Co-reporter:Amritraj Patra; Surajit Banerjee; Tracy L. Johnson Salyard; Chanchal K. Malik; Plamen P. Christov; Carmelo J. Rizzo; Michael P. Stone;Martin Egli
Journal of the American Chemical Society 2015 Volume 137(Issue 22) pp:7011-7014
Publication Date(Web):May 19, 2015
DOI:10.1021/jacs.5b02701
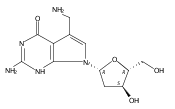
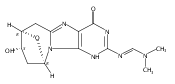
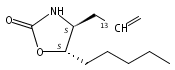
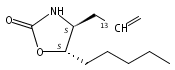
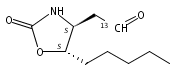
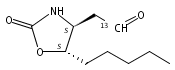
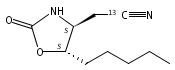
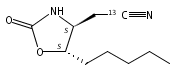
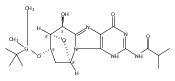
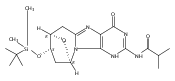
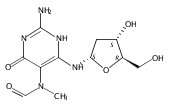
N6-(2-Deoxy-d-erythro-pentofuranosyl)-2,6-diamino-3,4-dihydro-4-oxo-5-N-methylformamidopyrimidine (MeFapy-dG) arises from N7-methylation of deoxyguanosine followed by imidazole ring opening. The lesion has been reported to persist in animal tissues. Previous in vitro replication bypass investigations of the MeFapy-dG adduct revealed predominant insertion of C opposite the lesion, dependent on the identity of the DNA polymerase (Pol) and the local sequence context. Here we report crystal structures of ternary Pol·DNA·dNTP complexes between MeFapy-dG-adducted DNA template:primer duplexes and the Y-family polymerases human Pol η and P2 Pol IV (Dpo4) from Sulfolobus solfataricus. The structures of the hPol η and Dpo4 complexes at the insertion and extension stages, respectively, are representative of error-free replication, with MeFapy-dG in the anti conformation and forming Watson–Crick pairs with dCTP or dC.
Co-reporter:Liang Li, Kyle L. Brown, Ruidan Ma, and Michael P. Stone
Chemical Research in Toxicology 2015 Volume 28(Issue 2) pp:225
Publication Date(Web):January 14, 2015
DOI:10.1021/tx5003832
Aflatoxin B1 (AFB1), a mycotoxin produced by Aspergillus flavus, is oxidized by cytochrome P450 enzymes to aflatoxin B1-8,9-epoxide, which alkylates DNA at N7-dG. Under basic conditions, this N7-dG adduct rearranges to yield the trans-8,9-dihydro-8-(2,6-diamino-4-oxo-3,4-dihydropyrimid-5-yl-formamido)-9-hydroxy aflatoxin B1 (AFB1–FAPY) adduct. The AFB1–FAPY adduct exhibits geometrical isomerism involving the formamide moiety. NMR analyses of duplex oligodeoxynucleotides containing the 5′-XA-3′, 5′-XC-3′, 5′-XT-3′, and 5′-XY-3′ sequences (X = AFB1–FAPY; Y = 7-deaza-dG) demonstrate that the equilibrium between E and Z isomers is controlled by major groove hydrogen bonding interactions. Structural analysis of the adduct in the 5′-XA-3′ sequence indicates the preference of the E isomer of the formamide group, attributed to formation of a hydrogen bond between the formyl oxygen and the N6 exocyclic amino group of the 3′-neighbor adenine. While the 5′-XA-3′ sequence exhibits the E isomer, the 5′-XC-3′ sequence exhibits a 7:3 E:Z ratio at equilibrium at 283 K. The E isomer is favored by a hydrogen bond between the formyl oxygen and the N4-dC exocyclic amino group of the 3′-neighbor cytosine. The 5′-XT-3′ and 5′-XY-3′ sequences cannot form such a hydrogen bond between the formyl oxygen and the 3′-neighbor T or Y, respectively, and in these sequence contexts the Z isomer is favored. Additional equilibria between α and β anomers and the potential to exhibit atropisomers about the C5–N5 bond do not depend upon sequence. In each of the four DNA sequences, the AFB1–FAPY adduct maintains the β deoxyribose configuration. Each of these four sequences feature the atropisomer of the AFB1 moiety that is intercalated above the 5′-face of the damaged guanine. This enforces the Ra axial conformation for the C5–N5 bond.
Co-reporter:Kallie M. Stavros, Edward K. Hawkins, Carmelo J. Rizzo, and Michael P. Stone
Chemical Research in Toxicology 2015 Volume 28(Issue 7) pp:1455
Publication Date(Web):June 17, 2015
DOI:10.1021/acs.chemrestox.5b00140
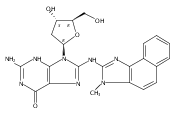
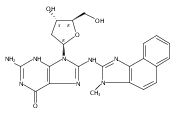
The conformation of an N2-dG adduct arising from the heterocyclic amine 2-amino-3-methylimidazo[4,5-f]quinoline (IQ), a potent food mutagen, was determined in 5′-d(C1T2C3X4G5C6G7C8C9A10T11C12)-3′:5′-d(G13A14T15G16G17C18G19C20C21G22A23G24)-3′; X = N2-dG-IQ, in which the modified nucleotide X4 corresponds to G1 in the 5′-d(G1G2CG3CC)-3′ NarI restriction endonuclease site. Circular dichroism (CD) revealed blue shifts relative to the unmodified duplex, consistent with adduct-induced twisting, and a hypochromic effect for the IQ absorbance in the near UV region. NMR revealed that the N2-dG-IQ adduct adopted a base-displaced intercalated conformation in which the modified guanine remained in the anti conformation about the glycosidic bond, the IQ moiety intercalated into the duplex, and the complementary base C21 was displaced into the major groove. The processing of the N2-dG-IQ lesion by hpol η is sequence-dependent; when placed at the reiterated G3 position, but not at the G1 position, this lesion exhibits a propensity for frameshift replication [Choi, J. Y., et al. (2006) J. Biol. Chem., 281, 25297–25306]. The structure of the N2-dG-IQ adduct at the nonreiterated G1 position was compared to that of the same adduct placed at the G3 position [Stavros, K. M., et al. (2014) Nucleic Acids Res., 42, 3450–3463]. CD indicted minimal spectral differences between the G1 vs G3 N2-dG-IQ adducts. NMR indicated that the N2-dG-IQ adduct exhibited similar base-displaced intercalated conformations at both the G1 and G3 positions. This result differed as compared to the corresponding C8-dG-IQ adducts placed at the same positions. The C8-dG-IQ adduct adopted a minor groove conformation when placed at position G1 but a base-displaced intercalated conformation when placed at position G3 in the NarI sequence. The present studies suggest that differences in lesion bypass by hpol η may be mediated by differences in the 3′-flanking sequences, perhaps modulating the ability to accommodate transient strand slippage intermediates.
Co-reporter:Dustin A. Politica, Chanchal K. Malik, Ashis K. Basu, and Michael P. Stone
Chemical Research in Toxicology 2015 Volume 28(Issue 12) pp:2253
Publication Date(Web):December 7, 2015
DOI:10.1021/acs.chemrestox.5b00277
3-Nitrobenzanthrone (3-NBA), an environmental mutagen found in diesel exhaust and a suspected carcinogen, undergoes metabolic reduction followed by reaction with DNA to form aminobenzanthrone (ABA) adducts, with the major alkylation product being N-(2′-deoxyguanosin-8-yl)-3-aminobenzanthrone (C8-dG-ABA). Site-specific synthesis of the C8-dG-ABA adduct in the oligodeoxynucleotide 5′-d(GTGCXTGTTTGT)-3′:5′-d(ACAAACACGCAC)-3′; X = C8-dG-ABA adduct, including codons 272–275 of the p53 gene, has allowed for investigation into the structural and thermodynamic properties of this adduct. The conformation of the C8-dG-ABA adduct was determined using NMR spectroscopy and was refined using molecular dynamics (MD) calculations restrained by experimentally determined interproton distance restraints obtained from NOE experiments. The refined structure revealed that the C8-dG-ABA adduct formed a base-displaced intercalated conformation. The adducted guanine was shifted into the syn conformation about the glycosidic bond. The 5′- and 3′-neighboring base pairs remained intact. While this facilitated π-stacking interactions between the ABA moiety and neighboring bases, the thermal melting temperature (Tm) of the adduct-containing duplex showed a decrease of 11 °C as compared to the corresponding unmodified oligodeoxynucleotide duplex. Overall, in this sequence, the base-displaced intercalated conformation of the C8-dG-ABA lesion bears similarity to structures of other arylamine C8-dG adducts. However, in this sequence, the base-displaced intercalated conformation for the C8-dG-ABA adduct differs from the conformation of the N2-dG-ABA adduct reported by de los Santos and co-workers, in which it is oriented in the minor groove toward the 5′ end of the duplex, with the modified guanine remaining in the anti conformation about the glyosidic torsion angle, and the complementary base remaining within the duplex. The results are discussed in relationship to differences between the C8-dG-ABA and N2-dG-ABA adducts with respect to susceptibility to nucleotide excision repair (NER).
