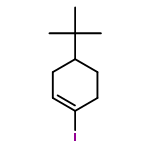
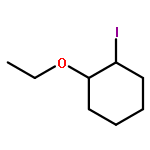
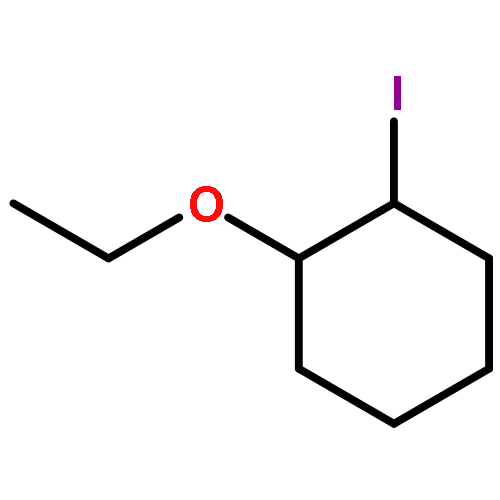
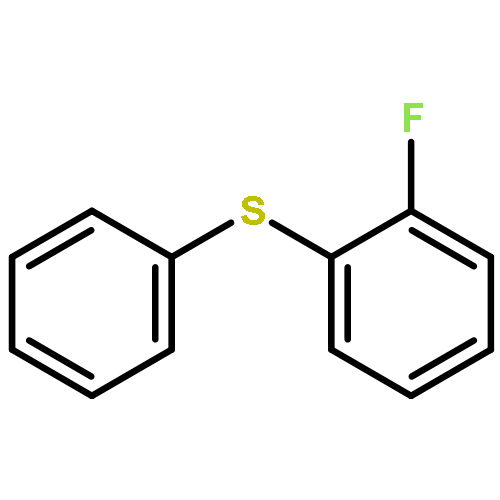
Co-reporter:Wei Zhao, Ryan P. Wurz, Jonas C. Peters, and Gregory C. Fu
Journal of the American Chemical Society September 6, 2017 Volume 139(Issue 35) pp:12153-12153
Publication Date(Web):August 25, 2017
DOI:10.1021/jacs.7b07546
The Curtius rearrangement is a classic, powerful method for converting carboxylic acids into protected amines, but its widespread use is impeded by safety issues (the need to handle azides). We have developed an alternative to the Curtius rearrangement that employs a copper catalyst in combination with blue-LED irradiation to achieve the decarboxylative coupling of aliphatic carboxylic acid derivatives (specifically, readily available N-hydroxyphthalimide esters) to afford protected amines under mild conditions. This C–N bond-forming process is compatible with a wide array of functional groups, including an alcohol, aldehyde, epoxide, indole, nitroalkane, and sulfide. Control reactions and mechanistic studies are consistent with the hypothesis that copper species are engaged in both the photochemistry and the key bond-forming step, which occurs through out-of-cage coupling of an alkyl radical.
Co-reporter:Jun Myun Ahn, Tanvi S. Ratani, Kareem I. Hannoun, Gregory C. Fu, and Jonas C. Peters
Journal of the American Chemical Society September 13, 2017 Volume 139(Issue 36) pp:12716-12716
Publication Date(Web):August 17, 2017
DOI:10.1021/jacs.7b07052
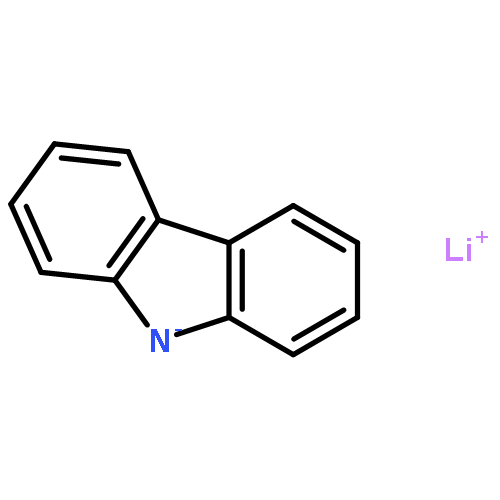
We have recently reported that a variety of couplings of nitrogen, sulfur, oxygen, and carbon nucleophiles with organic halides can be achieved under mild conditions (−40 to 30 °C) through the use of light and a copper catalyst. Insight into the various mechanisms by which these reactions proceed may enhance our understanding of chemical reactivity and facilitate the development of new methods. In this report, we apply an array of tools (EPR, NMR, transient absorption, and UV–vis spectroscopy; ESI–MS; X-ray crystallography; DFT calculations; reactivity, stereochemical, and product studies) to investigate the photoinduced, copper-catalyzed coupling of carbazole with alkyl bromides. Our observations are consistent with pathways wherein both an excited state of the copper(I) carbazolide complex ([CuI(carb)2]−) and an excited state of the nucleophile (Li(carb)) can serve as photoreductants of the alkyl bromide. The catalytically dominant pathway proceeds from the excited state of Li(carb), generating a carbazyl radical and an alkyl radical. The cross-coupling of these radicals is catalyzed by copper via an out-of-cage mechanism in which [CuI(carb)2]− and [CuII(carb)3]− (carb = carbazolide), both of which have been identified under coupling conditions, are key intermediates, and [CuII(carb)3]− serves as the persistent radical that is responsible for predominant cross-coupling. This study underscores the versatility of copper(II) complexes in engaging with radical intermediates that are generated by disparate pathways, en route to targeted bond constructions.
