Co-reporter:Yinping Tian, Wenhui Lv, Xuewei Li, Congying Wang, Dayuan Wang, Peng G. Wang, Jin Jin, Jie Shen
Bioorganic & Medicinal Chemistry Letters 2017 Volume 27, Issue 13(Issue 13) pp:
Publication Date(Web):1 July 2017
DOI:10.1016/j.bmcl.2017.05.004
Among 18 human histone deacetylases (HDAC), HDAC11 is least studied. MS275, a benzamide HDAC inhibitor (HDACi), was stereotypically considered to selectively target Class I HDACs. We verified this slow-binding inhibitor also targeted HDAC11. In a traditional enzyme based assay, MS275 at low concentrations surprisingly behaved as an agonist. This was attributed to the poor stability of HDAC11 which lost 40% activity in 3 h at 37 °C. By adding 0.2 μM SAHA, HDAC11 activity was stabilized during the 3-h assay period. Since 0.2 μM SAHA inhibited 50% HDAC11 activity, the apparent IC50′ of MS275 was adjusted to the true IC50 = 0.65 μM. Finally, the new method demonstrated its superiority in one-dose-screening assays by decreasing false negative results. This work highlighted an optimized strategy to assay slow-binding inhibitors of unstable proteins with known fast-binding inhibitors. It should be especially useful in a hit-discovery stage to find moderate potent compounds.Download high-res image (78KB)Download full-size image
Co-reporter:Yinping Tian, Jin Jin, Congying Wang, Wenhui Lv, Xuewei Li, Xiaona Che, Yanchao Gong, Yanjun Li, Quanli Li, Jingli Hou, Peng G. Wang, Jie Shen
Bioorganic & Medicinal Chemistry Letters 2016 Volume 26(Issue 10) pp:2434-2437
Publication Date(Web):15 May 2016
DOI:10.1016/j.bmcl.2016.03.116
This work demonstrated the high efficiency of a sub-milligram-synthesis based medicinal chemistry method. Totally 72 compounds, consisting a tri-substituted pyrrolidine core, were prepared. Around 0.1 mg of each compound was solid-phase synthesized. Based on the additive property of UV absorptions of unconjugated chromophores of a molecule, these compounds were quantified by UV measurement. A hit, whose IC50 value was 1.2 μM in HDAC11 inhibition assays, highlights the applicability of the approach reported here in future optimization works.
Co-reporter:Xuefeng Cao, Wenpeng Zhang, Xu Yan, Zhi Huang, Zhenqing Zhang, Peng Wang, Jie Shen
Bioorganic & Medicinal Chemistry Letters 2016 Volume 26(Issue 9) pp:2170-2173
Publication Date(Web):1 May 2016
DOI:10.1016/j.bmcl.2016.03.065
Poor pharmacokinetic stability is one of the issues of O-glucoside SGLT2 inhibitors in clinical trials, hence C-glucoside inhibitors have been developed and extensively applied. Herein, we provided an alternative approach to improve the pharmacokinetic stability of such inhibitors. Nine derivatives of Sergliflozin-A with modifications on the O-glucoside fragment were prepared, among which the 4-O-methyl derivative exhibited similar pharmacodynamics potency in excreted glucose urine test. Most attractively, significantly increased pharmacokinetic stability was observed for 4-O-methyl derivative of O-glucosides. This work proved that modification on the O-glucoside fragment could be a promising approach to the future SGLT2 inhibitor design.
Co-reporter:Dr. Tiehai Li;Hui Ye;Xuefeng Cao;Jiajia Wang;Yonghui Liu;Lifei Zhou;Qiang Liu;Wenjun Wang;Dr. Jie Shen;Dr. Wei Zhao; Peng Wang
ChemMedChem 2014 Volume 9( Issue 5) pp:1071-1080
Publication Date(Web):
DOI:10.1002/cmdc.201400019
Abstract
The anticoagulant pentasaccharide fondaparinux was synthesized using an improved and optimized synthetic strategy including a convergent [3+2] coupling approach, orthogonal protecting groups and various glycosyl donors. The new methods of glycosylation were also used for controlling the stereochemical configuration and improving the yield of the glycosylation. In addition, HPLC and NMR methods to monitor the process of total synthesis of fondaparinux were employed. This work provides a comprehensive elaboration for the synthesis and analysis of fondaparinux based on related literature, as well as abundant information for the synthesis of heparin-like oligosaccharides.
Co-reporter:Hui Ye, Ruihua Liu, Dongmei Li, Yonghui Liu, Haixin Yuan, Weikang Guo, Lifei Zhou, Xuefeng Cao, Hongqi Tian, Jie Shen, and Peng George Wang
Organic Letters 2013 Volume 15(Issue 1) pp:18-21
Publication Date(Web):December 14, 2012
DOI:10.1021/ol3028708
A facile approach to the diazotransfer reagent of imidazole-1-sulfonyl azide was reported. The procedure was well optimized to clarify potential explosion risks. A high production yield as well as small batch variation was achieved even without careful pretreatment of reagents and solvents. HPLC and NMR methods to monitor the process were provided. These features made this protocol suitable for large scale preparation in academia and industry as well.
Co-reporter:Yang Zou, Mengyang Xue, Wenjun Wang, Li Cai, Leilei Chen, Jun Liu, Peng George Wang, Jie Shen, Min Chen
Carbohydrate Research 2013 Volume 373() pp:76-81
Publication Date(Web):24 May 2013
DOI:10.1016/j.carres.2013.03.005
•A UTP-glucose-1-phosphate uridylyltransferase (GalUSpe4) and a galactokinase (GalKSpe4) were cloned from Streptococcus pneumoniae TIGR4 and were successfully used to synthesize UDP-galactose (UDP-Gal), UDP-glucose (UDP-Glc), and their derivatives in an efficient one-pot reaction system.•Six unnatural UDP-Gal derivatives, including UDP-2-deoxy-Galactose and UDP-GalN3 which were not accepted by other approach, can be synthesized efficiently in a one pot fashion.•This is the first time it has been reported that UDP-Glc can be synthesized in a simple one-pot three-enzyme synthesis reaction system.A UTP-glucose-1-phosphate uridylyltransferase (SpGalU) and a galactokinase (SpGalK) were cloned from Streptococcus pneumoniae TIGR4 and were successfully used to synthesize UDP-galactose (UDP-Gal), UDP-glucose (UDP-Glc), and their derivatives in an efficient one-pot reaction system. The reaction conditions for the one-pot multi-enzyme synthesis were optimized and nine UDP-Glc/Gal derivatives were synthesized. Using this system, six unnatural UDP-Gal derivatives, including UDP-2-deoxy-Galactose and UDP-GalN3 which were not accepted by other approach, can be synthesized efficiently in a one pot fashion. More interestingly, this is the first time it has been reported that UDP-Glc can be synthesized in a simpler one-pot three-enzyme synthesis reaction system.Graphical abstractEnzymatic synthesis of UDP-Glc/Gal and their derivatives by three-enzyme reaction system.