Co-reporter:Marta W. Szulik, Pradeep S. Pallan, Boguslaw Nocek, Markus Voehler, Surajit Banerjee, Sonja Brooks, Andrzej Joachimiak, Martin Egli, Brandt F. Eichman, and Michael P. Stone
Biochemistry 2015 Volume 54(Issue 5) pp:1294-1305
Publication Date(Web):January 29, 2015
DOI:10.1021/bi501534x
5-Hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC), and 5-carboxylcytosine (5caC) form during active demethylation of 5-methylcytosine (5mC) and are implicated in epigenetic regulation of the genome. They are differentially processed by thymine DNA glycosylase (TDG), an enzyme involved in active demethylation of 5mC. Three modified Dickerson–Drew dodecamer (DDD) sequences, amenable to crystallographic and spectroscopic analyses and containing the 5′-CG-3′ sequence associated with genomic cytosine methylation, containing 5hmC, 5fC, or 5caC placed site-specifically into the 5′-T8X9G10-3′ sequence of the DDD, were compared. The presence of 5caC at the X9 base increased the stability of the DDD, whereas 5hmC or 5fC did not. Both 5hmC and 5fC increased imino proton exchange rates and calculated rate constants for base pair opening at the neighboring base pair A5:T8, whereas 5caC did not. At the oxidized base pair G4:X9, 5fC exhibited an increase in the imino proton exchange rate and the calculated kop. In all cases, minimal effects to imino proton exchange rates occurred at the neighboring base pair C3:G10. No evidence was observed for imino tautomerization, accompanied by wobble base pairing, for 5hmC, 5fC, or 5caC when positioned at base pair G4:X9; each favored Watson–Crick base pairing. However, both 5fC and 5caC exhibited intranucleobase hydrogen bonding between their formyl or carboxyl oxygens, respectively, and the adjacent cytosine N4 exocyclic amines. The lesion-specific differences observed in the DDD may be implicated in recognition of 5hmC, 5fC, or 5caC in DNA by TDG. However, they do not correlate with differential excision of 5hmC, 5fC, or 5caC by TDG, which may be mediated by differences in transition states of the enzyme-bound complexes.
Co-reporter:Ewa A. Kowal, Uthpala Seneviratne, Susith Wickramaratne, Kathleen E. Doherty, Xiangkun Cao, Natalia Tretyakova, and Michael P. Stone
Chemical Research in Toxicology 2014 Volume 27(Issue 5) pp:805
Publication Date(Web):April 17, 2014
DOI:10.1021/tx400472p
1,3-Butadiene (BD) is an industrial and environmental chemical present in urban air and cigarette smoke, and is classified as a human carcinogen. It is oxidized by cytochrome P450 to form 1,2,3,4-diepoxybutane (DEB); DEB bis-alkylates the N6 position of adenine in DNA. Two enantiomers of bis-N6-dA adducts of DEB have been identified: R,R-N6,N6-(2,3-dihydroxybutan-1,4-diyl)-2′-deoxyadenosine (R,R-DHB-dA), and S,S-N6,N6-(2,3-dihydroxybutan-1,4-diyl)-2′-deoxyadenosine (S,S-DHB-dA) [Seneviratne, U., Antsypovich, S., Dorr, D. Q., Dissanayake, T., Kotapati, S., and Tretyakova, N. (2010) Chem. Res. Toxicol.23, 1556−1567]. Herein, the R,R-DHB-dA and S,S-DHB-dA adducts have been incorporated into the 5′-d(C1G2G3A4C5X6A7G8A9A10G11)-3′:5′-d(C12T13T14C15T16T17G18T19C20C21G22)-3′ duplex [X6 = R,R-DHB-dA (R6) or S,S-DHB-dA (S6)]. The structures of the duplexes were determined by molecular dynamics calculations, which were restrained by experimental distances obtained from NMR data. Both the R,R- and S,S-DHB-dA adducts are positioned in the major groove of DNA. In both instances, the bulky 3,4-dihydroxypyrrolidine rings are accommodated by an out-of-plane rotation about the C6-N6 bond of the bis-alkylated adenine. In both instances, the directionality of the dihydroxypyrrolidine ring is evidenced by the pattern of NOEs between the 3,4-dihydroxypyrrolidine protons and DNA. Also in both instances, the anti conformation of the glycosyl bond is maintained, which combined with the out-of-plane rotation about the C6-N6 bond, allows the complementary thymine, T17, to remain stacked within the duplex, and form one hydrogen bond with the modified base, between the imine nitrogen of the modified base and the T17 N3H imino proton. The loss of the second Watson–Crick hydrogen bonding interaction at the lesion sites correlates with the lower thermal stabilities of the R,R- and S,S-DHB-dA duplexes, as compared to the corresponding unmodified duplex. The reduced base stacking at the adduct sites may also contribute to the thermal instability.
Co-reporter:Ewa A. Kowal, Susith Wickramaratne, Srikanth Kotapati, Michael Turo, Natalia Tretyakova, and Michael P. Stone
Chemical Research in Toxicology 2014 Volume 27(Issue 10) pp:1675
Publication Date(Web):September 19, 2014
DOI:10.1021/tx500159w
1,3-Butadiene (BD) is an environmental and occupational toxicant classified as a human carcinogen. It is oxidized by cytochrome P450 monooxygenases to 1,2-epoxy-3-butene (EB), which alkylates DNA. BD exposures lead to large numbers of mutations at A:T base pairs even though alkylation of guanines is more prevalent, suggesting that one or more adenine adducts of BD play a role in BD-mediated genotoxicity. However, the etiology of BD-mediated genotoxicity at adenine remains poorly understood. EB alkylates the N6 exocyclic nitrogen of adenine to form N6-(hydroxy-3-buten-1-yl)-2′-dA ((2S)-N6-HB-dA) adducts (Tretyakova, N., Lin, Y., Sangaiah, R., Upton, P. B., and Swenberg, J. A. (1997) Carcinogenesis18, 137−147). The structure of the (2S)-N6-HB-dA adduct has been determined in the 5′-d(C1G2G3A4C5Y6A7G8A9A10G11)-3′:5′-d(C12T13T14C15T16T17G18T19 C20C21G22)-3′ duplex [Y = (2S)-N6-HB-dA] containing codon 61 (underlined) of the human N-ras protooncogene, from NMR spectroscopy. The (2S)-N6-HB-dA adduct was positioned in the major groove, such that the butadiene moiety was oriented in the 3′ direction. At the Cα carbon, the methylene protons of the modified nucleobase Y6 faced the 5′ direction, which placed the Cβ carbon in the 3′ direction. The Cβ hydroxyl group faced toward the solvent, as did carbons Cγ and Cδ. The Cβ hydroxyl group did not form hydrogen bonds with either T16 O4 or T17 O4. The (2S)-N6-HB-dA nucleoside maintained the anti conformation about the glycosyl bond, and the modified base retained Watson–Crick base pairing with the complementary base (T17). The adduct perturbed stacking interactions at base pairs C5:G18, Y6:T17, and A7:T16 such that the Y6 base did not stack with its 5′ neighbor C5, but it did with its 3′ neighbor A7. The complementary thymine T17 stacked well with both 5′ and 3′ neighbors T16 and G18. The presence of the (2S)-N6-HB-dA resulted in a 5 °C reduction in the Tm of the duplex, which is attributed to less favorable stacking interactions and adduct accommodation in the major groove.
Co-reporter:Ganesh Shanmugam, Irina G. Minko, Surajit Banerjee, Plamen P. Christov, Ivan D. Kozekov, Carmelo J. Rizzo, R. Stephen Lloyd, Martin Egli, and Michael P. Stone
Chemical Research in Toxicology 2013 Volume 26(Issue 9) pp:1348
Publication Date(Web):August 16, 2013
DOI:10.1021/tx400200b
Acrolein, a mutagenic aldehyde, reacts with deoxyguanosine (dG) to form 3-(2′-deoxy-β-d-erythro-pentofuranosyl)-5,6,7,8-tetrahydro-8-hydroxypyrimido[1,2-a] purin-10(3H)-one (γ-OH-PdG). When placed opposite deoxycytosine (dC) in DNA, γ-OH-PdG undergoes ring-opening to the N2-(3-oxopropyl)-dG. Ring-opening of the adduct has been hypothesized to facilitate nonmutagenic bypass, particularly by DNA polymerases of the Y family. This study examined the bypass of γ-OH-PdG by Sulfolobus solfataricus Dpo4, the prototypic Y-family DNA polymerase, using templates that contained the adduct in either the 5′-CXG-3′ or the 5′-TXG-3′ sequence context. Although γ-OH-PdG partially blocked Dpo4-catalyzed DNA synthesis, full primer extension was observed, and the majority of bypass products were error-free. Conversion of the adduct into an irreversibly ring-opened derivative prior to reaction facilitated bypass and further improved the fidelity. Structures of ternary Dpo4·DNA·dNTP complexes were determined with primers that either were positioned immediately upstream of the lesion (preinsertion complexes) or had a 3′-terminal dC opposite the lesion (postinsertion complexes); the incoming nucleotides, either dGTP or dATP, were complementary to the template 5′-neighbor nucleotide. In both postinsertion complexes, the adduct existed as ring-opened species, and the resulting base-pair featured Watson–Crick hydrogen bonding. The incoming nucleotide paired with the 5′-neighbor template, while the primer 3′-hydroxyl was positioned to facilitate extension. In contrast, γ-OH-PdG was in the ring-closed form in both preinsertion complexes, and the overall structure did not favor catalysis. These data provide insights into γ-OH-PdG chemistry during replication bypass by the Dpo4 DNA polymerase and may explain why γ-OH-PdG-induced mutations due to primer–template misalignment are uncommon.