Co-reporter:Tzu-Pin Lin and Jonas C. Peters
Journal of the American Chemical Society October 1, 2014 Volume 136(Issue 39) pp:13672-13683
Publication Date(Web):September 2, 2014
DOI:10.1021/ja504667f
New approaches toward the generation of late first-row metal catalysts that efficiently facilitate two-electron reductive transformations (e.g., hydrogenation) more typical of noble-metal catalysts is an important goal. Herein we describe the synthesis of a structurally unusual S = 1 bimetallic Co complex, [(CyPBP)CoH]2 (1), supported by bis(phosphino)boryl and bis(phosphino)hydridoborane ligands. This complex reacts reversibly with a second equivalent of H2 (1 atm) and serves as an olefin hydrogenation catalyst under mild conditions (room temperature, 1 atm H2). A bimetallic Co species is invoked in the rate-determining step of the catalysis according to kinetic studies. A structurally related NiINiI dimer, [(PhPBP)Ni]2 (3), has also been prepared. Like Co catalyst 1, Ni complex 3 displays reversible reactivity toward H2, affording the bimetallic complex [(PhPBHP)NiH]2 (4). This reversible behavior is unprecedented for NiI species and is attributed to the presence of a boryl–Ni bond. Lastly, a series of monomeric (tBuPBP)NiX complexes (X = Cl (5), OTf (6), H (7), OC(H)O (8)) have been prepared. The complex (tBuPBP)NiH (7) shows enhanced catalytic olefin hydrogenation activity when directly compared with its isoelectronic/isostructural analogues where the boryl unit is substituted by a phenyl or amine donor, a phenomenon that we posit is related to the strong trans influence exerted by the boryl ligand.
Co-reporter:Niklas B. Thompson, Michael T. Green, and Jonas C. Peters
Journal of the American Chemical Society November 1, 2017 Volume 139(Issue 43) pp:15312-15312
Publication Date(Web):October 9, 2017
DOI:10.1021/jacs.7b09364
Terminal iron nitrides (Fe≡N) have been proposed as intermediates of (bio)catalytic nitrogen fixation, yet experimental evidence to support this hypothesis has been lacking. In particular, no prior synthetic examples of terminal Fe≡N species have been derived from N2. Here we show that a nitrogen-fixing Fe–N2 catalyst can be protonated to form a neutral Fe(NNH2) hydrazido(2−) intermediate, which, upon further protonation, heterolytically cleaves the N–N bond to release [FeIV≡N]+ and NH3. These observations provide direct evidence for the viability of a Chatt-type (distal) mechanism for Fe-mediated N2-to-NH3 conversion. The physical oxidation state range of the Fe complexes in this transformation is buffered by covalency with the ligand, a feature of possible relevance to catalyst design in synthetic and natural systems that facilitate multiproton/multielectron redox processes.
Co-reporter:Matthew J. Chalkley, Trevor J. Del Castillo, Benjamin D. Matson, Joseph P. Roddy, and Jonas C. Peters
ACS Central Science March 22, 2017 Volume 3(Issue 3) pp:217-217
Publication Date(Web):February 14, 2017
DOI:10.1021/acscentsci.7b00014
We have recently reported on several Fe catalysts for N2-to-NH3 conversion that operate at low temperature (−78 °C) and atmospheric pressure while relying on a very strong reductant (KC8) and acid ([H(OEt2)2][BArF4]). Here we show that our original catalyst system, P3BFe, achieves both significantly improved efficiency for NH3 formation (up to 72% for e– delivery) and a comparatively high turnover number for a synthetic molecular Fe catalyst (84 equiv of NH3 per Fe site), when employing a significantly weaker combination of reductant (Cp*2Co) and acid ([Ph2NH2][OTf] or [PhNH3][OTf]). Relative to the previously reported catalysis, freeze-quench Mössbauer spectroscopy under turnover conditions suggests a change in the rate of key elementary steps; formation of a previously characterized off-path borohydrido–hydrido resting state is also suppressed. Theoretical and experimental studies are presented that highlight the possibility of protonated metallocenes as discrete PCET reagents under the present (and related) catalytic conditions, offering a plausible rationale for the increased efficiency at reduced driving force of this Fe catalyst system.
Co-reporter:Jonathan Rittle and Jonas C. Peters
Journal of the American Chemical Society March 1, 2017 Volume 139(Issue 8) pp:3161-3161
Publication Date(Web):January 31, 2017
DOI:10.1021/jacs.6b12861
Fe-mediated biological nitrogen fixation is thought to proceed via either a sequence of proton and electron transfer steps, concerted H atom transfer steps, or some combination thereof. Regardless of the specifics and whether the intimate mechanism for N2-to-NH3 conversion involves a distal pathway, an alternating pathway, or some hybrid of these limiting scenarios, Fe–NxHy intermediates are implicated that feature reactive N–H bonds. Thermodynamic knowledge of the N–H bond strengths of such species is scant, and is especially difficult to obtain for the most reactive early stage candidate intermediates (e.g., Fe–N═NH, Fe═N–NH2, Fe–NH═NH). Such knowledge is essential to considering various mechanistic hypotheses for biological (and synthetic) nitrogen fixation and to the rational design of improved synthetic N2 fixation catalysts. We recently reported several reactive complexes derived from the direct protonation of Fe–N2 and Fe–CN species at the terminal N atom (e.g., Fe═N–NH2, Fe–C≡NH, Fe≡C–NH2). These same Fe–N2 and Fe–CN systems are functionally active for N2-to-NH3 and CN-to-CH4/NH3 conversion, respectively, when subjected to protons and electrons, and hence provide an excellent opportunity for obtaining meaningful N–H bond strength data. We report here a combined synthetic, structural, and spectroscopic/analytic study to estimate the N–H bond strengths of several species of interest. We assess the reactivity profiles of species featuring reactive N–H bonds and estimate their homolytic N–H bond enthalpies (BDEN−H) via redox and acidity titrations. Very low N–H bond dissociation enthalpies, ranging from 65 (Fe–C≡NH) to ≤37 kcal/mol (Fe–N═NH), are determined. The collective data presented herein provide insight into the facile reactivity profiles of early stage protonated Fe–N2 and Fe–CN species.
