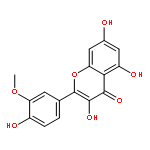
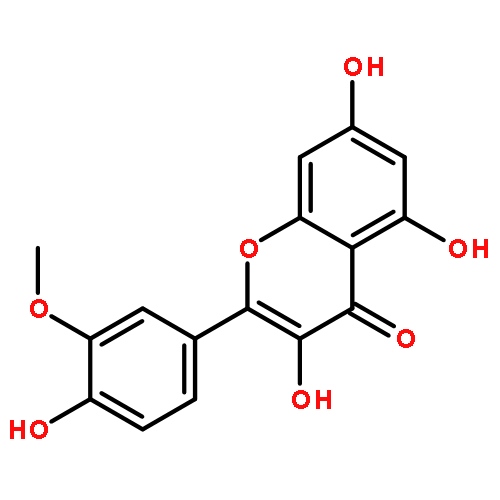
Co-reporter:Meng Chen, Yan Lou, Yaodong Wu, Zhaoke Meng, Liping Li, Lushan Yu, Su Zeng, Hui Zhou, Huidi Jiang
Journal of Pharmaceutical and Biomedical Analysis 2013 Volume 86() pp:161-168
Publication Date(Web):December 2013
DOI:10.1016/j.jpba.2013.08.008
•In this work, we collected and purified seven metabolites of anti-cancer furanodiene.•Their structures were identified by HPLC–MS and 1D and 2D NMR spectra.•Three novel structures were established based on these spectra.•After in vitro incubation, the metabolic pathways of furanodiene were proposed.•The major metabolic reaction was oxidation including hydroxylation and epoxidation.Furanodiene is an active ingredient of Rhizoma Curcumae, a very famous Traditional Chinese Medicine (TCM) widely used for the treatment of cancer. Although the anti-tumor effect of furanodiene has well been established, its metabolic profile in vivo and in vitro is still unclear. In the present study, the metabolites of furanodiene in rats were studied. After oral administration of furanodiene, the rats’ urine, feces and bile were collected and produced seven metabolites by the use of macroporous adsorption resin chromatography, and semi-preparative high performance liquid chromatography. Their structures were identified by mass spectrometry and NMR data including 1H, 13C, and two-dimensional NMR data. All of these metabolites were phase I metabolites, with three new compounds including 2β-hydroxyl-aeruginolactone (2), 14-hydroxyl-aeruginolactone (3), 1β,8β-dihydroxyeudesm-4,7(11)-dien-8α,12-olide (4a), and four known compounds, 1β,10α,4α,5β-diepoxy-8α-hydroxy-glechoman-8α,12-olide (1), 1β,8β-dihydroxyeudesm-4(14),7(11)-dien-8α,12-olide (4b), 1β,8β-dihydroxyeudesm-3,7(11)-dien-8α,12-olide (5) and aeruginolactone (6). Interestingly, the metabolite 6 was found to be a primary metabolite in urine, bile and feces. All metabolites were found to be both in urine and bile but only few metabolites except the metabolite 6 presented in feces after oral dose of furanodiene to rats. Furthermore, the metabolic pathways of furanodiene were proposed using an in vitro assay by incubation of furanodiene and its metabolites in vivo with rat liver S9 or liver microsomes. Clearly, aeruginolactone (6) seemed to be a major precursor for other metabolites.Structures of furanodiene metabolites in rat urine and the proposed metabolic pathways.
Co-reporter:Meijuan Tu, Siyuan Sun, Kai Wang, Xueying Peng, Ruihan Wang, Liping Li, Su Zeng, Hui Zhou, Huidi Jiang
Toxicology (15 September 2013) Volume 311(Issue 3) pp:225-230
Publication Date(Web):15 September 2013
DOI:10.1016/j.tox.2013.06.009
Monocrotaline (MCT) is a kind of toxic retronecine-type pyrrolizidine alkaloids (PAs) from plants of Crotalaria, which can be bio-activated by cytochrome P450 (CYP) enzymes in liver and then induce hepatotoxicity. Since CYPs are localized in the endoplasmic reticulum, the influx of MCT to the liver is the key step for its hepatotoxicity. The objective of the present study was to investigate the role of organic cation transporter 1 (OCT1), a transporter mainly expressed in liver, in the uptake of MCT and in hepatotoxicity induced by MCT. The results revealed that MCT markedly inhibited the uptake of 1-methyl-4-phenylpyridinium (MPP+), an OCT1 substrate, in Madin–Darby canine kidney (MDCK) cells stably expressing human OCT1 (MDCK-hOCT1) with the IC50 of 5.52 ± 0.56 μM. The uptake of MCT was significantly higher in MDCK-hOCT1 cells than in MDCK-mock cells, and MCT uptake in MDCK-hOCT1 cells followed Michaelis–Menten kinetics with the Km and Vmax values of 25.0 ± 6.7 μM and 266 ± 64 pmol/mg protein/min, respectively. Moreover, the OCT1 inhibitors, such as quinidine, d-tetrahydropalmatine (d-THP), obviously inhibited the uptake of MCT in MDCK-hOCT1 cells and isolated rat primary hepatocytes, and attenuated the viability reduction and LDH release of the primary cultured rat hepatocytes caused by MCT. In conclusion, OCT1 mediates the hepatic uptake of MCT and may play an important role in MCT induced-hepatotoxicity.Download full-size image





