Co-reporter:Minghui Li;Xu Liu;Xiaolin Sun;Zhenhua Wang;Weikang Guo;Fanlei Hu
Inflammation 2013 Volume 36( Issue 4) pp:888-896
Publication Date(Web):2013 August
DOI:10.1007/s10753-013-9616-0
The purpose of this study is to investigate the therapeutic effects of a novel histone deacetylase inhibitor (HDACi), NK-HDAC-1, on collagen-induced arthritis (CIA) and pathogenic fibroblast-like synoviocytes (FLSs) from patients with rheumatoid arthritis (RA). The proliferation and apoptosis of FLSs treated with NK-HDAC-1 were evaluated by flow cytometry and fluorescence staining. The effect of NK-HDAC-1 treatment on pro-inflammatory cytokine production was determined by ELISA. CIA was established in DBA/1 mice, and NK-HDAC-1 or vehicle was administered daily after the onset of arthritis. Clinical and histological scores were calculated to assess the therapeutic efficacy of NK-HDAC-1. NK-HDAC-1 significantly inhibited the proliferation of FLSs through cell cycle arrest at the G2/M checkpoint and enhanced apoptosis of FLSs. The activity of caspases was increased during NK-HDAC-1 treatment. IL-6 production by FLSs was also suppressed by NK-HDAC-1. Furthermore, the oral administration of NK-HDAC-1 significantly enhanced synoviocyte apoptosis in vivo and inhibited CIA progression. Compared with subcroylanilide hydroxamic acid which exhibited moderate prophylactic efficacy, NK-HDAC-1 demonstrated therapeutic efficacy in CIA. NK-HDAC-1 is a novel HDACi that may ameliorate inflammatory arthritis by regulating the activation, apoptosis, and inflammatory responses of FLSs. This is the first study to support that NK-HDAC-1 may be a potential therapeutic agent for RA.
Co-reporter:Guoxian Gu, Huihui Wang, Pi Liu, Chenzeng Fu, Zhonghua Li, Xuefeng Cao, Yunping Li, Qinghong Fang, Feng Xu, Jie Shen and Peng George Wang
Chemical Communications 2012 vol. 48(Issue 22) pp:2788-2790
Publication Date(Web):08 Feb 2012
DOI:10.1039/C1CC15851A
Novel bisaryl maleimide derivatives to mimic natural kinase inhibitors were prepared through click chemistry. A highly selective hit was discovered in a 124-kinase-assay, and docking studies revealed a π–π stacking interaction with the Phe67 at the P-loop of GSK-3β kinase.
Co-reporter:Yang Zou, Wenjun Wang, Li Cai, Leilei Chen, Mengyang Xue, Xiaomei Zhang, Jie Shen, Min Chen
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 10) pp:3540-3543
Publication Date(Web):15 May 2012
DOI:10.1016/j.bmcl.2012.03.095
Galactokinases (GalKs) have attracted significant research attention for their potential applications in the enzymatic synthesis of unique sugar phosphates. The galactokinase (GalKSpe4) cloned from Streptococcus pneumoniae TIGR4 presents a remarkably broad substrate range including 14 diverse natural and unnatural sugars. TLC and MS studies revealed that GalKSpe4 had relaxed activity towards galactose derivatives with modifications on the C-6, 4- or 2-positions. Additionally, GalKSpe4 can also tolerate glucose while glucose derivatives with modifications on the C-6, 4- or 2-positions were unacceptable. More interestingly, GalKSpe4 can phosphorylate l-mannose in moderate yield (43%), while other l-sugars such as l-Gal cannot be recognized by this enzyme. These results are very significant because there is rarely enzyme reported that can phosphorylate such uncommon substrates as l-mannose.
Co-reporter:Jingli Hou, Congran Feng, Zhonghua Li, Qinghong Fang, Huihui Wang, Guoxian Gu, Yikang Shi, Pi Liu, Feng Xu, Zheng Yin, Jie Shen, Peng Wang
European Journal of Medicinal Chemistry 2011 Volume 46(Issue 8) pp:3190-3200
Publication Date(Web):August 2011
DOI:10.1016/j.ejmech.2011.04.027
Previously, we reported a click-chemistry based approach to the synthesis of a novel class of histone deacetylase (HDAC) inhibitors [1]. The lead compound NSC746457 was found to be as potent as SAHA (Vorinostat). Further optimization of NSC746457 by using the HDAC2-TSA crystal structure is described herein. Docking of NSC746457 into HDAC2 binding domain suggested that the hydrophobic residue Phe210 flanking the cap-group binding-motif could be exploited for structural optimization. Substitution on the methylene group of cinnamic cap region led to identification of more potent HDAC inhibitors: isopropyl derivative 5 and tert-butyl derivative 6, with an IC50 value of 22 nM and 18 nM, respectively.Optimization of lead compound NSC746457 led to identification of two compounds with potent activity against HDAC enzymes.Highlights► The lead compound NSC746457 was optimized using the HDAC2-TSA crystal structure. ► Docking the NSC746457 into the histone deacetylase 2 suggested that Phe210 could be exploited for structure optimization. ► A series of novel HDAC inhibitors was designed and synthesized and two compounds showed strong activity against HDAC enzymes.
Co-reporter:Guoxian Gu, Huihui Wang, Pi Liu, Chenzeng Fu, Zhonghua Li, Xuefeng Cao, Yunping Li, Qinghong Fang, Feng Xu, Jie Shen and Peng George Wang
Chemical Communications 2012 - vol. 48(Issue 22) pp:NaN2790-2790
Publication Date(Web):2012/02/08
DOI:10.1039/C1CC15851A
Novel bisaryl maleimide derivatives to mimic natural kinase inhibitors were prepared through click chemistry. A highly selective hit was discovered in a 124-kinase-assay, and docking studies revealed a π–π stacking interaction with the Phe67 at the P-loop of GSK-3β kinase.

