Co-reporter:Laura A. Nguyen;Jimin Wang
PNAS 2017 Volume 114 (Issue 5 ) pp:1021-1026
Publication Date(Web):2017-01-31
DOI:10.1073/pnas.1611191114
Small self-cleaving ribozymes have been discovered in all evolutionary domains of life. They can catalyze site-specific RNA
cleavage, and as a result, they have relevance in gene regulation. Comparative genomic analysis has led to the discovery of
a new class of small self-cleaving ribozymes named Pistol. We report the crystal structure of Pistol at 2.97-Å resolution.
Our results suggest that the Pistol ribozyme self-cleavage mechanism likely uses a guanine base in the active site pocket
to carry out the phosphoester transfer reaction. The guanine G40 is in close proximity to serve as the general base for activating
the nucleophile by deprotonating the 2′-hydroxyl to initiate the reaction (phosphoester transfer). Furthermore, G40 can also
establish hydrogen bonding interactions with the nonbridging oxygen of the scissile phosphate. The proximity of G32 to the
O5′ leaving group suggests that G32 may putatively serve as the general acid. The RNA structure of Pistol also contains A-minor
interactions, which seem to be important to maintain its tertiary structure and compact fold. Our findings expand the repertoire
of ribozyme structures and highlight the conserved evolutionary mechanism used by ribozymes for catalysis.
Co-reporter:Anand T. Vaidya, Ivan B. Lomakin, Newlyn N. Joseph, Sergey E. Dmitriev, Thomas A. Steitz
Journal of Molecular Biology 2017 Volume 429, Issue 18(Volume 429, Issue 18) pp:
Publication Date(Web):1 September 2017
DOI:10.1016/j.jmb.2017.07.015
•eIF2D regulates the initiation of protein synthesis under a stress condition.•The structure of the C-terminal part of eIF2D was determined at 1.4-Å resolution.•eIF2D has an eIF1-like domain, crucial for scanning and the fidelity of AUG recognition.•Extensive atomic interaction between the domains imparts rigidity to the structure.Protein synthesis is a key process in all living organisms. In eukaryotes, initiation factor 2 (eIF2) plays an important role in translation initiation as it selects and delivers the initiator tRNA to the small ribosomal subunit. Under stress conditions, phosphorylation of the α-subunit of eIF2 downregulates cellular protein synthesis. However, translation of certain mRNAs continues via the eIF2D-dependent non-canonical initiation pathway. The molecular mechanism of this process remains elusive. In addition, eIF2D plays a role in translation re-initiation and ribosome recycling. Currently, there has been no structural information of eIF2D. We have now determined the crystal structure of the C-terminal domains of eIF2D at 1.4-Å resolution. One domain has the fold similar to that of eIF1, which is crucial for the scanning and initiation codon selection. The second domain has a known SWIB/MDM2 fold, which was not observed before in other translation initiation factors. Our structure reveals atomic details of inter-domain interactions in the C-terminal part of eIF2D and sheds light on the possible role of these domains in eIF2D during translation.Download high-res image (92KB)Download full-size image
Co-reporter:Ivan B. Lomakin, Elena A. Stolboushkina, Anand T. Vaidya, Chenguang Zhao, ... Thomas A. Steitz
Cell Reports 2017 Volume 20, Issue 3(Volume 20, Issue 3) pp:
Publication Date(Web):18 July 2017
DOI:10.1016/j.celrep.2017.06.025
•The DENR-MCT-1 dimer binds to the interface of the human 40S ribosomal subunit•The C-terminal domain of DENR shares both the fold and the 40S binding site with eIF1•The N-terminal domain of DENR forms binding interface with MCT-1•The binding site of MCT-1 on the 40S subunit overlaps with that of eIF3b RRMThe repertoire of the density-regulated protein (DENR) and the malignant T cell-amplified sequence 1 (MCT-1/MCTS1) oncoprotein was recently expanded to include translational control of a specific set of cancer-related mRNAs. DENR and MCT-1 form the heterodimer, which binds to the ribosome and operates at both translation initiation and reinitiation steps, though by a mechanism that is yet unclear. Here, we determined the crystal structure of the human small ribosomal subunit in complex with DENR-MCT-1. The structure reveals the location of the DENR-MCT-1 dimer bound to the small ribosomal subunit. The binding site of the C-terminal domain of DENR on the ribosome has a striking similarity with those of canonical initiation factor 1 (eIF1), which controls the fidelity of translation initiation and scanning. Our findings elucidate how the DENR-MCT-1 dimer interacts with the ribosome and have functional implications for the mechanism of unconventional translation initiation and reinitiation.Download high-res image (149KB)Download full-size image
Co-reporter:Matthieu G. Gagnon;Jinzhong Lin
PNAS 2016 Volume 113 (Issue 18 ) pp:4994-4999
Publication Date(Web):2016-05-03
DOI:10.1073/pnas.1522932113
During translation, a plethora of protein factors bind to the ribosome and regulate protein synthesis. Many of those factors
are guanosine triphosphatases (GTPases), proteins that catalyze the hydrolysis of guanosine 5′-triphosphate (GTP) to promote
conformational changes. Despite numerous studies, the function of elongation factor 4 (EF-4/LepA), a highly conserved translational
GTPase, has remained elusive. Here, we present the crystal structure at 2.6-Å resolution of the Thermus thermophilus 70S ribosome bound to EF-4 with a nonhydrolyzable GTP analog and A-, P-, and E-site tRNAs. The structure reveals the interactions
of EF-4 with the A-site tRNA, including contacts between the C-terminal domain (CTD) of EF-4 and the acceptor helical stem
of the tRNA. Remarkably, EF-4 induces a distortion of the A-site tRNA, allowing it to interact simultaneously with EF-4 and
the decoding center of the ribosome. The structure provides insights into the tRNA-remodeling function of EF-4 on the ribosome
and suggests that the displacement of the CCA-end of the A-site tRNA away from the peptidyl transferase center (PTC) is functionally
significant.