![(S)-2-(2-Chlorophenyl)-2-(6,7-dihydrothieno[3,2-c]pyridin-5(4H)-yl)acetic acid](http://img.cochemist.com/ccimg/144500/144457-28-3.png)
![(S)-2-(2-Chlorophenyl)-2-(6,7-dihydrothieno[3,2-c]pyridin-5(4H)-yl)acetic acid](http://img.cochemist.com/ccimg/144500/144457-28-3_b.png)
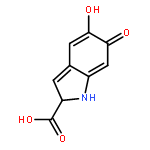

![Phenanthro[1,2-b]furan-10,11-dione, 1,2,8,9-tetrahydro-1,6-dimethyl-, (1R)-](/data/chemimg/449100/126979-84-8.png)
![Phenanthro[1,2-b]furan-10,11-dione, 1,2,8,9-tetrahydro-1,6-dimethyl-, (1R)-](/data/chemimg/449100/126979-84-8_b.png)
![2,4,4-Pyrrolidinetricarboxylicacid, 5-[3-hydroxy-5-(hydroxymethyl)-2-methyl-4-pyridinyl]-](http://img.cochemist.com/ccimg/126800/126706-35-2.png)
![2,4,4-Pyrrolidinetricarboxylicacid, 5-[3-hydroxy-5-(hydroxymethyl)-2-methyl-4-pyridinyl]-](http://img.cochemist.com/ccimg/126800/126706-35-2_b.png)
![4H-1-Benzopyran-4-one,2-[3,4-dihydroxy-2-(3-methyl-2-buten-1-yl)phenyl]-5,7-dihydroxy-3-methoxy-](http://img.cochemist.com/ccimg/107900/107882-43-9.png)
![4H-1-Benzopyran-4-one,2-[3,4-dihydroxy-2-(3-methyl-2-buten-1-yl)phenyl]-5,7-dihydroxy-3-methoxy-](http://img.cochemist.com/ccimg/107900/107882-43-9_b.png)
![Thieno[3,2-c]pyridine-5(4H)-aceticacid, a-(2-chlorophenyl)-6,7-dihydro-,methyl ester](http://img.cochemist.com/ccimg/90100/90055-48-4.png)
![Thieno[3,2-c]pyridine-5(4H)-aceticacid, a-(2-chlorophenyl)-6,7-dihydro-,methyl ester](http://img.cochemist.com/ccimg/90100/90055-48-4_b.png)


