Co-reporter:Yibin Wang, Heli Fan, Kumudha Balakrishnan, Zechao Lin, Sheng Cao, Wenbing Chen, Yukai Fan, Quibria A. Guthrie, Huabing Sun, Kelly A. Teske, Varsha Gandhi, Leggy A. Arnold, Xiaohua Peng
European Journal of Medicinal Chemistry 2017 Volume 133(Volume 133) pp:
Publication Date(Web):16 June 2017
DOI:10.1016/j.ejmech.2017.03.041
•Several H2O2-activated quinone methides precursors showed potent anticancer activity.•The biological activity depends on the aromatic substituents and the leaving group.•A meta donating group and/or bromo as a leaving group enhance the cellular activity.•The acetoxy group is not a suitable leaving group for H2O2-activated arylboronates.Quinone methide (QM) formation induced by endogenously generated H2O2 is attractive for biological and biomedical applications. To overcome current limitations due to low biological activity of H2O2-activated QM precursors, we are introducing herein several new arylboronates with electron donating substituents at different positions of benzene ring and/or different neutral leaving groups. The reaction rate of the arylboronate esters with H2O2 and subsequent bisquinone methides formation and DNA cross-linking was accelerated with the application of Br as a leaving group instead of acetoxy groups. Additionally, a donating group placed meta to the nascent exo-methylene group of the quinone methide greatly improves H2O2-induced DNA interstrand cross-link formation as well as enhances the cellular activity. Multiple donating groups decrease the stability and DNA cross-linking capability, which lead to low cellular activity. A cell-based screen demonstrated that compounds 2a and 5a with a OMe or OH group dramatically inhibited the growth of various tissue-derived cancer cells while normal cells were less affected. Induction of H2AX phosphorylation by these compounds in CLL lymphocytes provide evidence for a correlation between cell death and DNA damage. The compounds presented herein showed potent anticancer activities and selectivity, which represent a novel scaffold for anticancer drug development.Download high-res image (159KB)Download full-size image
Co-reporter:Yibin Wang, Shuo Liu, Zechao Lin, Yukai Fan, Yinsheng Wang, and Xiaohua Peng
Organic Letters 2016 Volume 18(Issue 11) pp:2544-2547
Publication Date(Web):May 18, 2016
DOI:10.1021/acs.orglett.6b00755
UV irradiation of several aryl boronates efficiently produced bifunctional benzyl cations that selectively form guanine-cytosine cross-links in DNA. Photoinduced homolysis of the C–Br bond took place with the aryl boronate bromides 3a and 4a, generating free radicals that were oxidized to benzyl cations via electron transfer. However, photoirradiation of the quaternary ammonium salts 3b and 4b led to heterolysis of C–N bond, directly producing benzyl cations. The electron-donating group in the aromatic ring greatly enhanced cross-linking efficiency.
Co-reporter:Yibin Wang;Dr. Zechao Lin;Heli Fan ; Xiaohua Peng
Chemistry - A European Journal 2016 Volume 22( Issue 30) pp:10382-10386
Publication Date(Web):
DOI:10.1002/chem.201601504
Abstract
Most photoinduced DNA cross-link formation by a bifunctional aryl derivative is through a bisquinone methide. DNA cross-linking via a bisarylcarbocation remains a less explored area. We designed and synthesized a series of naphthalene boronates that produce DNA interstrand cross-links via a carbocation upon UV irradiation. A free radical was generated from the naphthalene boronates with 350 nm irradiation and further converted to a carbocation by electron transfer. The activation mechanism was determined using the orthogonal traps, 2,2,6,6-tetramethylpiperidin-1-oxyl (TEMPO) and methoxyamine that react with either the free radical or the carbocation but not both. This represents a novel example of photoinduced DNA cross-link formation via carbocations generated from a bisaryl derivative. This work provides information useful for the design of novel photoactivated DNA cross-linking agents.