Co-reporter:Marta W. Szulik, Markus W. Voehler, Manjori Ganguly, Barry Gold, and Michael P. Stone
Biochemistry 2013 Volume 52(Issue 43) pp:
Publication Date(Web):October 16, 2013
DOI:10.1021/bi400695r
A cationic 7-aminomethyl-7-deaza-2′-deoxyguanosine (7amG) was incorporated site-specifically into the self-complementary duplex d(G1A2G3A4X5C6G7C8T9C10T11C12)2 (X = 7amG). This construct placed two positively charged amines adjacent to the major groove edges of two symmetry-related guanines, providing a model for probing how cation binding in the major groove modulates the structure and stability of DNA. Molecular dynamics calculations restrained by nuclear magnetic resonance (NMR) data revealed that the tethered cationic amines were in plane with the modified base pairs. The tethered amines did not form salt bridges to the phosphodiester backbone. There was also no indication of the amines being capable of hydrogen bonding to flanking DNA bases. NMR spectroscopy as a function of temperature revealed that the X5 imino resonance remained sharp at 55 °C. Additionally, two 5′-neighboring base pairs, A4:T9 and G3:C10, were stabilized with respect to the exchange of their imino protons with solvent. The equilibrium constant for base pair opening at the A4:T9 base pair determined by magnetization transfer from water in the absence and presence of added ammonia base catalyst decreased for the modified duplex compared to that of the A4:T9 base pair in the unmodified duplex, which confirmed that the overall fraction of the A4:T9 base pair in the open state of the modified duplex decreased. This was also observed for the G3:C10 base pair, where αKop for the G3:C10 base pair in the modified duplex was 3.0 × 106 versus 4.1 × 106 for the same base pair in the unmodified duplex. In contrast, equilibrium constants for base pair opening at the X5:C8 and C6:G7 base pairs did not change at 15 °C. These results argue against the notion that electrostatic interactions with DNA are entirely entropic and suggest that major groove cations can stabilize DNA via enthalpic contributions to the free energy of duplex formation.
Co-reporter:Hai Huang, Rajat S. Das, Ashis K. Basu, and Michael P. Stone
Chemical Research in Toxicology 2012 Volume 25(Issue 2) pp:478
Publication Date(Web):February 6, 2012
DOI:10.1021/tx2005053
Diastereomeric 8,5′-cyclopurine 2′-deoxynucleosides, containing a covalent bond between the deoxyribose and the purine base, are induced in DNA by ionizing radiation. They are suspected to play a role in the etiology of neurodegeneration in xeroderma pigmentosum patients. If not repaired, the S-8,5′-cyclo-2′-deoxyguanosine lesion (S-cdG) induces Pol V-dependent mutations at a frequency of 34% in Escherichia coli. Most are S-cdG → A transitions, suggesting mis-incorporation of dTTP opposite the lesion during replication bypass, although low levels of S-cdG → T transversions, arising from mis-incorporation of dATP, are also observed. We report the structures of 5′-d(GTGCXTGTTTGT)-3′·5′-d(ACAAACAYGCAC)-3′, where X denotes S-cdG and Y denotes either dA or dT, corresponding to the situation following mis-insertion of either dTTP or dATP opposite the S-cdG lesion. The S-cdG·dT mismatch pair adopts a wobble base pairing. This provides a plausible rationale for the S-cdG → A transitions. The S-cdG·dA mismatch pair differs in conformation from the dG·dA mismatch pair. For the S-cdG·dA mismatch pair, both S-cdG and dA intercalate, but no hydrogen bonding is observed between S-cdG and dA. This is consistent with the lower levels of S-cdG → T transitions in E. coli.
Co-reporter:Surajit Banerjee, Plamen P. Christov, Albena Kozekova, Carmelo J. Rizzo, Martin Egli, and Michael P. Stone
Chemical Research in Toxicology 2012 Volume 25(Issue 2) pp:422
Publication Date(Web):February 7, 2012
DOI:10.1021/tx200460j
trans-4-Hydroxynonenal (HNE) is the major peroxidation product of ω-6 polyunsaturated fatty acids in vivo. Michael addition of the N2-amino group of dGuo to HNE followed by ring closure of N1 onto the aldehyde results in four diastereomeric 1,N2-dGuo (1,N2-HNE-dGuo) adducts. The (6S,8R,11S)-HNE-1,N2-dGuo adduct was incorporated into the 18-mer templates 5′-d(TCATXGAATCCTTCCCCC)-3′ and d(TCACXGAATCCTTCCCCC)-3′, where X = (6S,8R,11S)-HNE-1,N2-dGuo adduct. These differed in the identity of the template 5′-neighbor base, which was either Thy or Cyt, respectively. Each of these templates was annealed with either a 13-mer primer 5′-d(GGGGGAAGGATTC)-3′ or a 14-mer primer 5′-d(GGGGGAAGGATTCC)-3′. The addition of dNTPs to the 13-mer primer allowed analysis of dNTP insertion opposite to the (6S,8R,11S)-HNE-1,N2-dGuo adduct, whereas the 14-mer primer allowed analysis of dNTP extension past a primed (6S,8R,11S)-HNE-1,N2-dGuo:dCyd pair. The Sulfolobus solfataricus P2 DNA polymerase IV (Dpo4) belongs to the Y-family of error-prone polymerases. Replication bypass studies in vitro reveal that this polymerase inserted dNTPs opposite the (6S,8R,11S)-HNE-1,N2-dGuo adduct in a sequence-specific manner. If the template 5′-neighbor base was dCyt, the polymerase inserted primarily dGTP, whereas if the template 5′-neighbor base was dThy, the polymerase inserted primarily dATP. The latter event would predict low levels of Gua → Thy mutations during replication bypass when the template 5′-neighbor base is dThy. When presented with a primed (6S,8R,11S)-HNE-1,N2-dGuo:dCyd pair, the polymerase conducted full-length primer extension. Structures for ternary (Dpo4-DNA-dNTP) complexes with all four template-primers were obtained. For the 18-mer:13-mer template-primers in which the polymerase was confronted with the (6S,8R,11S)-HNE-1,N2-dGuo adduct, the (6S,8R,11S)-1,N2-dGuo lesion remained in the ring-closed conformation at the active site. The incoming dNTP, either dGTP or dATP, was positioned with Watson–Crick pairing opposite the template 5′-neighbor base, dCyt or dThy, respectively. In contrast, for the 18-mer:14-mer template-primers with a primed (6S,8R,11S)-HNE-1,N2-dGuo:dCyd pair, ring opening of the adduct to the corresponding N2-dGuo aldehyde species occurred. This allowed Watson–Crick base pairing at the (6S,8R,11S)-HNE-1,N2-dGuo:dCyd pair.
Co-reporter:Hai Huang ; Hao Wang ; Albena Kozekova ; Carmelo J. Rizzo
Journal of the American Chemical Society 2011 Volume 133(Issue 40) pp:16101-16110
Publication Date(Web):September 14, 2011
DOI:10.1021/ja205145q
Michael addition of trans-4-hydroxynonenal (HNE) to deoxyguanosine yields diastereomeric 1,N2-dG adducts in DNA. When placed opposite dC in the 5′-CpG-3′ sequence, the (6S,8R,11S) diastereomer forms a N2-dG:N2-dG interstrand cross-link [Wang, H.; Kozekov, I. D.; Harris, T. M.; Rizzo, C. J. J. Am. Chem. Soc.2003, 125, 5687–5700]. We refined its structure in 5′-d(G1C2T3A4G5C6X7A8G9T10C11C12)-3′·5′-d(G13G14A15C16T17C18Y19C20T21A22G23C24)-3′ [X7 is the dG adjacent to the C6 carbon of the cross-link or the α-carbon of the (6S,8R,11S) 1,N2-dG adduct, and Y19 is the dG adjacent to the C8 carbon of the cross-link or the γ-carbon of the HNE-derived (6S,8R,11S) 1,N2-dG adduct; the cross-link is in the 5′-CpG-3′ sequence]. Introduction of 13C at the C8 carbon of the cross-link revealed one 13C8→H8 correlation, indicating that the cross-link existed predominantly as a carbinolamine linkage. The H8 proton exhibited NOEs to Y19 H1′, C20 H1′, and C20 H4′, orienting it toward the complementary strand, consistent with the (6S,8R,11S) configuration. An NOE was also observed between the HNE H11 proton and Y19 H1′, orienting the former toward the complementary strand. Imine and pyrimidopurinone linkages were excluded by observation of the Y19N2H and X7 N1H protons, respectively. A strong H8→H11 NOE and no 3J(13C→H) coupling for the 13C8–O–C11–H11 eliminated the tetrahydrofuran species derived from the (6S,8R,11S) 1,N2-dG adduct. The (6S,8R,11S) carbinolamine linkage and the HNE side chain were located in the minor groove. The X7N2 and Y19N2 atoms were in the gauche conformation with respect to the linkage, maintaining Watson–Crick hydrogen bonds at the cross-linked base pairs. A solvated molecular dynamics simulation indicated that the anti conformation of the hydroxyl group with respect to C6 of the tether minimized steric interaction and predicted hydrogen bonds involving O8H with C20O2 of the 5′-neighbor base pair G5·C20 and O11H with C18O2 of X7·C18. These may, in part, explain the stability of this cross-link and the stereochemical preference for the (6S,8R,11S) configuration.
Co-reporter:Hai Huang ; Rajat S. Das ; Ashis K. Basu
Journal of the American Chemical Society 2011 Volume 133(Issue 50) pp:20357-20368
Publication Date(Web):November 21, 2011
DOI:10.1021/ja207407n
Diastereomeric 8,5′-cyclopurine 2′-deoxynucleosides, containing a covalent bond between the deoxyribose and the purine base, represent an important class of DNA damage induced by ionizing radiation. The 8,5′-cyclo-2′-deoxyguanosine lesion (cdG) has been recently reported to be a strong block of replication and highly mutagenic in Escherichia coli. The 8,5′-cyclopurine-2′-deoxyriboses are suspected to play a role in the etiology of neurodegeneration in xeroderma pigmentosum patients. These lesions cannot be repaired by base excision repair, but they are substrates for nucleotide excision repair. The structure of an oligodeoxynucleotide duplex containing a site-specific S-cdG lesion placed opposite dC in the complementary strand was obtained by molecular dynamics calculations restrained by distance and dihedral angle restraints obtained from NMR spectroscopy. The S-cdG deoxyribose exhibited the O4′-exo (west) pseudorotation. Significant perturbations were observed for the β, γ, and χ torsion angles of the S-cdG nucleoside. Watson–Crick base pairing was conserved at the S-cdG·dC pair. However, the O4′-exo pseudorotation of the S-cdG deoxyribose perturbed the helical twist and base pair stacking at the lesion site and the 5′-neighbor dC·dG base pair. Thermodynamic destabilization of the duplex measured by UV melting experiments correlated with base stacking and structural perturbations involving the modified S-cdG·dC and 3′- neighbor dT·dA base pairs. These perturbations may be responsible for both the genotoxicity of this lesion and its ability to be recognized by nucleotide excision repair.