![Phenanthro[1,2-b]furan-10,11-dione, 1,2-dihydro-1,6-dimethyl-, (1R)-](/data/chemimg/1469400/87205-99-0.png)
![Phenanthro[1,2-b]furan-10,11-dione, 1,2-dihydro-1,6-dimethyl-, (1R)-](/data/chemimg/1469400/87205-99-0_b.png)







![(5aR)-9t-beta-D-Glucopyranosyloxy-5t-(4-hydroxy-3,5-dimethoxy-phenyl)-(5ar,8at)-5,8,8a,9-tetrahydro-5aH-furo[3',4';6,7]naphtho[2,3-d][1,3]dioxol-6-on](http://img.cochemist.com/ccimg/40600/40505-30-4.png)
![(5aR)-9t-beta-D-Glucopyranosyloxy-5t-(4-hydroxy-3,5-dimethoxy-phenyl)-(5ar,8at)-5,8,8a,9-tetrahydro-5aH-furo[3',4';6,7]naphtho[2,3-d][1,3]dioxol-6-on](http://img.cochemist.com/ccimg/40600/40505-30-4_b.png)





![Phenanthro[1,2-b]furan-6-carboxylicacid, 6,7,8,9,10,11-hexahydro-1,6-dimethyl-10,11-dioxo-, methyl ester, (6S)-](http://img.cochemist.com/ccimg/18900/18887-19-9.png)
![Phenanthro[1,2-b]furan-6-carboxylicacid, 6,7,8,9,10,11-hexahydro-1,6-dimethyl-10,11-dioxo-, methyl ester, (6S)-](http://img.cochemist.com/ccimg/18900/18887-19-9_b.png)
![9-hydroxy-10-methoxy-5,6-dihydro[1,3]dioxolo[4,5-g]isoquino[3,2-a]isoquinolin-7-ium](http://img.cochemist.com/ccimg/17400/17388-19-1.png)
![9-hydroxy-10-methoxy-5,6-dihydro[1,3]dioxolo[4,5-g]isoquino[3,2-a]isoquinolin-7-ium](http://img.cochemist.com/ccimg/17400/17388-19-1_b.png)



![(5aS,8aS,9R)-8-oxo-9-(3,4,5-trimethoxyphenyl)-5,5a,6,8,8a,9-hexahydrofuro[3',4':6,7]naphtho[2,3-d][1,3]dioxol-4-yl hexopyranoside](http://img.cochemist.com/ccimg/11100/11024-59-2.png)
![(5aS,8aS,9R)-8-oxo-9-(3,4,5-trimethoxyphenyl)-5,5a,6,8,8a,9-hexahydrofuro[3',4':6,7]naphtho[2,3-d][1,3]dioxol-4-yl hexopyranoside](http://img.cochemist.com/ccimg/11100/11024-59-2_b.png)


![[1,3]Dioxolo[4',5':4,5]benzo[1,2-c]phenanthridinium,2,3-dimethoxy-12-methyl-](http://img.cochemist.com/ccimg/6900/6872-57-7.png)
![[1,3]Dioxolo[4',5':4,5]benzo[1,2-c]phenanthridinium,2,3-dimethoxy-12-methyl-](http://img.cochemist.com/ccimg/6900/6872-57-7_b.png)
![(1R)-1-(4-hydroxy-3-{[(1R)-6-methoxy-1-(4-methoxybenzyl)-2-methyl-1,2,3,4-tetrahydroisoquinolin-7-yl]oxy}benzyl)-6-methoxy-2-methyl-1,2,3,4-tetrahydroisoquinolin-7-ol](http://img.cochemist.com/ccimg/6900/6817-41-0.png)
![(1R)-1-(4-hydroxy-3-{[(1R)-6-methoxy-1-(4-methoxybenzyl)-2-methyl-1,2,3,4-tetrahydroisoquinolin-7-yl]oxy}benzyl)-6-methoxy-2-methyl-1,2,3,4-tetrahydroisoquinolin-7-ol](http://img.cochemist.com/ccimg/6900/6817-41-0_b.png)