Co-reporter:Bin Liu;Yuhong Zuo
PNAS 2016 Volume 113 (Issue 15 ) pp:4051-4056
Publication Date(Web):2016-04-12
DOI:10.1073/pnas.1520555113
In bacteria, multiple σ factors compete to associate with the RNA polymerase (RNAP) core enzyme to form a holoenzyme that
is required for promoter recognition. During transcription initiation RNAP remains associated with the upstream promoter DNA
via sequence-specific interactions between the σ factor and the promoter DNA while moving downstream for RNA synthesis. As
RNA polymerase repetitively adds nucleotides to the 3′-end of the RNA, a pyrophosphate ion is generated after each nucleotide
incorporation. It is currently unknown how the release of pyrophosphate affects transcription. Here we report the crystal
structures of E. coli transcription initiation complexes (TICs) containing the stress-responsive σS factor, a de novo synthesized RNA oligonucleotide, and a complete transcription bubble (σS-TIC) at about 3.9-Å resolution. The structures show the 3D topology of the σS factor and how it recognizes the promoter DNA, including likely specific interactions with the template-strand residues of
the −10 element. In addition, σS-TIC structures display a highly stressed pretranslocated initiation complex that traps a pyrophosphate at the active site
that remains closed. The position of the pyrophosphate and the unusual phosphodiester linkage between the two terminal RNA
residues suggest an unfinished nucleotide-addition reaction that is likely at equilibrium between nucleotide addition and
pyrophosphorolysis. Although these σS-TIC crystals are enzymatically active, they are slow in nucleotide addition, as suggested by an NTP soaking experiment. Pyrophosphate
release completes the nucleotide addition reaction and is associated with extensive conformational changes around the secondary
channel but causes neither active site opening nor transcript translocation.
Co-reporter:Bin Liu;Yuhong Zuo
PNAS 2015 Volume 112 (Issue 7 ) pp:2006-2010
Publication Date(Web):2015-02-17
DOI:10.1073/pnas.1417152112
RNA polymerase (RNAP) loses activity during transcription as it stalls at various inactive states due to erratic translocation.
Reactivation of these stalled RNAPs is essential for efficient RNA synthesis. Here we report a 4.7-Å resolution crystal structure
of the Escherichia coli RNAP core enzyme in complex with ATPase RapA that is involved in reactivating stalled RNAPs. The structure reveals that RapA
binds at the RNA exit channel of the RNAP and makes the channel unable to accommodate the formation of an RNA hairpin. The
orientation of RapA on the RNAP core complex suggests that RapA uses its ATPase activity to propel backward translocation
of RNAP along the DNA template in an elongation complex. This structure provides insights into the reactivation of stalled
RNA polymerases and helps support ATP-driven backward translocation as a general mechanism for transcriptional regulation.
Co-reporter:Matthieu G. Gagnon;Jinzhong Lin;David Bulkley
Science 2014 Volume 345(Issue 6197) pp:684-687
Publication Date(Web):08 Aug 2014
DOI:10.1126/science.1253525
Better blood thinner, without bleeding
Blood thinners prevent heart attacks and strokes by making it harder for blood to clot, but these drugs can put patients at risk of dangerous bleeding. Now Moeckle et al. describe an enzyme that can prevent clots without this perilous side effect. They engineered the enzyme apyrase to remove the pro-clotting molecule ADP from the blood quickly. In dogs and mice with heart attacks, apyrase stopped blood cells from aggregating, the first step in forming a clot. At the highest dose, the animals suffered less heart damage and did not bleed excessively. In comparison, clopidogrel, a blood thinner used currently in patients, protected the heart less well and did cause excessive bleeding.