Co-reporter:Meaghan M. Deegan and Jonas C. Peters
Journal of the American Chemical Society February 22, 2017 Volume 139(Issue 7) pp:2561-2561
Publication Date(Web):February 3, 2017
DOI:10.1021/jacs.6b12444
One of the major challenges associated with developing molecular Fischer–Tropsch catalysts is the design of systems that promote the formation of C–H bonds from H2 and CO while also facilitating the release of the resulting CO-derived organic products. To this end, we describe the synthesis of reduced iron-hydride/carbonyl complexes that enable an electrophile-promoted hydride migration process, resulting in the reduction of coordinated CO to a siloxymethyl (LnFe-CH2OSiMe3) group. Intramolecular hydride-to-CO migrations are extremely rare, and to our knowledge the system described herein is the first example where such a process can be accessed from a thermally stable M(CO)(H) complex. Further addition of H2 to LnFe-CH2OSiMe3 releases CH3OSiMe3, demonstrating net four-electron reduction of CO to CH3OSiMe3 at a single Fe site.
Co-reporter:Sidney E. Creutz
Chemical Science (2010-Present) 2017 vol. 8(Issue 3) pp:2321-2328
Publication Date(Web):2017/02/28
DOI:10.1039/C6SC04805F
Hydrogen bonding and other types of secondary-sphere interactions are ubiquitous in metalloenzyme active sites and are critical to the transformations they mediate. Exploiting secondary sphere interactions in synthetic catalysts to study the role(s) they might play in biological systems, and to develop increasingly efficient catalysts, is an important challenge. Whereas model studies in this broad context are increasingly abundant, as yet there has been relatively little progress in the area of synthetic catalysts for nitrogen fixation that incorporate secondary sphere design elements. Herein we present our first study of Fe–NxHy complexes supported by new tris(phosphine)silyl ligands, abbreviated as [SiPNMe3] and [SiPiPr2PNMe], that incorporate remote tertiary amine hydrogen-bond acceptors within a tertiary phosphine/amine 6-membered ring. These remote amine sites facilitate hydrogen-bonding interactions via a boat conformation of the 6-membered ring when certain nitrogenous substrates (e.g., NH3 and N2H4) are coordinated to the apical site of a trigonal bipyramidal iron complex, and adopt a chair conformation when no H-bonding is possible (e.g., N2). Countercation binding at the cyclic amine is also observed for anionic {Fe–N2}− complexes. Reactivity studies in the presence of proton/electron sources show that the incorporated amine functionality leads to rapid generation of catalytically inactive Fe–H species, thereby substantiating a hydride termination pathway that we have previously proposed deactivates catalysts of the type [EPR3]FeN2 (E = Si, C).
Co-reporter:Trixia M. Buscagan;Dr. Paul H. Oyala; Jonas C. Peters
Angewandte Chemie 2017 Volume 129(Issue 24) pp:7025-7030
Publication Date(Web):2017/06/06
DOI:10.1002/ange.201703244
AbstractBridging iron hydrides are proposed to form at the active site of MoFe-nitrogenase during catalytic dinitrogen reduction to ammonia and may be key in the binding and activation of N2 via reductive elimination of H2. This possibility inspires the investigation of well-defined molecular iron hydrides as precursors for catalytic N2-to-NH3 conversion. Herein, we describe the synthesis and characterization of new P2P′PhFe(N2)(H)x systems that are active for catalytic N2-to-NH3 conversion. Most interestingly, we show that the yields of ammonia can be significantly increased if the catalysis is performed in the presence of mercury lamp irradiation. Evidence is provided to suggest that photo-elimination of H2 is one means by which the enhanced activity may arise.
Co-reporter:Trixia M. Buscagan;Dr. Paul H. Oyala; Jonas C. Peters
Angewandte Chemie International Edition 2017 Volume 56(Issue 24) pp:6921-6926
Publication Date(Web):2017/06/06
DOI:10.1002/anie.201703244
AbstractBridging iron hydrides are proposed to form at the active site of MoFe-nitrogenase during catalytic dinitrogen reduction to ammonia and may be key in the binding and activation of N2 via reductive elimination of H2. This possibility inspires the investigation of well-defined molecular iron hydrides as precursors for catalytic N2-to-NH3 conversion. Herein, we describe the synthesis and characterization of new P2P′PhFe(N2)(H)x systems that are active for catalytic N2-to-NH3 conversion. Most interestingly, we show that the yields of ammonia can be significantly increased if the catalysis is performed in the presence of mercury lamp irradiation. Evidence is provided to suggest that photo-elimination of H2 is one means by which the enhanced activity may arise.
Co-reporter:Jonathan Rittle
Journal of the American Chemical Society 2016 Volume 138(Issue 12) pp:4243-4248
Publication Date(Web):March 3, 2016
DOI:10.1021/jacs.6b01230
Biological N2 fixation to NH3 may proceed at one or more Fe sites in the active-site cofactors of nitrogenases. Modeling individual e–/H+ transfer steps of iron-ligated N2 in well-defined synthetic systems is hence of much interest but remains a significant challenge. While iron complexes have been recently discovered that catalyze the formation of NH3 from N2, mechanistic details remain uncertain. Herein, we report the synthesis and isolation of a diamagnetic, 5-coordinate Fe═NNH2+ species supported by a tris(phosphino)silyl ligand via the direct protonation of a terminally bound Fe-N2– complex. The Fe═NNH2+ complex is redox-active, and low-temperature spectroscopic data and DFT calculations evidence an accumulation of significant radical character on the hydrazido ligand upon one-electron reduction to S = 1/2 Fe═NNH2. At warmer temperatures, Fe═NNH2 rapidly converts to an iron hydrazine complex, Fe-NH2NH2+, via the additional transfer of proton and electron equivalents in solution. Fe-NH2NH2+ can liberate NH3, and the sequence of reactions described here hence demonstrates that an iron site can shuttle from a distal intermediate (Fe═NNH2+) to an alternating intermediate (Fe-NH2NH2+) en route to NH3 liberation from N2. It is interesting to consider the possibility that similar hybrid distal/alternating crossover mechanisms for N2 reduction may be operative in biological N2 fixation.
