in both Ti(N2)6+ and its neutral counterpart. The interaction between Ti and N2 weakens the N–N bond significantly. For neutral TiN12, the Ti(N3)4 azide, the N5TiN7 sandwich structure and the N6TiN6 structure are much higher in energy than the Ti(N2)6 complex. The DFT calculations predicted that the decomposition of Ti(N3)4, N5TiN7, and N6TiN6 into a Ti atom and six N2 molecules can release energies of about 139, 857, and 978 kJ mol−1 respectively.
Co-reporter:Gang Feng, Cheng-Wen Liu, Zhen Zeng, Gao-Lei Hou, Hong-Guang Xu and Wei-Jun Zheng
Physical Chemistry Chemical Physics 2017 - vol. 19(Issue 23) pp:NaN15569-15569
Publication Date(Web):2017/05/23
DOI:10.1039/C7CP02965A
To understand the initial hydration processes of MgCl2, we measured photoelectron spectra of MgCl2(H2O)n− (n = 0–6) and conducted ab initio calculations on MgCl2(H2O)n− and their neutral counterparts up to n = 7. A dramatic drop in the vertical detachment energy (VDE) was observed upon addition of the first water molecule to bare MgCl2−. This large variation in VDE can be associated with the charge-transfer-to-solvent (CTTS) effect occurring in the MgCl2(H2O)n− clusters, as hydration induces transfer of the excess electron of MgCl2− to the water molecules. Investigation of the separation of Cl−–Mg2+ ion pair shows that, in MgCl2(H2O)n− anions, breaking of the first Mg–Cl bond occurs at n = 4, while breaking of the second Mg–Cl bond takes place at n = 6. For neutral MgCl2(H2O)n clusters, breaking of the first Mg–Cl bond starts at n = 7.
Co-reporter:Hong-Guang Xu, Xiao-Na Li, Xiang-Yu Kong, Sheng-Gui He and Wei-Jun Zheng
Physical Chemistry Chemical Physics 2013 - vol. 15(Issue 40) pp:NaN17133-17133
Publication Date(Web):2013/08/14
DOI:10.1039/C3CP52823E
We investigated the interaction of TiO+ with water by conducting infrared photodissociation spectroscopy and density functional theory calculations on TiO(H2O)Ar+ and TiO(H2O)5–7+ clusters. The studies show that TiO(H2O)Ar+ has two isomers, Ti(OH)2Ar+ and (H2O)–TiOAr+, coexisting in our experiments. The structure of TiO(H2O)5+ is characterized by attaching four water molecules to a Ti(OH)2+ core with their O atoms interacting with the Ti atom directly. With the increasing number of water molecules, the additional water molecules start to form hydrogen bonds with the inner shell water molecules and the OH groups of Ti(OH)2+ instead of coordinating directly with the Ti atom. Therefore, the structures of TiO(H2O)6+ and TiO(H2O)7+ clusters are evolved from that of TiO(H2O)5+ by adding the sixth and seventh water molecules to the second solvent-shell. Our results demonstrate that a Ti(OH)2+ type of product is dominant when TiO+ interacts with water, especially when more water molecules are involved.
Co-reporter:Guo-Jin Cao, Hong-Guang Xu, Wei-Jun Zheng and Jun Li
Physical Chemistry Chemical Physics 2014 - vol. 16(Issue 7) pp:NaN2935-2935
Publication Date(Web):2013/12/02
DOI:10.1039/C3CP54478H
Combined anion photoelectron spectroscopy and relativistic quantum chemical studies are conducted on nucleobase–Au2− cluster anions. The vertical detachment energies of uracil–Au2− (UAu2−), thymine–Au2− (TAu2−), cytosine–Au2− (CAu2−), adenine–Au2− (AAu2−), guanine–Au2− (GAu2−) are determined to be 2.71 ± 0.08 eV, 2.74 ± 0.08 eV, 2.67 ± 0.08 eV, 2.65 ± 0.08 eV and 2.73 ± 0.08 eV, respectively, based on the measured photoelectron spectra. Through computational geometry optimizations we have identified the lowest-energy structures of these nucleobase–Au2− cluster anions. The structures are further confirmed by comparison of theoretically calculated vertical and adiabatic electron detachment energies with experimental measurements. The results reveal that the Au2− anion remains as an intact unit and interacts with the nucleobases through N–H⋯Au or C–H⋯Au nonconventional hydrogen bonds. The nucleobase–Au2− cluster anions have relatively weak N–H⋯Au hydrogen bonds and strong C–H⋯Au hydrogen bonds compared to those of nucleobase–Au− anions.
Co-reporter:Jin-Yun Yuan, Hong-Guang Xu and Wei-Jun Zheng
Physical Chemistry Chemical Physics 2014 - vol. 16(Issue 11) pp:NaN5439-5439
Publication Date(Web):2013/12/23
DOI:10.1039/C3CP54758B
ConC2− (n = 1–5) cluster anions were investigated using anion photoelectron spectroscopy. The adiabatic detachment energies (ADEs) and the vertical detachment energies (VDEs) of the ConC2− (n = 1–5) cluster anions were determined from their photoelectron spectra. Density functional calculations were performed for the ConC2 (n = 1–5) cluster anions and neutrals. Our studies show that the structures of ConC2− (n = 1–5) can be described as attaching C2 to the top sites, bridge sites, or hollow sites of the Con clusters. The C2 retains an integral structure unit in the ConC2 (n = 1–5) cluster anions and neutrals, rather than being separated by the Con clusters. The C2 unit in the ConC2 (n = 1–5) cluster anions and neutrals has the characteristics of a double-bond.
Co-reporter:Sheng-Jie Lu, Lian-Rui Hu, Xi-Ling Xu, Hong-Guang Xu, Hui Chen and Wei-Jun Zheng
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 30) pp:NaN20329-20329
Publication Date(Web):2016/03/24
DOI:10.1039/C6CP00373G
Gold-doped germanium clusters, AuGen− (n = 2–12), were investigated by using anion photoelectron spectroscopy in combination with ab initio calculations. Their geometric structures were determined by comparison of the theoretical calculations with the experimental results. The results show that the most stable isomers of AuGen− with n = 2–10 are all exohedral structures with the Au atom capping the vertex, edge or face of Gen clusters, while AuGe11− is found to be the critical size of the endohedral structure. Interestingly, AuGe12− has an Ih symmetric icosahedral structure with the Au atom located at the center. The molecular orbital analysis of the AuGe12− cluster suggests that the interactions between the 5d orbitals of the Au atom and the 4s4p hybridized orbitals of the Ge atoms may stabilize the Ih symmetric icosahedral cage and promote the Au atom to be encapsulated in the cage of Ge12. The NICS(0) and NICS(1) values are calculated to be −143.7 ppm and −36.3 ppm, respectively, indicating that the icosahedral AuGe12− cluster is significantly aromatic.