Co-reporter:Surajit Banerjee ; Kyle L. Brown ; Martin Egli
Journal of the American Chemical Society 2011 Volume 133(Issue 32) pp:12556-12568
Publication Date(Web):July 26, 2011
DOI:10.1021/ja2015668
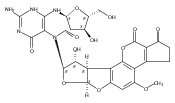
Aflatoxin B1 (AFB1) is oxidized to an epoxide in vivo, which forms an N7-dG DNA adduct (AFB1–N7-dG). The AFB1–N7-dG can rearrange to a formamidopyrimidine (AFB1–FAPY) derivative. Both AFB1–N7-dG and the β-anomer of the AFB1–FAPY adduct yield G→T transversions in Escherichia coli, but the latter is more mutagenic. We show that the Sulfolobus solfataricus P2 DNA polymerase IV (Dpo4) bypasses AFB1–N7-dG in an error-free manner but conducts error-prone replication past the AFB1–FAPY adduct, including misinsertion of dATP, consistent with the G→T mutations observed in E. coli. Three ternary (Dpo4–DNA–dNTP) structures with AFB1–N7-dG adducted template:primers have been solved. These demonstrate insertion of dCTP opposite the AFB1–N7-dG adduct, and correct vs incorrect insertion of dATP vs dTTP opposite the 5′-template neighbor dT from a primed AFB1–N7-dG:dC pair. The insertion of dTTP reveals hydrogen bonding between the template N3 imino proton and the O2 oxygen of dTTP, and between the template T O4 oxygen and the N3 imino proton of dTTP, perhaps explaining why this polymerase does not efficiently catalyze phosphodiester bond formation from this mispair. The AFB1–N7-dG maintains the 5′-intercalation of the AFB1 moiety observed in DNA. The bond between N7-dG and C8 of the AFB1 moiety remains in plane with the alkylated guanine, creating a 16° inclination of the AFB1 moiety with respect to the guanine. A binary (Dpo4–DNA) structure with an AFB1–FAPY adducted template:primer also maintains 5′-intercalation of the AFB1 moiety. The β-deoxyribose anomer is observed. Rotation about the FAPY C5–N5 bond orients the bond between N5 and C8 of the AFB1 moiety out of plane in the 5′-direction, with respect to the FAPY base. The formamide group extends in the 3′-direction. This improves stacking of the AFB1 moiety above the 5′-face of the FAPY base, as compared to the AFB1–N7-dG adduct. Ternary structures with AFB1–β-FAPY adducted template:primers show correct vs incorrect insertion of dATP vs dTTP opposite the 5′-template neighbor dT from a primed AFB1–β-FAPY:dC pair. For dATP, the oxygen atom of the FAPY formamide group participates in a water-mediated hydrogen bond with Arg332. The insertion of dTTP yields a structure similar to that observed for the AFB1–N7-dG adduct. The differential accommodation of these AFB1 adducts within the active site may, in part, modulate lesion bypass.
Co-reporter:Hai Huang, Hao Wang, Markus W. Voehler, Albena Kozekova, Carmelo J. Rizzo, Amanda K. McCullough, R. Stephen Lloyd, and Michael P. Stone
Chemical Research in Toxicology 2011 Volume 24(Issue 7) pp:1123
Publication Date(Web):May 11, 2011
DOI:10.1021/tx200113n
The γ-hydroxy-1,N2-propano-2′-deoxyguanosine adduct (γ-OH-PdG) was introduced into 5′-d(GCTAGCXAGTCC)-3′·5′-d(GGACTCGCTAGC)-3′ (X = γ-OH-PdG). In the presence of excess peptide KWKK, 13C isotope-edited NMR revealed the formation of two spectroscopically distinct DNA–KWKK conjugates. These involved the reaction of the KWKK N-terminal amino group with the N2-dG propylaldehyde tautomer of the γ-OH-PdG lesion. The guanine N1 base imino resonance at the site of conjugation was observed in isotope-edited 15N NMR experiments, suggesting that the conjugated guanine was inserted into the duplex and that the guanine imino proton was protected from exchange with water. The conjugates could be reduced in the presence of NaCNBH3, suggesting that they existed, in part, as imine (Schiff base) linkages. However, 13C isotope-edited NMR failed to detect the imine linkages, suggesting that these KWKK conjugates existed predominantly as diastereomeric carbinolamines, in equilibrium with trace amounts of the imines. The structures of the diastereomeric DNA–KWKK conjugates were predicted from potential energy minimization of model structures derived from the refined structure of the fully reduced cross-link [Huang, H., Kozekov, I. D., Kozekova, A., Rizzo, C. J., McCullough, A., Lloyd, R. S., and Stone, M. P. (2010) Biochemistry, 49, 6155−6164]. Molecular dynamics calculations carried out in explicit solvent suggested that the conjugate bearing the S-carbinolamine linkage was the major species due to its potential for intramolecular hydrogen bonding. These carbinolamine DNA–KWKK conjugates thermally stabilized duplex DNA. However, the DNA–KWKK conjugates were chemically reversible and dissociated when the DNA was denatured. In this 5′-CpX-3′ sequence, the DNA–KWKK conjugates slowly converted to interstrand N2-dG:N2-dG DNA cross-links and ring-opened γ-OH-PdG derivatives over a period of weeks.
Co-reporter:Ganesh Shanmugam, Ivan D. Kozekov, F. Peter Guengerich, Carmelo J. Rizzo, and Michael P. Stone
Chemical Research in Toxicology 2011 Volume 24(Issue 7) pp:1071
Publication Date(Web):June 16, 2011
DOI:10.1021/tx200089v
The oligodeoxynucleotide 5′-CGCATXGAATCC-3′·5′-GGATTCAATGCG-3′ containing 1,N2-etheno-2′-deoxyguanosine (1,N2-εdG) opposite deoxyadenosine (named the 1,N2-εdG·dA duplex) models the mismatched adenine product associated with error-prone bypass of 1,N2-εdG by the Sulfolobus solfataricus P2 DNA polymerase IV (Dpo4) and by Escherichia coli polymerases pol I exo– and pol II exo–. At pH 5.2, the Tm of this duplex was increased by 3 °C as compared to the duplex in which the 1,N2-εdG lesion is opposite dC, and it was increased by 2 °C compared to the duplex in which guanine is opposite dA (the dG·dA duplex). A strong NOE between the 1,N2-εdG imidazole proton and the anomeric proton of the attached deoxyribose, accompanied by strong NOEs to the minor groove A20 H2 proton and the mismatched A19 H2 proton from the complementary strand, establish that 1,N2-εdG rotated about the glycosyl bond from the anti to the syn conformation. The etheno moiety was placed into the major groove. This resulted in NOEs between the etheno protons and T5 CH3. A strong NOE between A20 H2 and A19 H2 protons established that A19, opposite to 1,N2-εdG, adopted the anti conformation and was directed toward the helix. The downfield shifts of the A19 amino protons suggested protonation of dA. Thus, the protonated 1,N2-εdG·dA base pair was stabilized by hydrogen bonds between 1,N2-εdG N1 and A19 N1H+ and between 1,N2-εdG O9 and A19N6H. The broad imino proton resonances for the 5′- and 3′-flanking bases suggested that both neighboring base pairs were perturbed. The increased stability of the 1,N2-εdG·dA base pair, compared to that of the 1,N2-εdG·dC base pair, correlated with the mismatch adenine product observed during the bypass of 1,N2-εdG by the Dpo4 polymerase, suggesting that stabilization of this mismatch may be significant with regard to the biological processing of 1,N2-εdG.
Co-reporter:Michael P. Stone;Hai Huang;Kyle L. Brown ;Ganesh Shanmugam
Chemistry & Biodiversity 2011 Volume 8( Issue 9) pp:1571-1615
Publication Date(Web):
DOI:10.1002/cbdv.201100033
Abstract
The formation of adducts by the reaction of chemicals with DNA is a critical step for the initiation of carcinogenesis. The structural analysis of various DNA adducts reveals that conformational and chemical rearrangements and interconversions are a common theme. Conformational changes are modulated both by the nature of adduct and the base sequences neighboring the lesion sites. Equilibria between conformational states may modulate both DNA repair and error-prone replication past these adducts. Likewise, chemical rearrangements of initially formed DNA adducts are also modulated both by the nature of adducts and the base sequences neighboring the lesion sites. In this review, we focus on DNA damage caused by a number of environmental and endogenous agents, and biological consequences.
Co-reporter:Ewa A. Kowal, Manjori Ganguly, Pradeep S. Pallan, Luis A. Marky, Barry Gold, Martin Egli, and Michael P. Stone
The Journal of Physical Chemistry B 2011 Volume 115(Issue 47) pp:13925-13934
Publication Date(Web):November 8, 2011
DOI:10.1021/jp207104w
As part of an ongoing effort to explore the effect of major groove electrostatics on the thermodynamic stability and structure of DNA, a 7-deaza-2′-deoxyadenosine:dT (7-deaza-dA:dT) base pair in the Dickerson–Drew dodecamer (DDD) was studied. The removal of the electronegative N7 atom on dA and the replacement with an electropositive C–H in the major groove was expected to have a significant effect on major groove electrostatics. The structure of the 7-deaza-dA:dT base pair was determined at 1.1 Å resolution in the presence of Mg2+. The 7-deaza-dA, which is isosteric for dA, had minimal effect on the base pairing geometry and the conformation of the DDD in the crystalline state. There was no major groove cation association with the 7-deaza-dA heterocycle. In solution, circular dichroism showed a positive Cotton effect centered at 280 nm and a negative Cotton effect centered at 250 nm that were characteristic of a right-handed helix in the B-conformation. However, temperature-dependent NMR studies showed increased exchange between the thymine N3 imino proton of the 7-deaza-dA:dT base pair and water, suggesting reduced stacking interactions and an increased rate of base pair opening. This correlated with the observed thermodynamic destabilization of the 7-deaza-dA modified duplex relative to the DDD. A combination of UV melting and differential scanning calorimetry experiments were conducted to evaluate the relative contributions of enthalpy and entropy in the thermodynamic destabilization of the DDD. The most significant contribution arose from an unfavorable enthalpy term, which probably results from less favorable stacking interactions in the modified duplex, which was accompanied by a significant reduction in the release of water and cations from the 7-deaza-dA modified DNA.