![Furo[3',4':6,7]naphtho[2,3-d]-1,3-dioxol-6(5aH)-one,5,8,8a,9-tetrahydro-10-hydroxy-5-(4-hydroxy-3,5-dimethoxyphenyl)-,(5R,5aR,8aR)-](http://img.cochemist.com/ccimg/600/568-53-6.png)
![Furo[3',4':6,7]naphtho[2,3-d]-1,3-dioxol-6(5aH)-one,5,8,8a,9-tetrahydro-10-hydroxy-5-(4-hydroxy-3,5-dimethoxyphenyl)-,(5R,5aR,8aR)-](http://img.cochemist.com/ccimg/600/568-53-6_b.png)
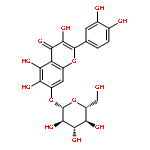
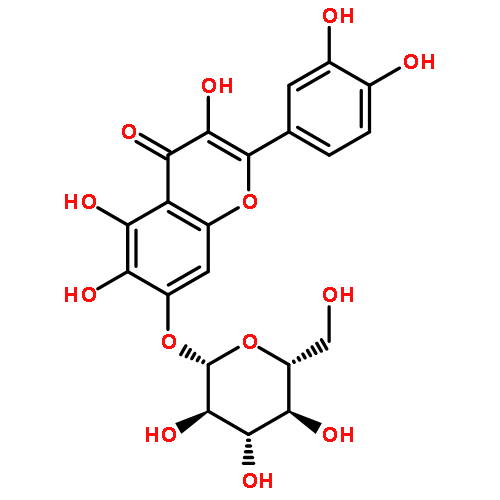


![2-Propen-1-one,3-(3,4-dihydroxyphenyl)-1-[4-(b-D-glucopyranosyloxy)-2,3-dihydroxyphenyl]-, (2E)-](http://img.cochemist.com/ccimg/600/535-96-6.png)
![2-Propen-1-one,3-(3,4-dihydroxyphenyl)-1-[4-(b-D-glucopyranosyloxy)-2,3-dihydroxyphenyl]-, (2E)-](http://img.cochemist.com/ccimg/600/535-96-6_b.png)
![(5R,5aR,8aR)-10-hydroxy-5-(3,4,5-trimethoxyphenyl)-5,8,8a,9-tetrahydrofuro[3',4':6,7]naphtho[2,3-d][1,3]dioxol-6(5aH)-one](http://img.cochemist.com/ccimg/600/518-29-6.png)
![(5R,5aR,8aR)-10-hydroxy-5-(3,4,5-trimethoxyphenyl)-5,8,8a,9-tetrahydrofuro[3',4':6,7]naphtho[2,3-d][1,3]dioxol-6(5aH)-one](http://img.cochemist.com/ccimg/600/518-29-6_b.png)


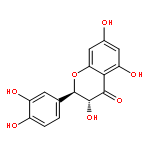
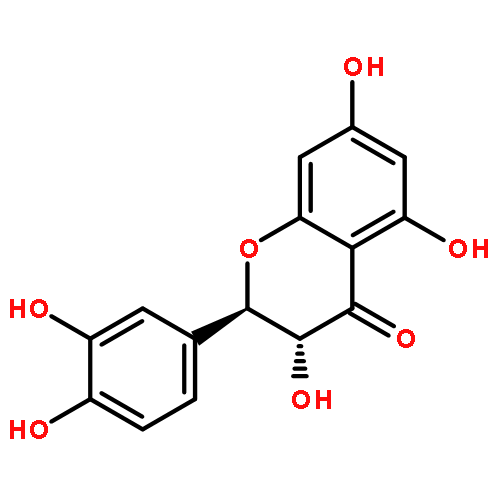
![Furo[3',4':6,7]naphtho[2,3-d]-1,3-dioxole-5,8-dione,5a,6,8a,9-tetrahydro-9-(3,4,5-trimethoxyphenyl)-, (5aR,8aR,9R)-](http://img.cochemist.com/ccimg/500/477-49-6.png)
![Furo[3',4':6,7]naphtho[2,3-d]-1,3-dioxole-5,8-dione,5a,6,8a,9-tetrahydro-9-(3,4,5-trimethoxyphenyl)-, (5aR,8aR,9R)-](http://img.cochemist.com/ccimg/500/477-49-6_b.png)