![A-d-glucopyranoside, Methyl 2-azido-4-o-[2-o-benzoyl-6-methyl-3-o-(phenylmethyl)-a-l-idopyranuronosyl]-2-deoxy-3-o-(phenylmethyl)-, 6-benzoate](http://img.cochemist.com/ccimg/501100/501089-97-0.png)
![A-d-glucopyranoside, Methyl 2-azido-4-o-[2-o-benzoyl-6-methyl-3-o-(phenylmethyl)-a-l-idopyranuronosyl]-2-deoxy-3-o-(phenylmethyl)-, 6-benzoate](http://img.cochemist.com/ccimg/501100/501089-97-0_b.png)
![2-Penten-4-ynamide, N-[(4-methoxyphenyl)methoxy]-, (2E)-](/data/chemimg/141300/1312592-14-5.png)
![2-Penten-4-ynamide, N-[(4-methoxyphenyl)methoxy]-, (2E)-](/data/chemimg/141300/1312592-14-5_b.png)

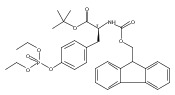





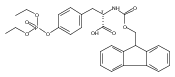

![2-Methyl-2-propanyl N-[(9H-fluoren-9-ylmethoxy)carbonyl]-L-tyrosinate](http://img.cochemist.com/ccimg/133900/133852-23-0.png)
![2-Methyl-2-propanyl N-[(9H-fluoren-9-ylmethoxy)carbonyl]-L-tyrosinate](http://img.cochemist.com/ccimg/133900/133852-23-0_b.png)
![[(2S,3R,4S,5R)-3,5,6-triacetoxy-4-benzyloxy-tetrahydropyran-2-yl]methyl acetate](http://img.cochemist.com/ccimg/118800/118711-49-2.png)
![[(2S,3R,4S,5R)-3,5,6-triacetoxy-4-benzyloxy-tetrahydropyran-2-yl]methyl acetate](http://img.cochemist.com/ccimg/118800/118711-49-2_b.png)