Co-reporter:Trevor J. Del Castillo; Niklas B. Thompson
Journal of the American Chemical Society 2016 Volume 138(Issue 16) pp:5341-5350
Publication Date(Web):March 29, 2016
DOI:10.1021/jacs.6b01706
The mechanisms of the few known molecular nitrogen-fixing systems, including nitrogenase enzymes, are of much interest but are not fully understood. We recently reported that Fe–N2 complexes of tetradentate P3E ligands (E = B, C) generate catalytic yields of NH3 under an atmosphere of N2 with acid and reductant at low temperatures. Here we show that these Fe catalysts are unexpectedly robust and retain activity after multiple reloadings. Nearly an order of magnitude improvement in yield of NH3 for each Fe catalyst has been realized (up to 64 equiv of NH3 produced per Fe for P3B and up to 47 equiv for P3C) by increasing acid/reductant loading with highly purified acid. Cyclic voltammetry shows the apparent onset of catalysis at the P3BFe–N2/P3BFe–N2– couple and controlled-potential electrolysis of P3BFe+ at −45 °C demonstrates that electrolytic N2 reduction to NH3 is feasible. Kinetic studies reveal first-order rate dependence on Fe catalyst concentration (P3B), consistent with a single-site catalyst model. An isostructural system (P3Si) is shown to be appreciably more selective for hydrogen evolution. In situ freeze-quench Mössbauer spectroscopy during turnover reveals an iron–borohydrido–hydride complex as a likely resting state of the P3BFe catalyst system. We postulate that hydrogen-evolving reaction activity may prevent iron hydride formation from poisoning the P3BFe system. This idea may be important to consider in the design of synthetic nitrogenases and may also have broader significance given that intermediate metal hydrides and hydrogen evolution may play a key role in biological nitrogen fixation.
Co-reporter:Pengfei Huo, Christopher Uyeda, Jason D. Goodpaster, Jonas C. Peters, and Thomas F. Miller III
ACS Catalysis 2016 Volume 6(Issue 9) pp:6114
Publication Date(Web):July 26, 2016
DOI:10.1021/acscatal.6b01387
A central challenge in the development of inorganic hydrogen evolution catalysts is to avoid deleterious coupling between the energetics of metal site reduction and the kinetics of metal hydride formation. In this work, we combine theoretical and experimental methods to investigate cobalt diimine-dioxime catalysts that show promise for achieving this aim by introducing an intramolecular proton shuttle via a pyridyl pendant group. Using over 200 coupled-cluster-level electronic structure calculations of the Co-based catalyst with a variety of pyridyl substituents, the energetic and kinetic barriers to hydrogen formation are investigated, revealing nearly complete decoupling of the energetics of Co reduction and the kinetics of intramolecular Co hydride formation. These calculations employ recently developed quantum embedding methods that allow for local regions of a molecule to be described using high-accuracy wavefunction methods (such as CCSD(T)), thus overcoming significant errors in the DFT-level description of transition-metal complexes. Experimental synthesis and cyclic voltammetry of the methyl-substituted form of the catalyst indicate that protonation of the pendant group leaves the Co reduction potential unchanged, which is consistent with the theoretical prediction that these catalysts can successfully decouple the electronic structures of the transition-metal and ligand-protonation sites. Additional computational analysis indicates that introduction of the pyridyl pendant group enhances the favorability of intramolecular proton shuttling in these catalysts by significantly reducing the energetic barrier for metal hydride formation relative to previously studied cobalt diimine-dioxime catalysts. These results demonstrate a promising proof of principle for achieving uncoupled and locally tunable intramolecular charge-transfer events in the context of homogeneous transition-metal catalysts.Keywords: cobalt diimine-dioxime catalysts; coupled-cluster theory; density functional theory; electrochemistry; embedding; hydrogen evolution; proton shuttle
Co-reporter:Miles W. Johnson, Kareem I. Hannoun, Yichen Tan, Gregory C. Fu and Jonas C. Peters
Chemical Science 2016 vol. 7(Issue 7) pp:4091-4100
Publication Date(Web):24 Feb 2016
DOI:10.1039/C5SC04709A
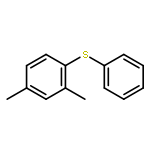
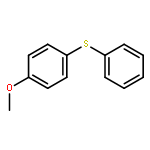
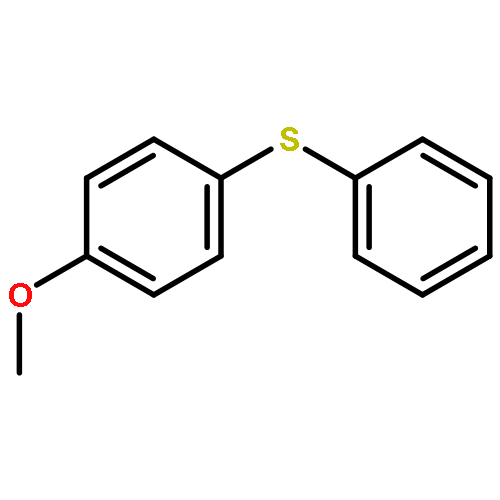
Photoinduced, copper-catalyzed cross-coupling can offer a complementary approach to thermal (non-photoinduced) methods for generating C–X (X = C, N, O, S, etc.) bonds. In this report, we describe the first detailed mechanistic investigation of one of the processes that we have developed, specifically, the (stoichiometric) coupling of a copper–thiolate with an aryl iodide. In particular, we focus on the chemistry of a discrete [CuI(SAr)2]− complex (Ar = 2,6-dimethylphenyl), applying a range of techniques, including ESI-MS, cyclic voltammetry, transient luminescence spectroscopy, optical spectroscopy, DFT calculations, Stern–Volmer analysis, EPR spectroscopy, actinometry, and reactivity studies. The available data are consistent with the viability of a pathway in which photoexcited [CuI(SAr)2]−* serves as an electron donor to an aryl iodide to afford an aryl radical, which then reacts in cage with the newly generated copper(II)–thiolate to furnish the cross-coupling product in a non-chain process.