![5-(2-Chlorobenzyl)-4,5,6,7-tetrahydrothieno[3,2-c]pyridine](http://img.cochemist.com/ccimg/55200/55142-85-3.png)
![5-(2-Chlorobenzyl)-4,5,6,7-tetrahydrothieno[3,2-c]pyridine](http://img.cochemist.com/ccimg/55200/55142-85-3_b.png)


![Dibenzo[a,g]quinolizinium,5,6-dihydro-](http://img.cochemist.com/ccimg/19800/19716-69-9.png)
![Dibenzo[a,g]quinolizinium,5,6-dihydro-](http://img.cochemist.com/ccimg/19800/19716-69-9_b.png)






![2,9,10-trimethoxy-5,6-dihydroisoquinolino[2,1-b]isoquinolin-7-ium-3-ol](http://img.cochemist.com/ccimg/3700/3621-38-3.png)
![2,9,10-trimethoxy-5,6-dihydroisoquinolino[2,1-b]isoquinolin-7-ium-3-ol](http://img.cochemist.com/ccimg/3700/3621-38-3_b.png)
![Bis[1,3]benzodioxolo[5,6-a:4',5'-g]quinolizinium,6,7-dihydro-](http://img.cochemist.com/ccimg/3500/3486-66-6.png)
![Bis[1,3]benzodioxolo[5,6-a:4',5'-g]quinolizinium,6,7-dihydro-](http://img.cochemist.com/ccimg/3500/3486-66-6_b.png)




![(13s,13ar)-2,3,9,10-tetramethoxy-13-methyl-6,8,13,13a-tetrahydro-5h-isoquinolino[2,1-b]isoquinoline](http://img.cochemist.com/ccimg/600/518-69-4.png)
![(13s,13ar)-2,3,9,10-tetramethoxy-13-methyl-6,8,13,13a-tetrahydro-5h-isoquinolino[2,1-b]isoquinoline](http://img.cochemist.com/ccimg/600/518-69-4_b.png)
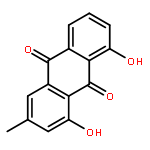
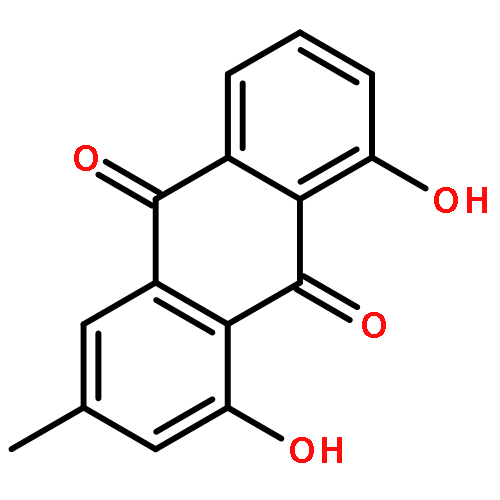
![[1,6]Dioxacyclododecino[2,3,4-gh]pyrrolizine-2,7-dione,3-ethylidene-3,4,5,6,9,11,13,14,14a,14b-decahydro-6-hydroxy-6-(hydroxymethyl)-5-methyl-,(3Z,5R,6S,14aR,14bR)-](http://img.cochemist.com/ccimg/500/480-54-6.png)
![[1,6]Dioxacyclododecino[2,3,4-gh]pyrrolizine-2,7-dione,3-ethylidene-3,4,5,6,9,11,13,14,14a,14b-decahydro-6-hydroxy-6-(hydroxymethyl)-5-methyl-,(3Z,5R,6S,14aR,14bR)-](http://img.cochemist.com/ccimg/500/480-54-6_b.png)