Science, this issue p. 684
Co-reporter:Jimin Wang;Daniel Eiler
PNAS 2014 Volume 111 (Issue 36 ) pp:13028-13033
Publication Date(Web):2014-09-09
DOI:10.1073/pnas.1414571111
Twister is a recently discovered RNA motif that is estimated to have one of the fastest known catalytic rates of any naturally
occurring small self-cleaving ribozyme. We determined the 4.1-Å resolution crystal structure of a twister sequence from an
organism that has not been cultured in isolation, and it shows an ordered scissile phosphate and nucleotide 5′ to the cleavage
site. A second crystal structure of twister from Orzyza sativa determined at 3.1-Å resolution exhibits a disordered scissile phosphate and nucleotide 5′ to the cleavage site. The core
of twister is stabilized by base pairing, a large network of stacking interactions, and two pseudoknots. We observe three
nucleotides that appear to mediate catalysis: a guanosine that we propose deprotonates the 2′-hydroxyl of the nucleotide 5′
to the cleavage site and a conserved adenosine. We suggest the adenosine neutralizes the negative charge on a nonbridging
phosphate oxygen atom at the cleavage site. The active site also positions the labile linkage for in-line nucleophilic attack,
and thus twister appears to simultaneously use three strategies proposed for small self-cleaving ribozymes. The twister crystal
structures (i) show its global structure, (ii) demonstrate the significance of the double pseudoknot fold, (iii) provide a possible hypothesis for enhanced catalysis, and (iv) illuminate the roles of all 10 highly conserved nucleotides of twister that participate in the formation of its small and
stable catalytic pocket.
Co-reporter:Angelita Simonetti;Jinzhong Lin;Daniel Eiler;Bruno P. Klaholz
PNAS 2013 Volume 110 (Issue 39 ) pp:15662-15667
Publication Date(Web):2013-09-24
DOI:10.1073/pnas.1309360110
The initiation of protein synthesis uses initiation factor 2 (IF2) in prokaryotes and a related protein named eukaryotic initiation
factor 5B (eIF5B) in eukaryotes. IF2 is a GTPase that positions the initiator tRNA on the 30S ribosomal initiation complex
and stimulates its assembly to the 50S ribosomal subunit to make the 70S ribosome. The 3.1-Å resolution X-ray crystal structures
of the full-length Thermus thermophilus apo IF2 and its complex with GDP presented here exhibit two different conformations (all of its domains except C2 domain
are visible). Unlike all other translational GTPases, IF2 does not have an effecter domain that stably contacts the switch
II region of the GTPase domain. The domain organization of IF2 is inconsistent with the “articulated lever” mechanism of communication
between the GTPase and initiator tRNA binding domains that has been proposed for eIF5B. Previous cryo-electron microscopy
reconstructions, NMR experiments, and this structure show that IF2 transitions from being flexible in solution to an extended
conformation when interacting with ribosomal complexes.
Co-reporter:Matthieu G. Gagnon;Sai V. Seetharaman;David Bulkley
Science 2012 Vol 335(6074) pp:1370-1372
Publication Date(Web):16 Mar 2012
DOI:10.1126/science.1217443
Co-reporter:Yury S. Polikanov;Gregor M. Blaha
Science 2012 Volume 336(Issue 6083) pp:915-918
Publication Date(Web):18 May 2012
DOI:10.1126/science.1218538
The Hibernating Ribosome
When bacteria enter stationary phase, their ribosomes are inactivated. In Escherichia coli, ribosome modulation factor (RMF) causes dimerization of the 70S ribosome and the dimer is stabilized by, hibernation promotion factor (HPF). Alternately, the stationary phase protein, YfiA, inactivates 70S ribosomes. Polikanov et al. (p. 915) present high-resolution structures of the Thermus thermophilus 70S ribosome bound to each of these three factors. The structures suggest that RMF binding inhibits protein synthesis by preventing initial messenger RNA (mRNA) binding and that HPF and YfiA have overlapping binding sites and would both interfere with binding of mRNA, transfer RNA, and initiation factors.
Co-reporter:David Bulkley;C. Axel Innis;Gregor Blaha
PNAS 2010 Volume 107 (Issue 40 ) pp:17158-17163
Publication Date(Web):2010-10-05
DOI:10.1073/pnas.1008685107
The increasing prevalence of antibiotic-resistant pathogens reinforces the need for structures of antibiotic-ribosome complexes
that are accurate enough to enable the rational design of novel ribosome-targeting therapeutics. Structures of many antibiotics
in complex with both archaeal and eubacterial ribosomes have been determined, yet discrepancies between several of these models
have raised the question of whether these differences arise from species-specific variations or from experimental problems.