Co-reporter:Sidney E. Creutz and Jonas C. Peters
Inorganic Chemistry 2016 Volume 55(Issue 8) pp:3894-3906
Publication Date(Web):April 4, 2016
DOI:10.1021/acs.inorgchem.6b00066
Low-coordinate transition-metal complexes that undergo spin crossover remain rare. We report here a series of four-coordinate, pseudo-tetrahedral P3FeII–X complexes supported by tris(phosphine)borate P3 ([PhBP3R]−) and phosphiniminato X-type ligands (−N═PR3′) that, in combination, tune the spin-crossover behavior of the system. Most of the reported iron complexes undergo spin crossover at temperatures near or above room temperature in solution and in the solid state. The change in spin state coincides with a significant change in the degree of π-bonding between Fe and the bound N atom of the phosphiniminato ligand. Spin crossover is accompanied by striking changes in the ultraviolet–visible (UV-vis) absorption spectra, which allows for quantitative modeling of the thermodynamic parameters of the spin equilibria. These spin equilibria have also been studied by numerous techniques including paramagnetic nuclear magnetic resonance (NMR), infrared, and Mössbauer spectroscopies; X-ray crystallography; and solid-state superconducting quantum interference device (SQUID) magnetometry. These studies allow qualitative correlations to be made between the steric and electronic properties of the ligand substituents and the enthalpy and entropy changes associated with the spin equilibria.
Co-reporter:Charles C. L. McCrory;Nathaniel K. Szymczak
Electrocatalysis 2016 Volume 7( Issue 1) pp:87-96
Publication Date(Web):2016 January
DOI:10.1007/s12678-015-0281-y
Molecular cobalt and nickel complexes are among the most promising homogeneous systems for electrocatalytic hydrogen evolution. However, there has been little exploration into the effect of gaseous co-additives such as CO and H2, which may be present in operating hydrogen-evolving or carbon-dioxide reduction systems, on the performance of these molecular electrocatalysts. In this report, we investigate the electrocatalytic activity of six cobalt and nickel complexes supported by tetraazamacrocyclic or diazadiphosphacyclooctane ligands for the reduction of p-toluenesulfonic acid to hydrogen in acetonitrile under inert atmosphere and in the presence of CO and H2. We present an elevated-pressure electrochemical apparatus capable of reaching CO and H2 pressures of ca. 15–520 pounds per square inch (psia) (∼1–35 atm), and we use this apparatus to determine binding constants for CO addition for each catalyst and study the inhibition of the electrocatalysis as a function of CO and H2 pressure. In the case of CO, the extent of catalytic inhibition is correlated to the binding constant, with the cobalt complexes showing a greater degree of catalyst inhibition compared to the nickel complexes. In the case of H2, no complex showed appreciable electrocatalytic inhibition even at H2 pressures of ca. 500 psia.
Co-reporter:Quirin M. Kainz;Susan L. Zultanski;Agnieszka Bartoszewicz;Carson D. Matier;Gregory C. Fu
Science 2016 Volume 351(Issue 6274) pp:681-684
Publication Date(Web):12 Feb 2016
DOI:10.1126/science.aad8313
Copper's light touch forges C-N bonds
Organic photochemistry has traditionally relied on excitation in the ultraviolet, where carbon-based compounds tend to absorb. Over the past decade, the field has undergone a renaissance as compounds that absorb visible light have proven to be versatile catalysts for organic reactions. For the most part, however, these catalysts have contained rare metals such as ruthenium or iridium. Kainz et al. now report a blue light-driven C-N bond-forming reaction catalyzed by Earth-abundant copper (see the Perspective by Greaney). Through coordination to a chiral ligand, the copper center couples alkyl chlorides to indoles and carbazoles with a high degree of enantioselectivity.
Science, this issue p. 681; see also p. 666
Co-reporter:Bridget A. Connor, Jonathan Rittle, David VanderVelde, and Jonas C. Peters
Organometallics 2016 Volume 35(Issue 5) pp:686-690
Publication Date(Web):February 22, 2016
DOI:10.1021/acs.organomet.5b00985
σ-adduct complexes of low-valent, late first-row metal complexes are highly unusual, and this is particularly true of d10 systems. We have discovered a nickel/phosphine/silyl system that undergoes reaction with H2 in solution to generate a species best described as Ni0(η2-(Si–H))(η2-H2) on the basis of multinuclear NMR studies. Theoretical calculations suggest that the Ni center facilitates H atom exchange between the η2-(Si–H) and η2-H2 ligands via interconversion with a higher valent NiII isomer. This exchange is exploited in the selective, catalytic deuteration of exogenous silanes.
Co-reporter:David C. Lacy; Gerri M. Roberts
Journal of the American Chemical Society 2015 Volume 137(Issue 14) pp:4860-4864
Publication Date(Web):March 23, 2015
DOI:10.1021/jacs.5b01838
Molecular cobalt-dmg (dmg = dimethylglyoxime) complexes are an important class of electrocatalysts used heavily in mechanistic model studies of the hydrogen evolution reaction (HER). Schrauzer’s early isolation of a phosphine-stabilized “[H-CoIII(dmgH)2P(nBu)3]” complex has long provided circumstantial support for the plausible intermediacy of Co(III)-H species in HER by cobaloximes in solution. Our investigation of this complex has led to a reassignment of its structure as [CoII(dmgH)2P(nBu)3], a complex that contains no hydride ligand and dimerizes to form an unsupported Co–Co bond in the solid state. A paramagnetic S = 3/2 impurity that forms during the synthesis of [CoII(dmgH)2P(nBu)3] when exposed to adventitious oxygen has also been characterized. This impurity features a 1H NMR resonance at −5.06 ppm that was recently but erroneously attributed to the hydride resonance of “[H-CoIII(dmgH)2P(nBu)3]”. We draw attention to this reassignment because of its relevance to cobaloxime hydrides and HER catalysis and because Schrauzer’s “hydridocobaloxime” is often cited as the primary example of a bona fide hydride that can be isolated and characterized on this widely studied HER platform.