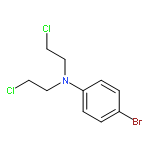
Co-reporter:Yanyan Han, Wenbing Chen, Yunyan Kuang, Huabing Sun, Zhiqiang Wang, and Xiaohua Peng
Chemical Research in Toxicology 2015 Volume 28(Issue 5) pp:919
Publication Date(Web):April 6, 2015
DOI:10.1021/tx500522r
Four novel photoactivated binitroimidazole prodrugs were synthesized. These agents produced DNA interstrand cross-links (ICLs) and direct strand breaks (DSB) upon UV irradiation, whereas no or very few DNA ICLs and DSBs were observed without UV treatment. Although these four molecules (1–4) contain the same binitroimidazole moiety, they bear four different leaving groups, which resulted in their producing different yields of DNA damage. Compound 4, with nitrogen mustard as a leaving group, showed the highest ICL yield. Surprisingly, compounds 1–3, without any alkylating functional group, also induced DNA ICL formation, although they did so with lower yields, which suggested that the binitroimidazole moiety released from UV irradiation of 1–3 is capable of cross-linking DNA. The DNA cross-linked products induced by these compounds were completely destroyed upon 1.0 M piperidine treatment at 90 °C (leading to cleavage at dG sites), which revealed that DNA cross-linking mainly occurred via alkylation of dGs. We proposed a possible mechanism by which alkylating agents were released from these compounds. HRMS and NMR analysis confirmed that free nitrogen mustards were generated by UV irradiation of 4. Suppression of DNA ICL and DSB formation by a radical trap, TEMPO, indicated the involvement of free radicals in the photo reactions of 3 and 4 with DNA. On the basis of these data, we propose that UV irradiation of compounds 1–4 generated a binitroimidazole intermediate that cross-links DNA. The higher ICL yield observed with 4 resulted from the amine effector nitrogen mustard released from UV irradiation.
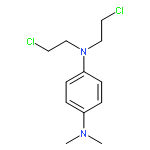
Co-reporter:Wenbing Chen ; Kumudha Balakrishnan ; Yunyan Kuang ; Yanyan Han ; Min Fu ; Varsha Gandhi
Journal of Medicinal Chemistry 2014 Volume 57(Issue 11) pp:4498-4510
Publication Date(Web):May 6, 2014
DOI:10.1021/jm401349g
Reducing host toxicity is one of the main challenges of cancer chemotherapy. Many tumor cells contain high levels of ROS that make them distinctively different from normal cells. We report a series of ROS-activated aromatic nitrogen mustards that selectively kill chronic lymphocytic leukemia (CLL) over normal lymphocytes. These agents showed powerful DNA cross-linking abilities when coupled with H2O2, one of the most common ROS in cancer cells, whereas little DNA cross-linking was detected without H2O2. Consistent with chemistry observation, in vitro cytotoxicity assay demonstrated that these agents induced 40–80% apoptosis in primary leukemic lymphocytes isolated from CLL patients but less than 25% cell death to normal lymphocytes from healthy donors. The IC50 for the most potent compound (2) was ∼5 μM in CLL cells, while the IC50 was not achieved in normal lymphocytes. Collectively, these data provide utility and selectivity of these agents that will inspire further and effective applications.
Co-reporter:Mohammad Mojibul Haque;Huabing Sun;Shuo Liu; Yinsheng Wang; Xiaohua Peng
Angewandte Chemie 2014 Volume 126( Issue 27) pp:7121-7125
Publication Date(Web):
DOI:10.1002/ange.201310609
Abstract
A coumarin-modified pyrimidine nucleoside (1) has been synthesized using a CuI-catalyzed click reaction and incorporated into oligodeoxynucleotides (ODNs). Interstrand cross-links are produced upon irradiation of ODNs containing 1 at 350 nm. Cross-linking occurs through a [2+2] cycloaddition reaction with the opposing thymidine, 2′-deoxycytidine, or 2′-deoxyadenosine. A much higher reactivity was observed with dT than dC or dA. Irradiation of the dT-1 and dC-1 cross-linked products at 254 nm leads to a reversible ring-opening reaction, while such phenomena were not observed with dA-1 adducts. The reversible reaction is ultrafast and complete within 50–90 s. Consistent photoswitching behavior was observed over 6 cycles of irradiation at 350 nm and 254 nm. To the best of our knowledge, this is the first example of photoswitchable interstrand cross-linking formation induced by a modified pyrimidine nucleoside.