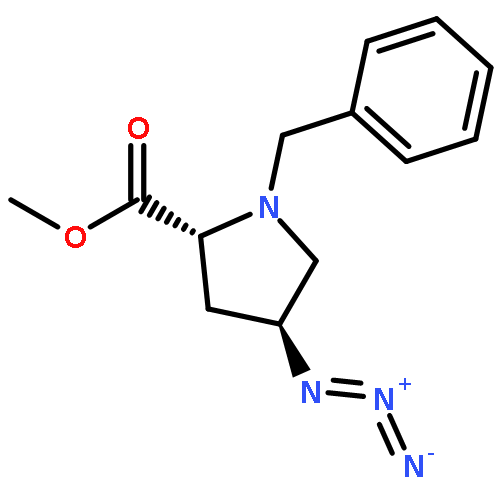
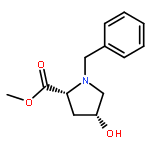
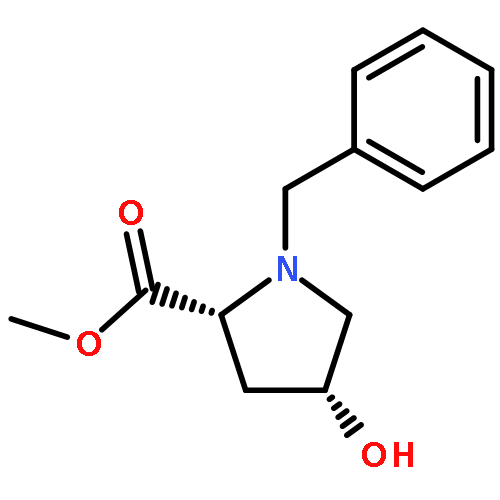


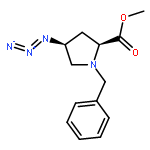







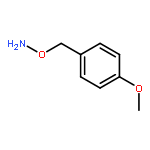
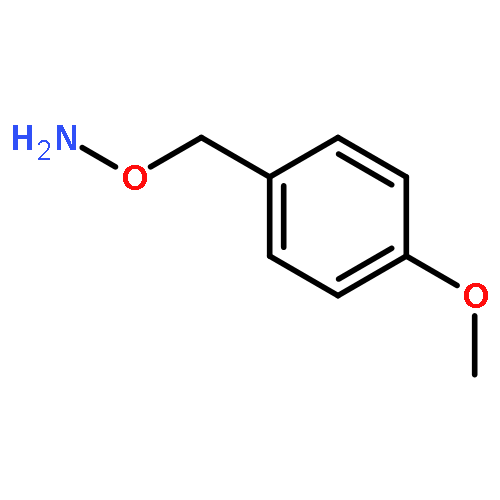


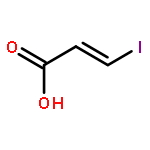
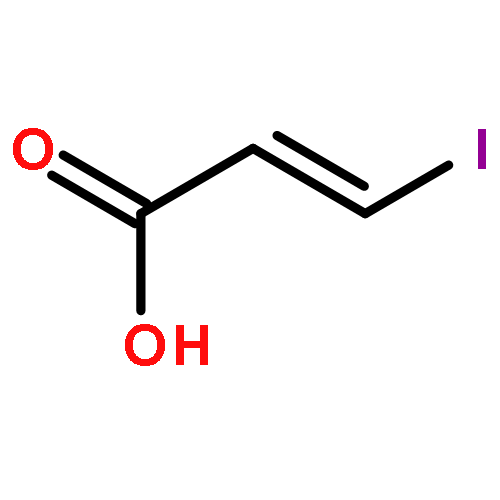
![1H-Isoindole-1,3(2H)-dione, 2-[(4-methoxyphenyl)methoxy]-](http://img.cochemist.com/ccimg/2100/2014-61-1.png)
![1H-Isoindole-1,3(2H)-dione, 2-[(4-methoxyphenyl)methoxy]-](http://img.cochemist.com/ccimg/2100/2014-61-1_b.png)