Co-reporter:Sidney E. Creutz
Journal of the American Chemical Society 2015 Volume 137(Issue 23) pp:7310-7313
Publication Date(Web):June 3, 2015
DOI:10.1021/jacs.5b04738
All known nitrogenase cofactors are rich in both sulfur and iron and are presumed capable of binding and reducing N2. Nonetheless, synthetic examples of transition metal model complexes that bind N2 and also feature sulfur donor ligands remain scarce. We report herein an unusual series of low-valent diiron complexes featuring thiolate and dinitrogen ligands. A new binucleating ligand scaffold is introduced that supports an Fe(μ-SAr)Fe diiron subunit that coordinates dinitrogen (N2-Fe(μ-SAr)Fe-N2) across at least three oxidation states (FeIIFeII, FeIIFeI, and FeIFeI). The (N2-Fe(μ-SAr)Fe-N2) system undergoes reduction of the bound N2 to produce NH3 (∼50% yield) and can efficiently catalyze the disproportionation of N2H4 to NH3 and N2. The present scaffold also supports dinitrogen binding concomitant with hydride as a co-ligand. Synthetic model complexes of these types are desirable to ultimately constrain hypotheses regarding Fe-mediated nitrogen fixation in synthetic and biological systems.
Co-reporter:John S. Anderson; George E. CutsailIII; Jonathan Rittle; Bridget A. Connor; William A. Gunderson; Limei Zhang; Brian M. Hoffman
Journal of the American Chemical Society 2015 Volume 137(Issue 24) pp:7803-7809
Publication Date(Web):May 22, 2015
DOI:10.1021/jacs.5b03432
The ability of certain transition metals to mediate the reduction of N2 to NH3 has attracted broad interest in the biological and inorganic chemistry communities. Early transition metals such as Mo and W readily bind N2 and mediate its protonation at one or more N atoms to furnish M(NxHy) species that can be characterized and, in turn, extrude NH3. By contrast, the direct protonation of Fe–N2 species to Fe(NxHy) products that can be characterized has been elusive. Herein, we show that addition of acid at low temperature to [(TPB)Fe(N2)][Na(12-crown-4)] results in a new S = 1/2 Fe species. EPR, ENDOR, Mössbauer, and EXAFS analysis, coupled with a DFT study, unequivocally assign this new species as [(TPB)Fe≡N–NH2]+, a doubly protonated hydrazido(2−) complex featuring an Fe-to-N triple bond. This unstable species offers strong evidence that the first steps in Fe-mediated nitrogen reduction by [(TPB)Fe(N2)][Na(12-crown-4)] can proceed along a distal or “Chatt-type” pathway. A brief discussion of whether subsequent catalytic steps may involve early or late stage cleavage of the N–N bond, as would be found in limiting distal or alternating mechanisms, respectively, is also provided.
Co-reporter:Tanvi S. Ratani; Shoshana Bachman; Gregory C. Fu
Journal of the American Chemical Society 2015 Volume 137(Issue 43) pp:13902-13907
Publication Date(Web):October 22, 2015
DOI:10.1021/jacs.5b08452
We have recently reported that, in the presence of light and a copper catalyst, nitrogen nucleophiles such as carbazoles and primary amides undergo C–N coupling with alkyl halides under mild conditions. In the present study, we establish that photoinduced, copper-catalyzed alkylation can also be applied to C–C bond formation, specifically, that the cyanation of unactivated secondary alkyl chlorides can be achieved at room temperature to afford nitriles, an important class of target molecules. Thus, in the presence of an inexpensive copper catalyst (CuI; no ligand coadditive) and a readily available light source (UVC compact fluorescent light bulb), a wide array of alkyl halides undergo cyanation in good yield. Our initial mechanistic studies are consistent with the hypothesis that an excited state of [Cu(CN)2]− may play a role, via single electron transfer, in this process. This investigation provides a rare example of a transition metal-catalyzed cyanation of an alkyl halide, as well as the first illustrations of photoinduced, copper-catalyzed alkylation with either a carbon nucleophile or a secondary alkyl chloride.
Co-reporter:Henry Fong and Jonas C. Peters
Inorganic Chemistry 2015 Volume 54(Issue 11) pp:5124-5135
Publication Date(Web):December 31, 2014
DOI:10.1021/ic502508p
Despite renewed interest in carbon dioxide (CO2) reduction chemistry, examples of homogeneous iron catalysts that hydrogenate CO2 are limited compared to their noble-metal counterparts. Knowledge of the thermodynamic properties of iron hydride complexes, including M–H hydricities (ΔGH–), could aid in the development of new iron-based catalysts. Here we present the experimentally determined hydricity of an iron hydride complex: (SiPiPr3)Fe(H2)(H), ΔGH– = 54.3 ± 0.9 kcal/mol [SiPiPr3 = [Si(o-C6H4PiPr2)3]−]. We also explore the CO2 hydrogenation chemistry of a series of triphosphinoiron complexes, each with a distinct apical unit on the ligand chelate (Si–, C–, PhB–, N, B). The silyliron (SiPR3)Fe (R = iPr and Ph) and boratoiron (PhBPiPr3)Fe (PhBPiPr3 = [PhB(CH2PiPr2)3]−) systems, as well as the recently reported (CPiPr3)Fe (CPiPr3 = [C(o-C6H4PiPr2)3]−), are also catalysts for CO2 hydrogenation in methanol and in the presence of triethylamine, generating methylformate and triethylammonium formate at up to 200 TON using (SiPPh3)FeCl as the precatalyst. Under stoichiometric conditions, the iron hydride complexes of this series react with CO2 to give formate complexes. Finally, the proposed mechanism of the (SiPiPr3)-Fe system proceeds through a monohydride intermediate (SiPiPr3)Fe(H2)(H), in contrast to that of the known and highly active tetraphosphinoiron, (tetraphos)Fe (tetraphos = P(o-C6H4PPh2)3), CO2 hydrogenation catalyst.