Co-reporter:Mohammad Mojibul Haque
Science China Chemistry 2014 Volume 57( Issue 2) pp:215-231
Publication Date(Web):2014 February
DOI:10.1007/s11426-013-5035-1
This review highlights the most recent advances in click chemistry associated with DNA. Cu[I]-catalyzed azides-alkynes Huisgen cycloadditions (CuAAC) and a strain-promoted alkyne-azide cycloaddition (SPAAC) are two popular click reactions that have great impact in DNA science. The simplicity, versatility, orthogonality, and high efficiency of click reaction along with a stable triazole product have been instrumental for the successful application of this reaction in the field of nucleic acid chemistry. CuAAC and SPAAC reactions have been widely used for DNA modification, including DNA labeling, metallization, conjugation, cross-linking, and ligation. Modified oligodeoxynucleotides obtained from click reaction have been extensively applied in the fields of drug discovery, nanotechnology, bio-conjugation, and material sciences, among others. The most recent advances in the synthesis and applications of clickable DNAs are discussed in detail in this article.
Co-reporter:Mohammad Mojibul Haque;Huabing Sun;Shuo Liu; Yinsheng Wang; Xiaohua Peng
Angewandte Chemie International Edition 2014 Volume 53( Issue 27) pp:7001-7005
Publication Date(Web):
DOI:10.1002/anie.201310609
Abstract
A coumarin-modified pyrimidine nucleoside (1) has been synthesized using a CuI-catalyzed click reaction and incorporated into oligodeoxynucleotides (ODNs). Interstrand cross-links are produced upon irradiation of ODNs containing 1 at 350 nm. Cross-linking occurs through a [2+2] cycloaddition reaction with the opposing thymidine, 2′-deoxycytidine, or 2′-deoxyadenosine. A much higher reactivity was observed with dT than dC or dA. Irradiation of the dT-1 and dC-1 cross-linked products at 254 nm leads to a reversible ring-opening reaction, while such phenomena were not observed with dA-1 adducts. The reversible reaction is ultrafast and complete within 50–90 s. Consistent photoswitching behavior was observed over 6 cycles of irradiation at 350 nm and 254 nm. To the best of our knowledge, this is the first example of photoswitchable interstrand cross-linking formation induced by a modified pyrimidine nucleoside.
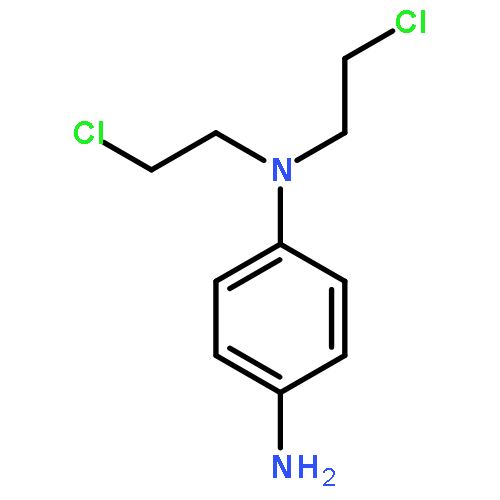
Co-reporter:Dr. Wenbing Chen;Dr. Yanyan Han ; Xiaohua Peng
Chemistry - A European Journal 2014 Volume 20( Issue 24) pp:7410-7418
Publication Date(Web):
DOI:10.1002/chem.201400090
Abstract
Three novel H2O2-activated aromatic nitrogen mustard prodrugs (6–8) are reported. These compounds contain a DNA alkylating agent connected to a H2O2-responsive trigger by different electron-withdrawing linkers so that they are inactive towards DNA but can be triggered by H2O2 to release active species. The activity and selectivity of these compounds towards DNA were investigated by measuring DNA interstrand cross-link (ICL) formation in the presence or absence of H2O2. An electron-withdrawing linker unit, such as a quaternary ammonia salt (6), a carboxyamide (7), and a carbonate group (8), is sufficient to deactivate the aromatic nitrogen mustard resulting in less than 1.5 % cross-linking formation. However, H2O2 can restore the activity of the effectors by converting a withdrawing group to a donating group, therefore increasing the cross-linking efficiency (>20 %). The stability and reaction sites of the ICL products were determined, which revealed that alkylation induced by 7 