Our structure of chloramphenicol in complex with the 70S ribosome from Thermus thermophilus suggests a model for chloramphenicol bound to the large subunit of the bacterial ribosome that is radically different from
the prevailing model. Further, our structures of the macrolide antibiotics erythromycin and azithromycin in complex with a
bacterial ribosome are indistinguishable from those determined of complexes with the 50S subunit of Haloarcula marismortui, but differ significantly from the models that have been published for 50S subunit complexes of the eubacterium Deinococcus radiodurans. Our structure of the antibiotic telithromycin bound to the T. thermophilus ribosome reveals a lactone ring with a conformation similar to that observed in the H. marismortui and D. radiodurans complexes. However, the alkyl-aryl moiety is oriented differently in all three organisms, and the contacts observed with
the T. thermophilus ribosome are consistent with biochemical studies performed on the Escherichia coli ribosome. Thus, our results support a mode of macrolide binding that is largely conserved across species, suggesting that
the quality and interpretation of electron density, rather than species specificity, may be responsible for many of the discrepancies
between the models.
Co-reporter:Thomas A. Steitz;Yong Xiong;Baocheng Pan
Science 2010 Volume 330(Issue 6006) pp:937-940
Publication Date(Web):12 Nov 2010
DOI:10.1126/science.1194985
Co-reporter:C. Axel Innis;Birgit Seidelt;Marco Gartmann;Daniel N. Wilson;Jean-Paul Armache;Elizabeth Villa;Leonardo G. Trabuco;Thomas Becker;Thorsten Mielke;Klaus Schulten;Roland Beckmann
Science 2009 Volume 326(Issue 5958) pp:1412-1415
Publication Date(Web):04 Dec 2009
DOI:10.1126/science.1177662
Co-reporter:Thomas A. Steitz;Gregor Blaha;Robin E. Stanley
Science 2009 Volume 325(Issue 5943) pp:966-970
Publication Date(Web):21 Aug 2009
DOI:10.1126/science.1175800
Co-reporter:Hitesh Sharma;Shaoning Yu;Jilie Kong;Jimin Wang
PNAS 2009 Volume 106 (Issue 39 ) pp:16604-16609
Publication Date(Web):2009-09-29
DOI:10.1073/pnas.0908380106
The binding of cAMP to the Escherichia coli catabolite gene activator protein (CAP) produces a conformational change that enables it to bind specific DNA sequences and
regulate transcription, which it cannot do in the absence of the nucleotide. The crystal structures of the unliganded CAP
containing a D138L mutation and the unliganded WT CAP were determined at 2.3 and 3.6 Å resolution, respectively, and reveal
that the two DNA binding domains have dimerized into one rigid body and their two DNA recognition helices become buried. The
WT structure shows multiple orientations of this rigid body relative to the nucleotide binding domain supporting earlier biochemical
data suggesting that the inactive form exists in an equilibrium among different conformations. Comparison of the structures
of the liganded and unliganded CAP suggests that cAMP stabilizes the active DNA binding conformation of CAP through the interactions
that the N6 of the adenosine makes with the C-helices. These interactions are associated with the reorientation and elongation of the
C-helices that precludes the formation of the inactive structure.
Co-reporter:Kimberly J. Durniak;Scott Bailey
Science 2008 Vol 322(5901) pp:553-557
Publication Date(Web):24 Oct 2008
DOI:10.1126/science.1163433
Abstract
Structural studies of the T7 bacteriophage DNA-dependent RNA polymerase (T7 RNAP) have shown that the conformation of the amino-terminal domain changes substantially between the initiation and elongation phases of transcription, but how this transition is achieved remains unclear. We report crystal structures of T7 RNAP bound to promoter DNA containing either a 7- or an 8-nucleotide (nt) RNA transcript that illuminate intermediate states along the transition pathway. The amino-terminal domain comprises the C-helix subdomain and the promoter binding domain (PBD), which consists of two segments separated by subdomain H. The structures of the intermediate complex reveal that the PBD and the bound promoter rotate by ∼45° upon synthesis of an 8-nt RNA transcript. This allows the promoter contacts to be maintained while the active site is expanded to accommodate a growing heteroduplex. The C-helix subdomain moves modestly toward its elongation conformation, whereas subdomain H remains in its initiation- rather than its elongation-phase location, more than 70 angstroms away.