Co-reporter:Ganesh Shanmugam, Ivan D. Kozekov, F. Peter Guengerich, Carmelo J. Rizzo and Michael P. Stone
Biochemistry 2010 Volume 49(Issue 12) pp:
Publication Date(Web):March 4, 2010
DOI:10.1021/bi901516d
The structure of the 1,N2-ethenodeoxyguanosine lesion (1,N2-εdG) has been characterized in 5′-d(CGCATXGAATCC)-3′·5′-d(GGATTCATGCG)-3′ (X = 1,N2-εdG), in which there is no dC opposite the lesion. This duplex (named the 1-BD duplex) models the product of translesion bypass of 1,N2-εdG by Sulfolobus solfataricus P2 DNA polymerase IV (Dpo4) [Zang, H., Goodenough, A. K., Choi, J. Y., Irimia, A., Loukachevitch, L. V., Kozekov, I. D., Angel, K. C., Rizzo, C. J., Egli, M., and Guengerich, F. P. (2005) J. Biol. Chem. 280, 29750−29764], leading to a one-base deletion. The Tm of this duplex is 6 °C higher than that of the duplex in which dC is present opposite the 1,N2-εdG lesion and 8 °C higher than that of the unmodified 1-BD duplex. Analysis of NOEs between the 1,N2-εdG imidazole and deoxyribose H1′ protons and between the 1,N2-εdG etheno H6 and H7 protons and DNA protons establishes that 1,N2-εdG adopts the anti conformation about the glycosyl bond and that the etheno moiety is accommodated within the helix. The resonances of the 1,N2-εdG H6 and H7 etheno protons shift upfield relative to the monomer 1,N2-εdG, attributed to ring current shielding, consistent with their intrahelical location. NMR data reveal that Watson−Crick base pairing is maintained at both the 5′ and 3′ neighbor base pairs. The structure of the 1-BD duplex has been refined using molecular dynamics calculations restrained by NMR-derived distance and dihedral angle restraints. The increased stability of the 1,N2-εdG lesion in the absence of the complementary dC correlates with the one-base deletion extension product observed during the bypass of the 1,N2-εdG lesion by the Dpo4 polymerase, suggesting that stabilization of this bulged intermediate may be significant with regard to the biological processing of the lesion.
Co-reporter:Hai Huang, Ivan D. Kozekov, Albena Kozekova, Carmelo J. Rizzo, Amanda K. McCullough, R. Stephen Lloyd and Michael P. Stone
Biochemistry 2010 Volume 49(Issue 29) pp:
Publication Date(Web):July 6, 2010
DOI:10.1021/bi100364f
DNA−protein conjugates are potentially repaired via proteolytic digestion to DNA−peptide conjugates. The latter have been modeled with the amino-terminal lysine of the peptide KWKK conjugated via a trimethylene linkage to the N2-dG amine positioned in 5′-d(GCTAGCXAGTCC)-3′·5′-d(GGACTCGCTAGC)-3′ (X = N2-dG−trimethylene link−KWKK). This linkage is a surrogate for the reversible linkage formed by the γ-OH-1,N2-propanodeoxyguanosine (γ-OH-PdG) adduct. This conjugated KWKK stabilizes the DNA. Amino acids K26, W27, K28, and K29 are in the minor groove. The W27 indolyl group does not intercalate into the DNA. The G7 N2 amine and the K26 N-terminal amine nitrogens are in the trans configuration with respect to the Cα or Cγ of the trimethylene tether, respectively. The structure of this DNA−KWKK conjugate is discussed in the context of its biological processing.
Co-reporter:Hai Huang, Hao Wang, R. Stephen Lloyd, Carmelo J. Rizzo and Michael P. Stone
Chemical Research in Toxicology 2009 Volume 22(Issue 1) pp:187
Publication Date(Web):December 3, 2008
DOI:10.1021/tx800320m
The (6S,8R,11S) 1,N2-HNE-dGuo adduct of trans-4-hydroxynonenal (HNE) was incorporated into the duplex 5′-d(GCTAGCXAGTCC)-3′·5′-d(GGACTAGCTAGC)-3′ [X = (6S,8R,11S) HNE-dG], in which the lesion was mismatched opposite dAdo. The (6S,8R,11S) adduct maintained the ring-closed 1,N2-HNE-dG structure. This was in contrast to when this adduct was correctly paired with dCyd, conditions under which it underwent ring opening and rearrangement to diastereomeric minor groove cyclic hemiacetals [Huang, H., Wang, H., Qi, N., Lloyd, R. S., Harris, T. M., Rizzo, C. J., and Stone, M. P. (2008) J. Am. Chem. Soc.130, 10898−10906]. The (6S,8R,11S) adduct exhibited a syn/anti conformational equilibrium about the glycosyl bond. The syn conformation was predominant in acidic solution. Structural analysis of the syn conformation revealed that X7 formed a distorted base pair with the complementary protonated A18. The HNE moiety was located in the major groove. Structural perturbations were observed at the neighbor C6·G19 and A8·T17 base pairs. At basic pH, the anti conformation of X7 was the major species. The 1,N2-HNE-dG intercalated and displaced the complementary A18 in the 5′-direction, resulting in a bulge at the X7·A18 base pair. The HNE aliphatic chain was oriented toward the minor groove. The Watson−Crick hydrogen bonding of the neighboring A8·T17 base pair was also disrupted.
Co-reporter:Irina G. Minko, Ivan D. Kozekov, Thomas M. Harris, Carmelo J. Rizzo, R. Stephen Lloyd and Michael P. Stone
Chemical Research in Toxicology 2009 Volume 22(Issue 5) pp:759
Publication Date(Web):April 27, 2009
DOI:10.1021/tx9000489
The α,β-unsaturated aldehydes (enals) acrolein, crotonaldehyde, and trans-4-hydroxynonenal (4-HNE) are products of endogenous lipid peroxidation, arising as a consequence of oxidative stress. The addition of enals to dG involves Michael addition of the N2-amine to give N2-(3-oxopropyl)-dG adducts, followed by reversible cyclization of N1 with the aldehyde, yielding 1,N2-dG exocyclic products. The 1,N2-dG exocyclic adducts from acrolein, crotonaldehyde, and 4-HNE exist in human and rodent DNA. The enal-induced 1,N2-dG lesions are repaired by the nucleotide excision repair pathway in both Escherichia coli and mammalian cells. Oligodeoxynucleotides containing structurally defined 1,N2-dG adducts of acrolein, crotonaldehyde, and 4-HNE were synthesized via a postsynthetic modification strategy. Site-specific mutagenesis of enal adducts has been carried out in E. coli and various mammalian cells. In all cases, the predominant mutations observed are G→T transversions, but these adducts are not strongly miscoding. When placed into duplex DNA opposite dC, the 1,N2-dG exocyclic lesions undergo ring opening to the corresponding N2-(3-oxopropyl)-dG derivatives. Significantly, this places a reactive aldehyde in the minor groove of DNA, and the adducted base possesses a modestly perturbed Watson−Crick face. Replication bypass studies in vitro indicate that DNA synthesis past the ring-opened lesions can be catalyzed by pol η, pol ι, and pol κ. It also can be accomplished by a combination of Rev1 and pol ζ acting sequentially. However, efficient nucleotide insertion opposite the 1,N2-dG ring-closed adducts can be carried out only by pol ι and Rev1, two DNA polymerases that do not rely on the Watson−Crick pairing to recognize the template base. The N2-(3-oxopropyl)-dG adducts can undergo further chemistry, forming interstrand DNA cross-links in the 5′-CpG-3′ sequence, intrastrand DNA cross-links, or DNA−protein conjugates. NMR and mass spectrometric analyses indicate that the DNA interstand cross-links contain a mixture of carbinolamine and Schiff base, with the carbinolamine forms of the linkages predominating in duplex DNA. The reduced derivatives of the enal-mediated N2-dG:N2-dG interstrand cross-links can be processed in mammalian cells by a mechanism not requiring homologous recombination. Mutations are rarely generated during processing of these cross-links. In contrast, the reduced acrolein-mediated N2-dG peptide conjugates can be more mutagenic than the corresponding monoadduct. DNA polymerases of the DinB family, pol IV in E. coli and pol κ in human, are implicated in error-free bypass of model acrolein-mediated N2-dG secondary adducts, the interstrand cross-links, and the peptide conjugates.