and 8 not only occurred at the purine sites but also at the pyrimidine site. For the first time, we isolated and characterized the monomer adducts formed between the canonical nucleosides and the aromatic nitrogen mustard (15) which supported that nitrogen mustards reacted with dG, dA, and dC. The activation mechanism was studied by NMR spectroscopic analysis. An in vitro cytotoxicity assay demonstrated that compound 7 with a carboxyamide linker dramatically inhibited the growth of various cancer cells with a GI50 of less than 1 μM, whereas compound 6 with a charged linker did not show any obvious toxicity in all cell lines tested. These data indicated that a neutral carboxyamide linker is preferable for developing nitrogen mustard prodrugs. Our results showed that 7 is a potent anticancer prodrug that can serve as a model compound for further development. We believe these novel aromatic nitrogen mustards will inspire further and effective applications.
Co-reporter:Sheng Cao, Yibin Wang, and Xiaohua Peng
The Journal of Organic Chemistry 2014 Volume 79(Issue 2) pp:501-508
Publication Date(Web):December 30, 2013
DOI:10.1021/jo401901x
We evaluated the effects of the benzylic leaving group and core structure of arylboronates on H2O2-induced formation of bisquinone methides for DNA interstrand cross-linking. The mechanism of DNA cross-linking induced by these arylboronates involves generation of phenol intermediates followed by departure of benzylic leaving groups leading to QMs which directly cross-link DNA via alkylation. The QM formation is the rate-determining step for DNA cross-linking. A better leaving group (Br) and stepwise bisquinone methide formation increased interstrand cross-linking efficiency. These findings provide essential guidelines for designing novel anticancer prodrugs.
Co-reporter:Huabing Sun, Heli Fan, and Xiaohua Peng
The Journal of Organic Chemistry 2014 Volume 79(Issue 23) pp:11359-11369
Publication Date(Web):November 5, 2014
DOI:10.1021/jo5014756
The coumarin analogues have been widely utilized in medicine, biology, biochemistry, and material sciences. Here, we report a detailed study on the reactivity of coumarins toward DNA. A series of coumarin analogues were synthesized and incorporated into oligodeoxynucleotides. A photoinduced [2 + 2] cycloaddition occurs between the coumarin moiety and the thymidine upon 350 nm irradiation forming both syn- and anti-cyclobutane adducts (17 and 18), which are photoreversible by 254/350 nm irradiation in DNA. Quantitative DNA interstrand cross-link (ICL) formation was observed with the coumarin moieties containing a flexible two-carbon or longer chain. DNA cross-linking by coumarins shows a kinetic preference when flanked by an A:T base pair as opposed to a G:C pair. An efficient photoinduced electron transfer between coumarin and dG slows down ICL formation. ICL formation quenches the fluorescence of coumarin, which, for the first time, enables fast, easy, and real-time monitoring of DNA cross-linking and photoreversibility via fluorescence spectroscopy. It can be used to detect the transversion mutation between pyrimidines and purines. Overall, this work provides new insights into the biochemical properties and possible toxicity of coumarins. A quantitative, fluorescence-detectable, and photoswitchable DNA cross-linking reaction of the coumarin moieties can potentially serve as mechanistic probes and tools for bioresearch without disrupting native biological environment.