Co-reporter:Nicolai Lehnert
Inorganic Chemistry 2015 Volume 54(Issue 19) pp:9229-9233
Publication Date(Web):October 5, 2015
DOI:10.1021/acs.inorgchem.5b02124
Co-reporter:Dr. Gaël Ung ; Jonas C. Peters
Angewandte Chemie International Edition 2015 Volume 54( Issue 2) pp:532-535
Publication Date(Web):
DOI:10.1002/anie.201409454
Abstract
The two-coordinate [(CAAC)2Fe] complex [CAAC=cyclic (alkyl)(amino)carbene] binds dinitrogen at low temperature (T<−80 °C). The resulting putative three-coordinate N2 complex, [(CAAC)2Fe(N2)], was trapped by one-electron reduction to its corresponding anion [(CAAC)2FeN2]− at low temperature. This complex was structurally characterized and features an activated dinitrogen unit which can be silylated at the β-nitrogen atom. The redox-linked complexes [(CAAC)2FeI][BArF4], [(CAAC)2Fe0], and [(CAAC)2Fe−IN2]− were all found to be active for the reduction of dinitrogen to ammonia upon treatment with KC8 and HBArF4⋅2 Et2O at −95 °C [up to (3.4±1.0) equivalents of ammonia per Fe center]. The N2 reduction activity is highly temperature dependent, with significant N2 reduction to NH3 only occurring below −78 °C. This reactivity profile tracks with the low temperatures needed for N2 binding and an otherwise unavailable electron-transfer step to generate reactive [(CAAC)2FeN2]−.
Co-reporter:Mark A. Nesbit, Daniel L. M. Suess, and Jonas C. Peters
Organometallics 2015 Volume 34(Issue 19) pp:4741-4752
Publication Date(Web):August 28, 2015
DOI:10.1021/acs.organomet.5b00530
An exciting challenge in transition metal catalyst design is to explore whether earth-abundant base metals such as Fe, Co, and Ni can mediate two-electron reductive transformations that their precious metal counterparts (e.g., Ru, Rh, Ir, and Pd) are better known to catalyze. Organometallic metalloboranes are an interesting design concept in this regard because they can serve as organometallic frustrated Lewis pairs. To build on prior studies with nickel metalloboranes featuring the DPB and PhDPBMes ligands in the context of H2 and silane activation and catalysis (DPB = bis(o-diisopropylphosphinophenyl)phenylborane, PhDPBMes = bis(o-diphenylphosphinophenyl)mesitylborane), we now explore the reactivity of iron, [(DPB)Fe]2(N2), 1, and cobalt, (DPB)Co(N2), 2, metalloboranes toward a series of substrates with E–H bonds (E = O, S, C, N) including phenol, thiophenol, benzo[h]quinoline, and 8-aminoquinoline. In addition to displaying high stoichiometric E–H bond activation reactivity, complexes 1 and 2 prove to be more active catalysts for the hydrosilylation of ketones and aldehydes with diphenylsilane relative to (PhDPBMes)Ni. Indeed, 2 appears to be the most active homogeneous cobalt catalyst reported to date for the hydrosilylation of acetophenone under the conditions studied.
Co-reporter:M. Zhang
The Journal of Physical Chemistry C 2015 Volume 119(Issue 9) pp:4645-4654
Publication Date(Web):February 12, 2015
DOI:10.1021/jp5127738
In situ FT-IR measurements and electronic structure calculations are reported for the reduction of CO2 catalyzed by the macrocyclic complex [CoIIN4H]2+ (N4H = 2,12-dimethyl-3,7,11,17-tetraazabicyclo-[11.3.1]-heptadeca-1(17),2,11,13,15-pentaene). Beginning from the [CoIIN4H]2+ resting state of the complex in wet acetonitrile solution, two different visible light sensitizers with substantially different reducing power are employed to access reduced states. Accessing reduced states of the complex with a [Ru(bpy)3]2+ sensitizer yields an infrared band at 1670 cm–1 attributed to carboxylate, which is also observed for an authentic sample of the one-electron reduced complex [CoN4H(MeCN)]+ in CO2 saturated acetonitrile solution. The results are interpreted based on calculations using the pure BP86 functional that correctly reproduces experimental geometries. Continuum solvation effects are also included. The calculations show that Co is reduced to CoI in the first reduction, which is consistent with experimental d–d spectra of square Co(I) macrocycle complexes. The energy of the CO2 adduct of the one-electron reduced catalyst complex is essentially the same as for [CoN4H(MeCN)]+, which implies that only a fraction of the latter forms an adduct with CO2. By contrast, the calculations indicate a crucial role for redox noninnocence of the macrocyclic ligand in the doubly reduced state, [CoI(N4H) –•], and show that [CoI(N4H) –•] binds partially reduced CO2 fairly strongly. Experimentally accessing [CoI(N4H) –•] with an Ir(bpy)3 sensitizer with greater reducing power closes the catalytic cycle as FT-IR spectroscopy shows CO production. Use of isotopically substituted C18O2 also shows clear evidence for 18O-substituted byproducts from CO2 reduction to CO.
Co-reporter:Dr. Gaël Ung ; Jonas C. Peters
Angewandte Chemie 2015 Volume 127( Issue 2) pp:542-545
Publication Date(Web):
DOI:10.1002/ange.201409454
Abstract
The two-coordinate [(CAAC)2Fe] complex [CAAC=cyclic (alkyl)(amino)carbene] binds dinitrogen at low temperature (T<−80 °C). The resulting putative three-coordinate N2 complex, [(CAAC)2Fe(N2)], was trapped by one-electron reduction to its corresponding anion [(CAAC)2FeN2]− at low temperature. This complex was structurally characterized and features an activated dinitrogen unit which can be silylated at the β-nitrogen atom. The redox-linked complexes [(CAAC)2FeI][BArF4], [(CAAC)2Fe0], and [(CAAC)2Fe−IN2]− were all found to be active for the reduction of dinitrogen to ammonia upon treatment with KC8 and HBArF4⋅2 Et2O at −95 °C [up to (3.4±1.0) equivalents of ammonia per Fe center]. The N2 reduction activity is highly temperature dependent, with significant N2 reduction to NH3 only occurring below −78 °C. This reactivity profile tracks with the low temperatures needed for N2 binding and an otherwise unavailable electron-transfer step to generate reactive [(CAAC)2FeN2]−.