Co-reporter:Hai Huang, Patricia A. Dooley, Constance M. Harris, Thomas M. Harris and Michael P. Stone
Chemical Research in Toxicology 2009 Volume 22(Issue 11) pp:1810
Publication Date(Web):October 9, 2009
DOI:10.1021/tx900225c
Synthetically derived trimethylene interstrand DNA cross-links have been used as surrogates for the native cross-links that arise from the 1,N2-deoxyguanosine adducts derived from α,β-unsaturated aldehydes. The native enal-mediated cross-linking occurs in the 5′-CpG-3′ sequence context but not in the 5′-GpC-3′ sequence context. The ability of the native enal-derived 1,N2-dG adducts to induce interstrand DNA cross-links in the 5′-CpG-3′ sequence as opposed to the 5′-GpC-3′ sequence is attributed to the destabilization of the DNA duplex in the latter sequence context. Here, we report higher accuracy solution structures of the synthetically derived trimethylene cross-links, which are refined from NMR data with the AMBER force field. When the synthetic trimethylene cross-links are placed into either the 5′-CpG-3′ or the 5′-GpC-3′ sequence contexts, the DNA duplex maintains B-DNA geometry with structural perturbations confined to the cross-linked base pairs. Watson−Crick hydrogen bonding is conserved throughout the duplexes. Although different from canonical B-DNA stacking, the cross-linked and the neighbor base pairs stack in the 5′-CpG-3′ sequence. In contrast, the stacking at the cross-linked base pairs in the 5′-GpC-3′ sequence is greatly perturbed. The π-stacking interactions between the cross-linked and the neighbor base pairs are reduced. This is consistent with remarkable chemical shift perturbations of the C5 H5 and H6 nucleobase protons that shifted downfield by 0.4−0.5 ppm. In contrast, these chemical shift perturbations in the 5′-CpG-3′ sequence are not remarkable, consistent with the stacked structure. The differential stacking of the base pairs at the cross-linking region probably explains the difference in stabilities of the trimethylene cross-links in the 5′-CpG-3′ and 5′-GpC-3′ sequence contexts and might, in turn, account for the sequence selectivity of the interstrand cross-link formation induced by the native enal-derived 1,N2-dG adducts.
Co-reporter:Kyle L. Brown, Ashis K. Basu and Michael P. Stone
Biochemistry 2009 Volume 48(Issue 41) pp:
Publication Date(Web):September 22, 2009
DOI:10.1021/bi900695e
Oxidative damage to 5-methylcytosine in DNA, followed by deamination, yields thymine glycol (Tg), 5,6-dihydroxy-5,6-dihydrothymine, mispaired with deoxyguanosine. The structure of the 5R Tg·G mismatch pair has been refined using a combination of simulated annealing and isothermal molecular dynamics calculations restrained by NMR-derived distance restraints and torsion angle restraints in 5′-d(G1T2G3C4G5Tg6G7T8T9T10G11T12)-3′·5′-d(A13C14A15A16A17C18G19C20G21C22A23C24)-3′; Tg = 5R Tg. In this duplex the cis-5R,6S:trans-5R,6R equilibrium favors the cis-5R,6S epimer [Brown, K. L., Adams, T., Jasti, V. P., Basu, A. K., and Stone, M. P. (2008) J. Am. Chem. Soc. 130, 11701−11710]. The cis-5R,6S Tg lesion is in the wobble orientation such that Tg6 O2 is proximate to G19 N1H and Tg6 N3H is proximate to G19 O6. Both Tg6 and the mismatched nucleotide G19 remain stacked in the helix. The Tg6 nucleotide shifts toward the major groove and stacks below the 5′-neighbor base G5, while its complement G19 stacks below the 5′-neighbor C20. In the 3′-direction, stacking between Tg6 and the G7·C18 base pair is disrupted. The solvent-accessible surface area of the Tg nucleotide increases as compared to the native Watson−Crick hydrogen-bonded T·A base pair. An increase in T2 relaxation rates for the Tg6 base protons is attributed to puckering of the Tg base, accompanied by increased disorder at the Tg·G mismatch pair. The axial vs equatorial conformation of the Tg6 CH3 group cannot be determined with certainty from the NMR data. The rMD trajectories suggest that in either the axial or equatorial conformations the cis-5R,6S Tg lesion does not form strong intrastrand hydrogen bonds with the imidazole N7 atom of the 3′-neighbor purine G7. The wobble pairing and disorder of the Tg·G mismatch correlate with the reduced thermodynamic stability of the mismatch and likely modulate its recognition by DNA base excision repair systems.
Co-reporter:Michael P. Stone, Young-Jin Cho, Hai Huang, Hye-Young Kim, Ivan D. Kozekov, Albena Kozekova, Hao Wang, Irina G. Minko, R. Stephen Lloyd, Thomas M. Harris and Carmelo J. Rizzo
Accounts of Chemical Research 2008 Volume 41(Issue 7) pp:793
Publication Date(Web):May 24, 2008
DOI:10.1021/ar700246x
Significant levels of the 1,N2-γ-hydroxypropano-dG adducts of the α,β-unsaturated aldehydes acrolein, crotonaldehyde, and 4-hydroxy-2E-nonenal (HNE) have been identified in human DNA, arising from both exogenous and endogenous exposures. They yield interstrand DNA cross-links between guanines in the neighboring C·G and G·C base pairs located in 5′-CpG-3′ sequences, as a result of opening of the 1,N2-γ-hydroxypropano-dG adducts to form reactive aldehydes that are positioned within the minor groove of duplex DNA. Using a combination of chemical, spectroscopic, and computational methods, we have elucidated the chemistry of cross-link formation in duplex DNA. NMR spectroscopy revealed that, at equilibrium, the acrolein and crotonaldehyde cross-links consist primarily of interstrand carbinolamine linkages between the exocyclic amines of the two guanines located in the neighboring C·G and G·C base pairs located in 5′-CpG-3′ sequences, that maintain the Watson−Crick hydrogen bonding of the cross-linked base pairs. The ability of crotonaldehyde and HNE to form interstrand cross-links depends upon their common relative stereochemistry at the C6 position of the 1,N2-γ-hydroxypropano-dG adduct. The stereochemistry at this center modulates the orientation of the reactive aldehyde within the minor groove of the double-stranded DNA, either facilitating or hindering the cross-linking reactions; it also affects the stabilities of the resulting diastereoisomeric cross-links. The presence of these cross-links in vivo is anticipated to interfere with DNA replication and transcription, thereby contributing to the etiology of human disease. Reduced derivatives of these cross-links are useful tools for studying their biological processing.
Co-reporter:Feng Wang, Feng Li, Manjori Ganguly, Luis A. Marky, Barry Gold, Martin Egli and Michael P. Stone
Biochemistry 2008 Volume 47(Issue 27) pp:
Publication Date(Web):June 13, 2008
DOI:10.1021/bi800375m
Site-specific insertion of 5-(3-aminopropyl)-2′-deoxyuridine (Z3dU) and 7-deaza-dG into the Dickerson−Drew dodecamers 5′-d(C1G2C3G4A5A6T7T8C9Z10C11G12)-3′·5′-d(C13G14C15G16A17A18T19T20C21Z22C23G24)-3′ (named DDDZ10) and 5′-d(C1G2C3G4A5A6T7X8C9Z10C11G12)-3′·5′-d(C13G14C15G16A17A18T19X20C21Z22C23G24)-3′ (named DDD2+Z10) (X = Z3dU; Z = 7-deaza-dG) suggests a mechanism underlying the formation of interstrand N+2 DNA cross-links by nitrogen mustards, e.g., melphalan and mechlorethamine. Analysis of the DDD2+Z10 duplex reveals that the tethered cations at base pairs A5·X20 and X8·A17 extend within the major groove in the 3′-direction, toward conserved Mg2+ binding sites located adjacent to N+2 base pairs C3·Z22 and Z10·C15. Bridging waters located between the tethered amines and either Z10 or Z22 O6 stabilize the tethered cations and allow interactions with the N + 2 base pairs without DNA bending. Incorporation of 7-deaza-dG into the DDD2+Z10 duplex weakens but does not eliminate electrostatic interactions between tethered amines and Z10 O6 and Z22 O6. The results suggest a mechanism by which tethered N7-dG aziridinium ions, the active species involved in formation of interstrand 5′-GNC-3′ cross-links by nitrogen mustards, modify the electrostatics of the major groove and position the aziridinium ions proximate to the major groove edge of the N+2 C·G base pair, facilitating interstrand cross-linking.
Co-reporter:Yazhen Wang, Sarah K. Musser, Sam Saleh, Lawrence J. Marnett, Martin Egli and Michael P. Stone
Biochemistry 2008 Volume 47(Issue 28) pp:
Publication Date(Web):June 19, 2008
DOI:10.1021/bi800152j
1,N2-Propanodeoxyguanosine (PdG) is a stable structural analogue for the 3-(2′-deoxy-β-d-erythro-pentofuranosyl)pyrimido[1,2-α]purin-10(3H)-one (M1dG) adduct derived from exposure of DNA to base propenals and to malondialdehyde. The structures of ternary polymerase−DNA−dNTP complexes for three template−primer DNA sequences were determined, with the Y-family Sulfolobus solfataricus DNA polymerase IV (Dpo4), at resolutions between 2.4 and 2.7 Å. Three template 18-mer−primer 13-mer sequences, 5′-d(TCACXAAATCCTTCCCCC)-3′·5′-d(GGGGGAAGGATTT)-3′ (template I), 5′-d(TCACXGAATCCTTCCCCC)-3′·5′-d(GGGGGAAGGATTC)-3′ (template II), and 5′-d(TCATXGAATCCTTCCCCC)-3′·5′-d(GGGGGAAGGATTC)-3′ (template III), where X is PdG, were analyzed. With templates I and II, diffracting ternary complexes including dGTP were obtained. The dGTP did not pair with PdG, but instead with the 5′-neighboring template dC, utilizing Watson−Crick geometry. Replication bypass experiments with the template−primer 5′-TCACXAAATCCTTACGAGCATCGCCCCC-3′·5′-GGGGGCGATGCTCGTAAGGATTT-3′, where X is PdG, which includes PdG in the 5′-CXA-3′ template sequence as in template I, showed that the Dpo4 polymerase inserted dGTP and dATP when challenged by the PdG adduct. For template III, in which the template sequence was 5′-TXG-3′, a diffracting ternary complex including dATP was obtained. The dATP did not pair with PdG, but instead with the 5′-neighboring T, utilizing Watson−Crick geometry. Thus, all three ternary complexes were of the “type II” structure described for ternary complexes with native DNA [Ling, H., Boudsocq, F., Woodgate, R., and Yang, W. (2001) Cell 107, 91−102]. The PdG adduct remained in the anti conformation about the glycosyl bond in each of these threee ternary complexes. These results provide insight into how −1 frameshift mutations might be generated for the PdG adduct, a structural model for the exocylic M1dG adduct formed by malondialdehyde.