Co-reporter:Huabing Sun and Xiaohua Peng
Bioconjugate Chemistry 2013 Volume 24(Issue 7) pp:1226
Publication Date(Web):June 11, 2013
DOI:10.1021/bc4001678
A novel nonfluorescent alkyne-modified coumarin phosphoramidite was synthesized and successfully incorporated into oligonucleotides, which were then used in highly efficient DNA interstrand cross-linking and ligation reactions via “click” chemistry. The template-directed fluorogenic ligation “click” chemistry reaction was used for single nucleotide polymorphism analysis, where the target DNA catalyzes the ligation of two nonfluorescent probes to generate a fluorescent product. The upstream oligonucleotide probe is a nonfluorescent alkyne-modified coumarin and the downstream probe is an azide-modified oligonucleotide. When bound to a fully complementary template, the oligonucleotides ligated to produce a fluorescent product with a fluorophore at the ligation point. Wild-type and mutant p53 alleles were used to demonstrate that template-directed fluorogenic oligonucleotide ligation is sequence-specific and is capable of single nucleotide discrimination under mild conditions, even without the removal of unreacted probes.
Co-reporter:Dr. Sheng Cao;Robin Christiansen ; Xiaohua Peng
Chemistry - A European Journal 2013 Volume 19( Issue 27) pp:9050-9058
Publication Date(Web):
DOI:10.1002/chem.201300539
Abstract
A series of arylboronic esters containing different aromatic substituents and various benzylic leaving groups (Br or N+Me3Br−) have been synthesized. The substituent effects on their reactivity with H2O2 and formation of quinone methide (QM) have been investigated. NMR spectroscopy and ethyl vinyl ether (EVE) trapping experiments were used to determine the reaction mechanism and QM formation, respectively. QMs were not generated during oxidative cleavage of the boronic esters but by subsequent transformation of the phenol products under physiological conditions. The oxidative deboronation is facilitated by electron-withdrawing substituents, such as aromatic F, NO2, or benzylic N+Me3Br−, whereas electron-donating substituents or a better leaving group favor QM generation. Compounds containing an aromatic CH3 or OMe group, or a good leaving group (Br), efficiently generate QMs under physiological conditions. Finally, a quantitative relationship between the structure and activity has been established for the arylboronic esters by using a Hammett plot. The reactivity of the arylboronic acids/esters and the inhibition or facilitation of QM formation can now be predictably adjusted. This adjustment is important as some applications may benefit and others may be limited by QM generation.
Co-reporter:Dr. Sheng Cao;Yibin Wang ; Xiaohua Peng
Chemistry - A European Journal 2012 Volume 18( Issue 13) pp:3850-3854
Publication Date(Web):
DOI:10.1002/chem.201200075
Co-reporter:Dr. Yunyan Kuang;Dr. Huabing Sun;Dr. J. Craig Blain; Xiaohua Peng
Chemistry - A European Journal 2012 Volume 18( Issue 40) pp:12609-12613
Publication Date(Web):
DOI:10.1002/chem.201201960
Co-reporter:Yunyan Kuang ; Kumudha Balakrishnan ; Varsha Gandhi
Journal of the American Chemical Society 2011 Volume 133(Issue 48) pp:19278-19281
Publication Date(Web):October 28, 2011
DOI:10.1021/ja2073824
The major concern for anticancer chemotherapeutic agents is the host toxicity. The development of anticancer prodrugs targeting the unique biochemical alterations in cancer cells is an attractive approach to achieve therapeutic activity and selectivity. We designed and synthesized a new type of nitrogen mustard prodrug that can be activated by high level of reactive oxygen species (ROS) found in cancer cells to release the active chemotherapy agent. The activation mechanism was determined by NMR analysis. The activity and selectivity of these prodrugs toward ROS was determined by measuring DNA interstrand cross-links and/or DNA alkylations. These compounds showed 60–90% inhibition toward various cancer cells, while normal lymphocytes were not affected. To the best of our knowledge, this is the first example of H2O2-activated anticancer prodrugs.