Co-reporter:Yuhong Zuo, Yeming Wang, Thomas A. Steitz
Molecular Cell (9 May 2013) Volume 50(Issue 3) pp:430-436
Publication Date(Web):9 May 2013
DOI:10.1016/j.molcel.2013.03.020
Guanosine tetraphosphate (ppGpp) is an alarmone that enables bacteria to adapt to their environment. It has been known for years that ppGpp acts directly on RNA polymerase (RNAP) to alter the rate of transcription, but its exact target site is still under debate. Here we report a crystal structure of Escherichia coli RNAP holoenzyme in complex with ppGpp at 4.5 Å resolution. The structure reveals that ppGpp binds at an interface between the shelf and core modules on the outer surface of RNAP, away from the catalytic center and the nucleic acid binding path. Bound ppGpp connects these two pivotal modules that may restrain the opening of the RNAP cleft. A detailed mechanism of action of ppGpp is proposed in which ppGpp prevents the closure of the active center that is induced by the binding of NTP, which could slow down nucleotide addition cycles and destabilize the initial transcription complexes.Highlights► A cocrystal structure of the E. coli RNA polymerase in complex with ppGpp ► No ppGpp binding seen in the vicinity of the RNAP active site ► ppGpp binds at an interface between modules on the outer surface of the RNAP ► An allosteric mechanism is proposed for ppGpp regulation of RNAP activity
Co-reporter:Yuhong Zuo, Thomas A. Steitz
Molecular Cell (7 May 2015) Volume 58(Issue 3) pp:534-540
Publication Date(Web):7 May 2015
DOI:10.1016/j.molcel.2015.03.010
•Crystal structures of transcription initiation complexes with a complete DNA bubble•σ4 domain rotates to accommodate promoter spacer variations•A bridging interaction between the nascent RNA 5′-triphosphate and the σ3.2 loop•Template-strand DNA scrunching presses on the σ3 globular domainDuring transcription initiation, RNA polymerase binds to promoter DNA to form an initiation complex containing a DNA bubble and enters into abortive cycles of RNA synthesis before escaping the promoter to transit into the elongation phase for processive RNA synthesis. Here we present the crystal structures of E. coli transcription initiation complexes containing a complete transcription bubble and de novo synthesized RNA oligonucleotides at about 6-Å resolution. The structures show how RNA polymerase recognizes DNA promoters that contain spacers of different lengths and reveal a bridging interaction between the 5′-triphosphate of the nascent RNA and the σ factor that may function to stabilize the short RNA-DNA hybrids during the early stage of transcription initiation. The conformation of the RNA oligonucleotides and the paths of the DNA strands in the complete initiation complexes provide insights into the mechanism that controls both the abortive and productive RNA synthesis.Download high-res image (153KB)Download full-size image
Co-reporter:Gregor Blaha, Güliz Gürel, Susan J. Schroeder, Peter B. Moore, Thomas A. Steitz
Journal of Molecular Biology (6 June 2008) Volume 379(Issue 3) pp:505-519
Publication Date(Web):6 June 2008
DOI:10.1016/j.jmb.2008.03.075
Eleven mutations that make Haloarcula marismortui resistant to anisomycin, an antibiotic that competes with the amino acid side chains of aminoacyl tRNAs for binding to the A-site cleft of the large ribosomal unit, have been identified in 23S rRNA. The correlation observed between the sensitivity of H. marismortui to anisomycin and the affinity of its large ribosomal subunits for the drug indicates that its response to anisomycin is determined primarily by the binding of the drug to its large ribosomal subunit. The structures of large ribosomal subunits containing resistance mutations show that these mutations can be divided into two classes: (1) those that interfere with specific drug–ribosome interactions and (2) those that stabilize the apo conformation of the A-site cleft of the ribosome relative to its drug-bound conformation. The conformational effects of some mutations of the second kind propagate through the ribosome for considerable distances and are reversed when A-site substrates bind to the ribosome.
Co-reporter:Yury S. Polikanov, Agata L. Starosta, Manuel F. Juette, Roger B. Altman, ... Daniel N. Wilson
Molecular Cell (4 June 2015) Volume 58(Issue 5) pp:832-844
Publication Date(Web):4 June 2015
DOI:10.1016/j.molcel.2015.04.014
•X-ray structures of antibiotics Hygromycin A and A201A on the ribosome•Structural basis for Hygromycin A and A201A resistance mechanisms•tRNA accommodation intermediates with Hygromycin A/A201A observed by FRET•X-ray structures of partially accommodated A-tRNAs with Hygromycin A/A201AThe increase in multi-drug-resistant bacteria is limiting the effectiveness of currently approved antibiotics, leading to a renewed interest in antibiotics with distinct chemical scaffolds. We have solved the structures of the Thermus thermophilus 70S ribosome with A-, P-, and E-site tRNAs bound and in complex with either the aminocyclitol-containing antibiotic hygromycin A (HygA) or the nucleoside antibiotic A201A. Both antibiotics bind at the peptidyl transferase center and sterically occlude the CCA-end of the A-tRNA from entering the A site of the peptidyl transferase center. Single-molecule Förster resonance energy transfer (smFRET) experiments reveal that HygA and A201A specifically interfere with full accommodation of the A-tRNA, leading to the presence of tRNA accommodation intermediates and thereby inhibiting peptide bond formation. Thus, our results provide not only insight into the mechanism of action of HygA and A201A, but also into the fundamental process of tRNA accommodation during protein synthesis.Download high-res image (270KB)Download full-size image