Co-reporter:Hai Huang, Hao Wang, Nan Qi, R. Stephen Lloyd, Carmelo J. Rizzo and Michael P. Stone
Biochemistry 2008 Volume 47(Issue 44) pp:
Publication Date(Web):October 11, 2008
DOI:10.1021/bi8011143
The trans-4-hydroxynonenal (HNE)-derived exocyclic 1,N2-dG adduct with (6S,8R,11S) stereochemistry forms interstrand N2-dG−N2-dG cross-links in the 5′-CpG-3′ DNA sequence context, but the corresponding adduct possessing (6R,8S,11R) stereochemistry does not. Both exist primarily as diastereomeric cyclic hemiacetals when placed into duplex DNA [Huang, H., Wang, H., Qi, N., Kozekova, A., Rizzo, C. J., and Stone, M. P. (2008) J. Am. Chem. Soc. 130, 10898−10906]. To explore the structural basis for this difference, the HNE-derived diastereomeric (6S,8R,11S) and (6R,8S,11R) cyclic hemiacetals were examined with respect to conformation when incorporated into 5′-d(GCTAGCXAGTCC)-3′·5′-d(GGACTCGCTAGC)-3′, containing the 5′-CpX-3′ sequence [X = (6S,8R,11S)- or (6R,8S,11R)-HNE−dG]. At neutral pH, both adducts exhibited minimal structural perturbations to the DNA duplex that were localized to the site of the adduction at X7·C18 and its neighboring base pair, A8·T17. Both the (6S,8R,11S) and (6R,8S,11R) cyclic hemiacetals were located within the minor groove of the duplex. However, the respective orientations of the two cyclic hemiacetals within the minor groove were dependent upon (6S) versus (6R) stereochemistry. The (6S,8R,11S) cyclic hemiacetal was oriented in the 5′-direction, while the (6R,8S,11R) cyclic hemiacetal was oriented in the 3′-direction. These cyclic hemiacetals effectively mask the reactive aldehydes necessary for initiation of interstrand cross-link formation. From the refined structures of the two cyclic hemiacetals, the conformations of the corresponding diastereomeric aldehydes were predicted, using molecular mechanics calculations. Potential energy minimizations of the duplexes containing the two diastereomeric aldehydes predicted that the (6S,8R,11S) aldehyde was oriented in the 5′-direction while the (6R,8S,11R) aldehyde was oriented in the 3′-direction. These stereochemical differences in orientation suggest a kinetic basis that explains, in part, why the (6S,8R,11S) stereoisomer forms interchain cross-links in the 5′-CpG-3′ sequence whereas the (6R,8S,11R) stereoisomer does not.
Co-reporter:Yazhen Wang, Nathalie C. Schnetz-Boutaud, Heiko Kroth, Haruhiko Yagi, Jane M. Sayer, Subodh Kumar, Donald M. Jerina and Michael P. Stone
Chemical Research in Toxicology 2008 Volume 21(Issue 7) pp:1348
Publication Date(Web):June 13, 2008
DOI:10.1021/tx7004103
The conformation of the 1R,2S,3R,4S-benzo[c]phenanthrene-N2-dG adduct, arising from trans opening of the (+)-1S,2R,3R,4S-anti-benzo[c]phenanthrene diol epoxide, was examined in 5′- d(ATCGCXCGGCATG)-3′·5′-d(CATGCCGCGCGAT)-3′, where X = 1R,2S,3R,4S-B[c]P-N2-dG. This duplex, derived from the hisD3052 frameshift tester strain of Salmonella typhimurium, contains a (CG)3 iterated repeat, a hotspot for frameshift mutagenesis. NMR experiments showed a disconnection in sequential NOE connectivity between X4 and C5, and in the complementary strand, they showed another disconnection between G18 and C19. In the imino region of the 1H NMR spectrum, a resonance was observed at the adducted base pair X4·C19. The X4 N1H and G18 N1H resonances shifted upfield as compared to the other guanine imino proton resonances. NOEs were observed between X4 N1H and C19 N4H and between C5 N4H and G18 N1H, indicating that base pairs X4·C19 and C5·G18 maintained Watson−Crick hydrogen bonding. No NOE connectivity was observed between X4 and G18 in the imino region of the spectrum. Chemical shift perturbations of greater than 0.1 ppm were localized at nucleotides X4 and C5 in the modified strand and G18 and C19 in the complementary strand. A total of 13 NOEs between the protons of the 1R-B[c]Ph moiety and the DNA were observed between B[c]Ph and major groove aromatic or amine protons at base pairs X4·C19 and 3′-neighbor C5·G18. Structural refinement was achieved using molecular dynamics calculations restrained by interproton distances and torsion angle restraints obtained from NMR data. The B[c]Ph moiety intercalated on the 3′-face of the X4·C19 base pair such that the terminal ring of 1R-B[c]Ph threaded the duplex and faced into the major groove. The torsion angle α′ [X4]−N3−C2−N2−B[c]Ph]−C1 was calculated to be −177°, maintaining an orientation in which the X4 exocyclic amine remained in plane with the purine. The torsion angle β′ [X4]−C2−N2−[B[c]Ph]−C1−C2 was calculated to be 75°. This value governed the 3′-orientation of the B[c]Ph moiety with respect to X4. The helical rise between base pairs X4·C19 and C5·G18 increased and resulted in unwinding of the right-handed helix. The aromatic rings of the B[c]Ph moiety were below the Watson−Crick hydrogen-bonding face of the modified base pair X4·C19. The B[c]Ph moiety was stacked above nucleotide G18, in the complementary strand.
Co-reporter:Ganesh Shanmugam, Ivan D. Kozekov, F. Peter Guengerich, Carmelo J. Rizzo and Michael P. Stone
Chemical Research in Toxicology 2008 Volume 21(Issue 9) pp:1795
Publication Date(Web):August 12, 2008
DOI:10.1021/tx8001466
The exocyclic 1,N2-ethenodeoxyguanosine (1,N2-ϵdG) adduct, arising from the reaction of vinyl halides and other vinyl monomers, including chloroacetaldehyde, and lipid peroxidation products with dG, was examined at pH 5.2 in the oligodeoxynucleotide duplex 5′-d(CGCATXGAATCC)-3′·5′-d(GGATTCCATGCG)-3′ (X = 1,N2-ϵdG). Previously, X(anti)·C(anti) pairing was established in this duplex, containing the 5′-TXG-3′ sequence context, at pH 8.6 [Shanmugam, G., Goodenough, A. K., Kozekov, I. D., Harris, T. M., Guengerich, F. P., Rizzo, C. J., and Stone, M. P. (2007) Chem. Res. Toxicol.20, 1601−1611]. At pH 5.2, the 1,N2-ϵdG adduct decreased the thermal stability of the duplex by ∼13 °C. The 1,N2-ϵdG adduct rotated about the glycosyl bond from the anti to the syn conformation. This resulted in the observation of a strong nuclear Overhauser effect (NOE) between the imidazole proton of 1,N2-ϵdG and the anomeric proton of the attached deoxyribose, accompanied by an NOE to the minor groove A20 H2 proton from the complementary strand. The syn conformation of the glycosyl bond at 1,N2-ϵdG placed the exocyclic etheno moiety into the major groove. This resulted in the observation of NOEs between the etheno protons and the major groove protons of the 5′-neighboring thymine. The 1,N2-ϵdG adduct formed a Hoogsteen pair with the complementary cytosine, characterized by downfield shifts of the amino protons of the cytosine complementary to the exocyclic adduct. The pattern of chemical shift perturbations indicated that the lesion introduced a localized structural perturbation involving the modified base pair and its 3′- and 5′-neighbor base pairs. A second conformational equilibrium was observed, in which both the modified base pair and its 3′-neighboring G·C base pair formed tandem Hoogsteen pairs. The results support the conclusion that at neutral pH, in the 5′-TXG-3′ sequence, the 1,N2-ϵdG adduct exists as a blend of conformations in duplex DNA. These involve the interconversion of the glycosyl torsion angle between the anti and the syn conformations, occurring at an intermediate rate on the NMR time scale.
Co-reporter:Yazhen Wang, Nathalie C. Schnetz-Boutaud, Sam Saleh, Lawrence J. Marnett and Michael P. Stone
Chemical Research in Toxicology 2007 Volume 20(Issue 8) pp:1200
Publication Date(Web):July 24, 2007
DOI:10.1021/tx700121j
The OPdG adduct N2-(3-oxo-1-propenyl)dG, formed in DNA exposed to malondialdehyde, was introduced into 5′-d(ATCGCXCGGCATG)-3′·5′-d(CATGCCGCGAT)-3′ at pH 7 (X = OPdG). The OPdG adduct is the base-catalyzed rearrangement product of the M1dG adduct, 3-(β-d-ribofuranosyl)pyrimido[1,2-a]purin-10(3H)-one. This duplex, named the OPdG-2BD oligodeoxynucleotide, was derived from a frameshift hotspot of the Salmonella typhimuium hisD3052 gene and contained a two-base deletion in the complementary strand. NMR spectroscopy revealed that the OPdG-2BD oligodeoxynucleotide underwent rapid bulge migration. This hindered its conversion to the M1dG-2BD duplex, in which the bulge was localized and consisted of the M1dG adduct and the 3′-neighbor dC [Schnetz-Boutaud, N. C., Saleh, S., Marnett, L. J., and Stone, M. P. (2001) Biochemistry40, 15638–15649]. The spectroscopic data suggested that bulge migration transiently positioned OPdG opposite dC in the complementary strand, hindering formation of the M1dG-2BD duplex, or alternatively, reverting rapidly formed intermediates in the OPdG to M1dG reaction pathway when dC was placed opposite from OPdG. The approach of initially formed M1dG-2BD or OPdG-2BD duplexes to an equilibrium mixture of the M1dG-2BD and OPdG-2BD duplexes was monitored as a function of time, using NMR spectroscopy. Both samples attained equilibrium in ∼140 days at pH 7 and 25 °C.