Co-reporter:Xiaohua Peng;Hong Li;Michael Seidman
European Journal of Organic Chemistry 2010 Volume 2010( Issue 22) pp:4194-4197
Publication Date(Web):
DOI:10.1002/ejoc.201000615
Abstract
A highly efficient chemical ligation was developed for quantitative conjugation of PNA with DNA (PNA or peptide) by using the copper-catalyzed azide–alkyne cycloaddition reaction. Whereas PNAs with an alkyne at the C-terminus and an azide at the N-terminus have been used, an efficient click–click reaction occurs. The PNA click ligation is sequence specific and capable of single nucleotide discrimination.

![N,N-bis(2-chloroethyl)-N-methyl-N-[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]methanaminium bromide](http://img.cochemist.com/ccimg/1347000/1346936-27-3.png)
![N,N-bis(2-chloroethyl)-N-methyl-N-[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]methanaminium bromide](http://img.cochemist.com/ccimg/1347000/1346936-27-3_b.png)

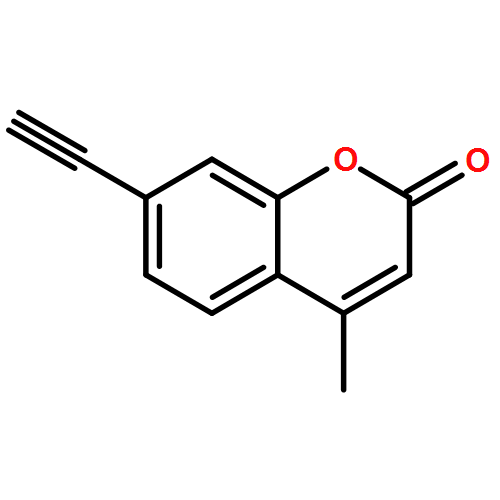
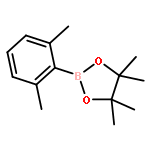
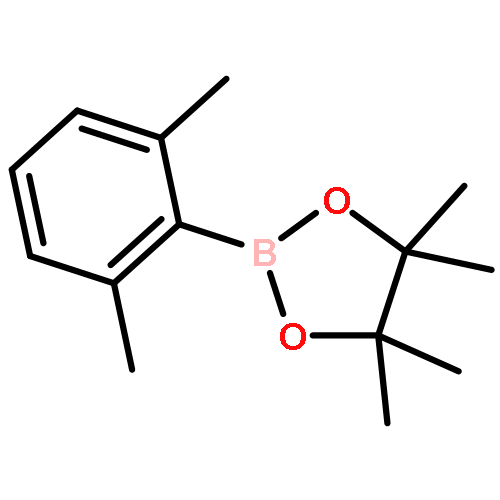










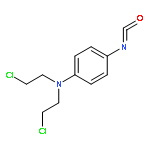






![5-(azidomethyl)-1-[4-hydroxy-5-(hydroxymethyl)oxolan-2-yl]pyrimidine-2,4-dione](http://img.cochemist.com/ccimg/59100/59090-48-1.png)
![5-(azidomethyl)-1-[4-hydroxy-5-(hydroxymethyl)oxolan-2-yl]pyrimidine-2,4-dione](http://img.cochemist.com/ccimg/59100/59090-48-1_b.png)




![2,2'-[(4-BROMOPHENYL)IMINO]DIETHANOL](/data/chemimg/416700/13165-33-8.png)
![2,2'-[(4-BROMOPHENYL)IMINO]DIETHANOL](/data/chemimg/416700/13165-33-8_b.png)




![Phenol,4-[bis(2-chloroethyl)amino]-](http://img.cochemist.com/ccimg/1300/1204-69-9.png)
![Phenol,4-[bis(2-chloroethyl)amino]-](http://img.cochemist.com/ccimg/1300/1204-69-9_b.png)