Co-reporter:Yury S. Polikanov, Teresa Szal, Fuyan Jiang, Pulkit Gupta, ... Alexander S. Mankin
Molecular Cell (20 November 2014) Volume 56(Issue 4) pp:541-550
Publication Date(Web):20 November 2014
DOI:10.1016/j.molcel.2014.09.021
•Negamycin inhibits translation by binding to the small ribosomal subunit•The drug interacts simultaneously with rRNA and aminoacyl-tRNA•By stimulating tRNA binding, negamycin inhibits translocation and induces miscodingNegamycin (NEG) is a ribosome-targeting antibiotic that exhibits clinically promising activity. Its binding site and mode of action have remained unknown. We solved the structure of the Thermus thermophilus ribosome bound to mRNA and three tRNAs, in complex with NEG. The drug binds to both small and large ribosomal subunits at nine independent sites. Resistance mutations in the 16S rRNA unequivocally identified the binding site in the vicinity of the conserved helix 34 (h34) in the small subunit as the primary site of antibiotic action in the bacterial and, possibly, eukaryotic ribosome. At this site, NEG contacts 16S rRNA as well as the anticodon loop of the A-site tRNA. Although the NEG site of action overlaps with that of tetracycline (TET), the two antibiotics exhibit different activities: while TET sterically hinders binding of aminoacyl-tRNA to the ribosome, NEG stabilizes its binding, thereby inhibiting translocation and stimulating miscoding.Download high-res image (872KB)Download full-size image
Co-reporter:Yury S. Polikanov, Ilya A. Osterman, Teresa Szal, Vadim N. Tashlitsky, ... Petr V. Sergiev
Molecular Cell (20 November 2014) Volume 56(Issue 4) pp:531-540
Publication Date(Web):20 November 2014
DOI:10.1016/j.molcel.2014.09.020
•The antibiotic amicoumacin A is a potent inhibitor of protein synthesis•Amicoumacin A binds to the ribosome and interacts with 16S rRNA and mRNA•By stabilizing mRNA-ribosome interaction, the antibiotic inhibits translocationWe demonstrate that the antibiotic amicoumacin A (AMI) is a potent inhibitor of protein synthesis. Resistance mutations in helix 24 of the 16S rRNA mapped the AMI binding site to the small ribosomal subunit. The crystal structure of bacterial ribosome in complex with AMI solved at 2.4 Å resolution revealed that the antibiotic makes contacts with universally conserved nucleotides of 16S rRNA in the E site and the mRNA backbone. Simultaneous interactions of AMI with 16S rRNA and mRNA and the in vivo experimental evidence suggest that it may inhibit the progression of the ribosome along mRNA. Consistent with this proposal, binding of AMI interferes with translocation in vitro. The inhibitory action of AMI can be partly compensated by mutations in the translation elongation factor G.Download high-res image (738KB)Download full-size image
Co-reporter:David Bulkley, Francis Johnson, Thomas A. Steitz
Journal of Molecular Biology (2 March 2012) Volume 416(Issue 4) pp:571-578
Publication Date(Web):2 March 2012
DOI:10.1016/j.jmb.2011.12.055
Thermorubin is a small-molecule inhibitor of bacterial protein synthesis, but relatively little is known about the molecular mechanism by which it blocks translation. The structure of the complex between thermorubin and the 70S ribosome from Thermus thermophilus reported here shows that thermorubin interacts with the ribosome in a way that is distinct from any other known class of ribosome inhibitor. Though it is structurally similar to tetracycline, it binds to the ribosome at an entirely different location—the interface between the small and large subunits that is formed by inter-subunit bridge B2a. This region of the ribosome is known to play a role in the initiation of translation, and thus, the binding site we observe is consistent with evidence suggesting that thermorubin inhibits the initiation stage of protein synthesis. The binding of thermorubin induces a rearrangement of two bases on helix 69 of the 23S rRNA, and presumably, this rearrangement blocks the binding of an A-site tRNA, thereby inhibiting peptide bond formation. Due in part to its low solubility in aqueous media, thermorubin has not been used clinically, although it is a potent antibacterial agent with low toxicity (Therapeutic Index > 200). The interactions between thermorubin and the ribosome, as well as its adjacency to the observed binding sites of three other antibiotic classes, may enable the design of novel derivatives that share thermorubin's mode of action but possess improved pharmacodynamic properties.Download high-res image (216KB)Download full-size imageHighlights► Thermorubin inhibits bacterial, but not eukaryotic, protein synthesis. ► Thermorubin binds in the A-site at the interface between the large and small subunits. ► Thermorubin induces rearrangement of the bases of C1914 and A1913 on the large subunit. ► A-site tRNA would clash with the 23S rRNA when thermorubin is bound to the ribosome.