Co-reporter:Ganesh Shanmugam, Angela K. Goodenough, Ivan D. Kozekov, F. Peter Guengerich, Carmelo J. Rizzo and Michael P. Stone
Chemical Research in Toxicology 2007 Volume 20(Issue 11) pp:1601
Publication Date(Web):October 18, 2007
DOI:10.1021/tx7001788
The structure of the 1,N2-etheno-2′-deoxyguanosine (1,N2-εdG) adduct, arising from the reaction of vinyl chloride with dG, was determined in the oligonucleotide duplex 5′-d(CGCATXGAATCC)-3′·5′-d(GGATTCCATGCG)-3′ (X = 1,N2-εdG) at pH 8.6 using high resolution NMR spectroscopy. The exocyclic lesion prevented Watson–Crick base-pairing capability at the adduct site and resulted in an ∼17 °C decrease in Tm of the oligodeoxynucleotide duplex. At neutral pH, conformational exchange resulted in spectral line broadening near the adducted site, and it was not possible to determine the structure. However, at pH 8.6, it was possible to obtain well-resolved 1H NMR spectra. This enabled a total of 385 NOE-based distance restraints to be obtained, consisting of 245 intra- and 140 inter-nucleotide distances. The 31P NMR spectra exhibited two downfield-shifted resonances, suggesting a localized perturbation of the DNA backbone. The two downfield 31P resonances were assigned to G7 and C19. The solution structure was refined by molecular dynamics calculations restrained by NMR-derived distance and dihedral angle restraints, using a simulated annealing protocol. The generalized Born approximation was used to simulate solvent. The emergent structures indicated that the 1,N2-εdG-induced structural perturbation was localized at the X6·C19 base pair, and its 5′-neighbor T5·A20. Both 1,N2-εdG and the complementary dC adopted the anti conformation about the glycosyl bonds. The 1,N2-εdG adduct was inserted into the duplex but was shifted towards the minor groove as compared to dG in a normal Watson–Crick C·G base pair. The complementary cytosine was displaced toward the major groove. The 5′-neighbor T5·A20 base pair was destabilized with respect to Watson–Crick base pairing. The refined structure predicted a bend in the helical axis associated with the adduct site.
.jpg)
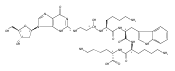
![Formamide,N-(2,6-diamino-1,4-dihydro-4-oxo-5-pyrimidinyl)-N-[(6aS,8R,9R,9aR)-1,2,3,6a,8,9,9a,11-octahydro-9-hydroxy-4-methoxy-1,11-dioxocyclopenta[c]furo[3',2':4,5]furo[2,3-h][1]benzopyran-8-yl]-](http://img.cochemist.com/ccimg/65400/65386-83-6.png)
![Formamide,N-(2,6-diamino-1,4-dihydro-4-oxo-5-pyrimidinyl)-N-[(6aS,8R,9R,9aR)-1,2,3,6a,8,9,9a,11-octahydro-9-hydroxy-4-methoxy-1,11-dioxocyclopenta[c]furo[3',2':4,5]furo[2,3-h][1]benzopyran-8-yl]-](http://img.cochemist.com/ccimg/65400/65386-83-6_b.png)
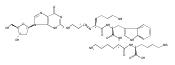
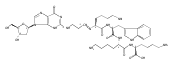
![Guanosine, 2'-deoxy-8-[(7-oxo-7H-benz[de]anthracen-3-yl)amino]-](http://img.cochemist.com/ccimg/604000/603994-31-6.png)
![Guanosine, 2'-deoxy-8-[(7-oxo-7H-benz[de]anthracen-3-yl)amino]-](http://img.cochemist.com/ccimg/604000/603994-31-6_b.png)
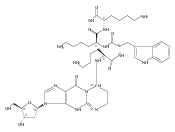
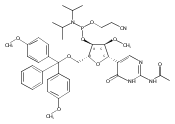
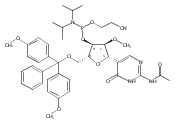
![N-(2-amino-3-methyl-3H-imidazo[4,5-f]quinolin-5-yl)-2'-deoxyguanosine](http://img.cochemist.com/ccimg/142100/142038-29-7.png)
![N-(2-amino-3-methyl-3H-imidazo[4,5-f]quinolin-5-yl)-2'-deoxyguanosine](http://img.cochemist.com/ccimg/142100/142038-29-7_b.png)






























































































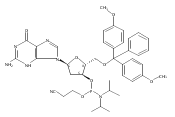
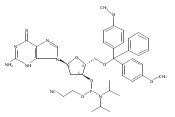


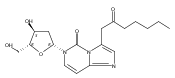

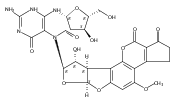





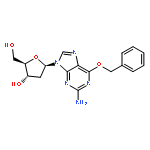
![O6-Benzyl-8-bromo-N9-[3’,5’-O-(1,1,3,3-tetrakis(isopropyl)-1,3-disiloxanediyl)--D-2'-deoxyribofuranosyl]guanine](http://img.cochemist.com/ccimg/328400/328394-26-9.png)
![O6-Benzyl-8-bromo-N9-[3’,5’-O-(1,1,3,3-tetrakis(isopropyl)-1,3-disiloxanediyl)--D-2'-deoxyribofuranosyl]guanine](http://img.cochemist.com/ccimg/328400/328394-26-9_b.png)

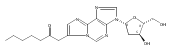


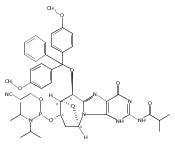
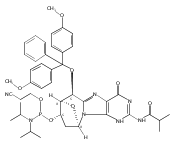

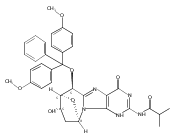







![Guanosine,2'-deoxy-8-[(3-methyl-3H-imidazo[4,5-f]quinolin-2-yl)amino]-](http://img.cochemist.com/ccimg/115800/115747-35-8.png)
![Guanosine,2'-deoxy-8-[(3-methyl-3H-imidazo[4,5-f]quinolin-2-yl)amino]-](http://img.cochemist.com/ccimg/115800/115747-35-8_b.png)


![2-AMINO-3-METHYL-3H-IMIDAZO[4,5-F]QUINOLINE](http://img.cochemist.com/ccimg/76200/76180-96-6.png)
![2-AMINO-3-METHYL-3H-IMIDAZO[4,5-F]QUINOLINE](http://img.cochemist.com/ccimg/76200/76180-96-6_b.png)
![Cyclopent[c]oxireno[4',5']furo[3',2':4,5]furo[2,3-h][1]benzopyran-1,10-dione,2,3,6a,7a,8a,8b-hexahydro-4-methoxy-, (6aS,7aS,8aR,8bR)-](http://img.cochemist.com/ccimg/42600/42583-46-0.png)
![Cyclopent[c]oxireno[4',5']furo[3',2':4,5]furo[2,3-h][1]benzopyran-1,10-dione,2,3,6a,7a,8a,8b-hexahydro-4-methoxy-, (6aS,7aS,8aR,8bR)-](http://img.cochemist.com/ccimg/42600/42583-46-0_b.png)
![3-methyl-3H-naphtho[1,2-d]imidazol-2-amine](http://img.cochemist.com/ccimg/35200/35199-58-7.png)
![3-methyl-3H-naphtho[1,2-d]imidazol-2-amine](http://img.cochemist.com/ccimg/35200/35199-58-7_b.png)




![7H-Pyrrolo[2,3-d]pyrimidin-4-amine,7-b-D-ribofuranosyl-](http://img.cochemist.com/ccimg/100/69-33-0.png)
![7H-Pyrrolo[2,3-d]pyrimidin-4-amine,7-b-D-ribofuranosyl-](http://img.cochemist.com/ccimg/100/69-33-0_b.png)

![3H-Imidazo[2,1-i]purine,3-(2-deoxy-b-D-erythro-pentofuranosyl)-](http://img.cochemist.com/ccimg/68500/68498-25-9.png)
![3H-Imidazo[2,1-i]purine,3-(2-deoxy-b-D-erythro-pentofuranosyl)-](http://img.cochemist.com/ccimg/68500/68498-25-9_b.png)
![7H-Pyrrolo[2,3-d]pyrimidin-4-amine,7-(2-deoxy-b-D-erythro-pentofuranosyl)-](http://img.cochemist.com/ccimg/60200/60129-59-1.png)
![7H-Pyrrolo[2,3-d]pyrimidin-4-amine,7-(2-deoxy-b-D-erythro-pentofuranosyl)-](http://img.cochemist.com/ccimg/60200/60129-59-1_b.png)


![9H-Imidazo[1,2-a]purin-9-one,3-(2-deoxy-b-D-erythro-pentofuranosyl)-3,4-dihydro-](http://img.cochemist.com/ccimg/109000/108929-11-9.png)
![9H-Imidazo[1,2-a]purin-9-one,3-(2-deoxy-b-D-erythro-pentofuranosyl)-3,4-dihydro-](http://img.cochemist.com/ccimg/109000/108929-11-9_b.png)