Co-reporter:Bin Liu, Jinzhong Lin, Thomas A. Steitz
Structure (2 April 2013) Volume 21(Issue 4) pp:658-664
Publication Date(Web):2 April 2013
DOI:10.1016/j.str.2013.02.002
The C-terminal domain (CTD) of the τ subunit of the clamp loader (τc) binds to both the DnaB helicase and the DNA polymerase III α subunit (PolIIIα), and determines their relative positions and orientations on the leading and lagging strands. Here, we present a 3.2 Å resolution structure of Thermus aquaticus PolIIIα in complex with τc and a DNA substrate. The structure reveals that the CTD of τc interacts with the CTD of PolIIIα through its C-terminal helix and the adjacent loop. Additionally, in this complex PolIIIα displays an open conformation that includes the reorientations of the oligonucleotide-binding fold and the thumb domain, which may be an indirect result of crystal packing due to the presence of the τc. Nevertheless, the position of the τc on PolIIIα allows us to suggest an approximate model for how the PolIIIα is oriented and positioned on the DnaB helicase.Graphical AbstractDownload high-res image (457KB)Download full-size imageHighlights► Crystal structure of the DNA polymerase IIIα in complex with τc and a DNA substrate ► τc interacts with the C-terminal domain of PolIIIα through its C-terminal helix and the loop that follows it ► PolIIIα in complex with τc and DNA displays an open form ► The structure of this complex suggests an atomic model of the bacterial replisome
Co-reporter:Richard A. Wing, Scott Bailey, Thomas A. Steitz
Journal of Molecular Biology (17 October 2008) Volume 382(Issue 4) pp:859-869
Publication Date(Web):17 October 2008
DOI:10.1016/j.jmb.2008.07.058
The crystal structure of the catalytic α−subunit of the DNA polymerase III (PolIIIα) holoenzyme bound to primer–template DNA and an incoming deoxy-nucleoside 5′-triphosphate has been determined at 4.6-Å resolution. The polymerase interacts with the sugar–phosphate backbone of the DNA across its minor groove, which is made possible by significant movements of the thumb, finger, and β-binding domains relative to their orientations in the unliganded polymerase structure. Additionally, the DNA and incoming nucleotide are bound to the active site of PolIIIα nearly identically as they are in their complex with DNA polymerase β, thereby proving that the eubacterial replicating polymerase, but not the eukaryotic replicating polymerase, is homologous to DNA polymerase β. Finally, superimposing a recent structure of the clamp bound to DNA on this PolIIIα complex with DNA places a loop of the β-binding domain into the appropriate clamp cleft and supports a mechanism of polymerase switching.
.jpg)
![N~5~-(diaminomethylidene)ornithyl-N-[13,22-di(butan-2-yl)-4-({1-carboxy-4-[(diaminomethylidene)amino]butyl}carbamoyl)-10,25-bis{3-[(diaminomethylidene)amino]propyl}-6,9,12,15,18,21,24,27-octaoxo-7,16,19-tri(propan-2-yl)-1,2-dithia-5,8,11,14,17,20,23,26-oc](http://img.cochemist.com/ccimg/116300/116229-36-8.png)
![N~5~-(diaminomethylidene)ornithyl-N-[13,22-di(butan-2-yl)-4-({1-carboxy-4-[(diaminomethylidene)amino]butyl}carbamoyl)-10,25-bis{3-[(diaminomethylidene)amino]propyl}-6,9,12,15,18,21,24,27-octaoxo-7,16,19-tri(propan-2-yl)-1,2-dithia-5,8,11,14,17,20,23,26-oc](http://img.cochemist.com/ccimg/116300/116229-36-8_b.png)
![4-AMINO-2,3-DIHYDROXY-N-[1-(8-HYDROXY-1-OXO-3,4-DIHYDROISOCHROMEN-3-YL)-3-METHYLBUTYL]HEXANEDIAMIDE;HYDROCHLORIDE](http://img.cochemist.com/ccimg/78700/78683-77-9.png)
![4-AMINO-2,3-DIHYDROXY-N-[1-(8-HYDROXY-1-OXO-3,4-DIHYDROISOCHROMEN-3-YL)-3-METHYLBUTYL]HEXANEDIAMIDE;HYDROCHLORIDE](http://img.cochemist.com/ccimg/78700/78683-77-9_b.png)




![L-Lysine, N6-[(3R)-1,5-didehydro-3-methyl-D-prolyl]-](/data/chemimg/106600/448235-52-7.png)
![L-Lysine, N6-[(3R)-1,5-didehydro-3-methyl-D-prolyl]-](/data/chemimg/106600/448235-52-7_b.png)


![Adenosine,3'-deoxy-3'-[[(2E)-3-[4-[[(4Z)-6-O-(6-deoxy-3,4-di-O-methyl-a-D-mannopyranosyl)-5-O-methyl-b-D-threo-hex-4-enofuranosyl]oxy]phenyl]-2-methyl-1-oxo-2-propenyl]amino]-N,N-dimethyl-(9CI)](http://img.cochemist.com/ccimg/37400/37305-78-5.png)
![Adenosine,3'-deoxy-3'-[[(2E)-3-[4-[[(4Z)-6-O-(6-deoxy-3,4-di-O-methyl-a-D-mannopyranosyl)-5-O-methyl-b-D-threo-hex-4-enofuranosyl]oxy]phenyl]-2-methyl-1-oxo-2-propenyl]amino]-N,N-dimethyl-(9CI)](http://img.cochemist.com/ccimg/37400/37305-78-5_b.png)





![Benzoic acid,2-hydroxy-6-methyl-,[(1S,2R,3R,4S,5S)-5-[(3-acetylphenyl)amino]-4-amino-3-[[(dimethylamino)carbonyl]amino]-1,2-dihydroxy-3-[(1S)-1-hydroxyethyl]-2-methylcyclopentyl]methylester](http://img.cochemist.com/ccimg/23700/23668-11-3.png)
![Benzoic acid,2-hydroxy-6-methyl-,[(1S,2R,3R,4S,5S)-5-[(3-acetylphenyl)amino]-4-amino-3-[[(dimethylamino)carbonyl]amino]-1,2-dihydroxy-3-[(1S)-1-hydroxyethyl]-2-methylcyclopentyl]methylester](http://img.cochemist.com/ccimg/23700/23668-11-3_b.png)










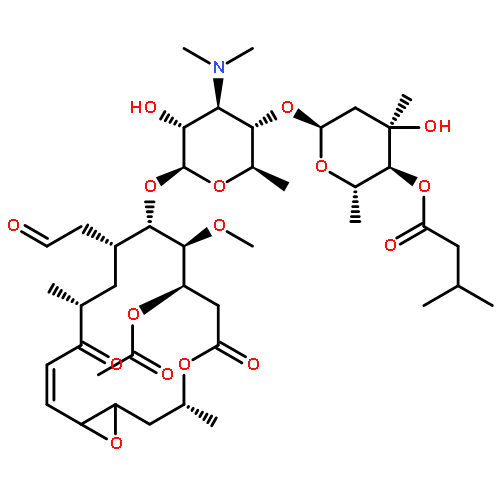











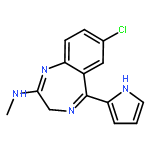
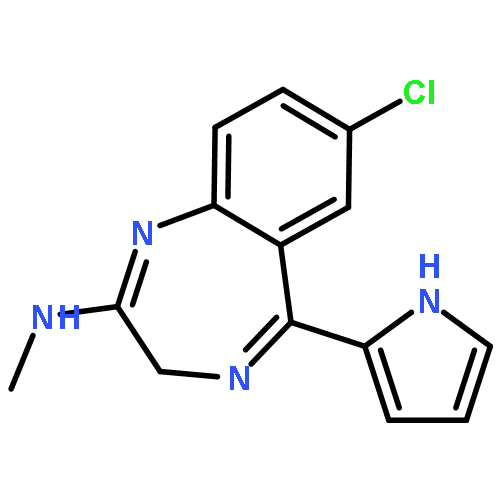
![2-[[(3,6-DIAMINO-5-HYDROXYHEXANOYL)AMINO]-METHYLAMINO]ACETIC ACID](http://img.cochemist.com/ccimg/33500/33404-78-3.png)
![2-[[(3,6-DIAMINO-5-HYDROXYHEXANOYL)AMINO]-METHYLAMINO]ACETIC ACID](http://img.cochemist.com/ccimg/33500/33404-78-3_b.png)
![Glycine,3-amino-N-[(3S)-3,6-diamino-1-oxohexyl]-L-alanyl-L-seryl-L-seryl-(2Z)-3-[(aminocarbonyl)amino]-2,3-didehydroalanyl-2-[(4R,6S)-2-amino-1,4,5,6-tetrahydro-6-hydroxy-4-pyrimidinyl]-,(5®13)-lactam, (2S)-](http://img.cochemist.com/ccimg/33000/32988-50-4.png)
![Glycine,3-amino-N-[(3S)-3,6-diamino-1-oxohexyl]-L-alanyl-L-seryl-L-seryl-(2Z)-3-[(aminocarbonyl)amino]-2,3-didehydroalanyl-2-[(4R,6S)-2-amino-1,4,5,6-tetrahydro-6-hydroxy-4-pyrimidinyl]-,(5®13)-lactam, (2S)-](http://img.cochemist.com/ccimg/33000/32988-50-4_b.png)
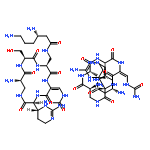

![D-neo-Inositol,5-deoxy-5-[[(2E)-3-[4-[(6-deoxy-b-D-arabino-hexofuranos-5-ulos-1-yl)oxy]-3-hydroxyphenyl]-2-methyl-1-oxo-2-propen-1-yl]amino]-1,2-O-methylene-](http://img.cochemist.com/ccimg/6400/6379-56-2.png)
![D-neo-Inositol,5-deoxy-5-[[(2E)-3-[4-[(6-deoxy-b-D-arabino-hexofuranos-5-ulos-1-yl)oxy]-3-hydroxyphenyl]-2-methyl-1-oxo-2-propen-1-yl]amino]-1,2-O-methylene-](http://img.cochemist.com/ccimg/6400/6379-56-2_b.png)