Co-reporter:Jonathan Rittle ; Charles C. L. McCrory
Journal of the American Chemical Society 2014 Volume 136(Issue 39) pp:13853-13862
Publication Date(Web):September 3, 2014
DOI:10.1021/ja507217v
Transient hydride ligands bridging two or more iron centers purportedly accumulate on the iron–molybdenum cofactor (FeMoco) of nitrogenase, and their role in the reduction of N2 to NH3 is unknown. One role of these ligands may be to facilitate N2 coordination at an iron site of FeMoco. Herein, we consider this hypothesis and describe the preparation of a series of diiron complexes supported by two bridging hydride ligands. These compounds bind either one or two molecules of N2 depending on the redox state of the Fe2(μ-H)2 unit. An unusual example of a mixed-valent FeII(μ-H)2FeI is described that displays a 106-fold enhancement of N2 binding affinity over its oxidized congener, quantified by spectroscopic and electrochemical techniques. Furthermore, these compounds show promise as functional models of nitrogenase as substantial amounts of NH3 are produced upon exposure to proton and electron equivalents. The Fe(μ-H)Fe(N2) sub-structure featured herein was previously unknown. This subunit may be relevant to consider in nitrogenases during turnover.
Co-reporter:Hien-Quang Do ; Shoshana Bachman ; Alex C. Bissember ; Jonas C. Peters ;Gregory C. Fu
Journal of the American Chemical Society 2014 Volume 136(Issue 5) pp:2162-2167
Publication Date(Web):January 21, 2014
DOI:10.1021/ja4126609
The development of a mild and general method for the alkylation of amides with relatively unreactive alkyl halides (i.e., poor substrates for SN2 reactions) is an ongoing challenge in organic synthesis. We describe herein a versatile transition-metal-catalyzed approach: in particular, a photoinduced, copper-catalyzed monoalkylation of primary amides. A broad array of alkyl and aryl amides (as well as a lactam and a 2-oxazolidinone) couple with unactivated secondary (and hindered primary) alkyl bromides and iodides using a single set of comparatively simple and mild conditions: inexpensive CuI as the catalyst, no separate added ligand, and C–N bond formation at room temperature. The method is compatible with a variety of functional groups, such as an olefin, a carbamate, a thiophene, and a pyridine, and it has been applied to the synthesis of an opioid receptor antagonist. A range of mechanistic observations, including reactivity and stereochemical studies, are consistent with a coupling pathway that includes photoexcitation of a copper–amidate complex, followed by electron transfer to form an alkyl radical.
Co-reporter:Yichen Tan, José María Muñoz-Molina, Gregory C. Fu and Jonas C. Peters
Chemical Science 2014 vol. 5(Issue 7) pp:2831-2835
Publication Date(Web):22 Apr 2014
DOI:10.1039/C4SC00368C
Most copper-catalyzed cross-couplings require an elevated reaction temperature. Recently, a photoinduced variant has been developed that enables C–X bond-forming reactions of certain nitrogen and sulfur nucleophiles to proceed under unusually mild conditions (−40 °C to room temperature). In view of the importance of carbon–oxygen bond construction in organic chemistry, the expansion of this photochemical approach to oxygen nucleophiles is an important objective. In this report, we establish that, in the presence of light and an inexpensive copper pre-catalyst (CuI), a wide array of phenols and aryl iodides can be coupled to generate diaryl ethers under mild conditions (room temperature) in the presence of a variety of functional groups. Our studies indicate that a Cu(I)–phenoxide complex is a viable intermediate in photoinduced C–O bond-formation.
Co-reporter:Samantha N. MacMillan, W. Hill Harman and Jonas C. Peters
Chemical Science 2014 vol. 5(Issue 2) pp:590-597
Publication Date(Web):22 Oct 2013
DOI:10.1039/C3SC52626G
Metal–borane complexes are emerging as promising systems for study in the context of bifunctional catalysis. Herein we describe diphosphineborane nickel complexes that activate Si–H bonds and catalyze the hydrosilylation of aldehydes. Treatment of [MesDPBPh]Ni (1) ([MesDPBPh] = MesB(o-Ph2PC6H4)2) with organosilanes affords the complexes [MesDPBPh](μ-H)NiE (E = SiH2Ph (3), SiHPh2 (4)). Complex 4 is in solution equilibrium with 1 and the thermodynamic and kinetic parameters of their exchange have been characterized by NMR spectroscopy. Complex 1 is a catalyst for the hydrosilylation of a range of para-substituted benzaldehydes. Mechanistic studies on this reaction via multinuclear NMR spectroscopy are consistent with the intermediacy of a borohydrido-Ni-siloxyalkyl species.
Co-reporter:David C. Lacy, Charles C. L. McCrory, and Jonas C. Peters
Inorganic Chemistry 2014 Volume 53(Issue 10) pp:4980-4988
Publication Date(Web):April 28, 2014
DOI:10.1021/ic403122j
The cobalt complex [CoIIIN4H(Br)2]+ (N4H = 2,12-dimethyl-3,7,11,17-tetraazabicyclo-[11.3.1]-heptadeca-1(7),2,11,13,15-pentaene) was used for electrocatalytic CO2 reduction in wet MeCN with a glassy carbon working electrode. When water was employed as the proton source (10 M in MeCN), CO was produced (fCO= 45% ± 6.4) near the CoI/0 redox couple for [CoIIIN4H(Br)2]+ (E1/2 = −1.88 V FeCp2+/0) with simultaneous H2 evolution (fH2= 30% ± 7.8). Moreover, we successfully demonstrated that the catalytically active species is homogeneous through the use of control experiments and XPS studies of the working glassy-carbon electrodes. As determined by cyclic voltammetry, CO2 catalysis occurred near the formal CoI/0redox couple, and attempts were made to isolate the triply reduced compound (“[Co0N4H]”). Instead, the doubly reduced (“CoI”) compounds [CoN4] and [CoN4H(MeCN)]+ were isolated and characterized by X-ray crystallography. Their molecular structures prompted DFT studies to illuminate details regarding their electronic structure. The results indicate that reducing equivalents are stored on the ligand, implicating redox noninnocence in the ligands for H2 evolution and CO2 reduction electrocatalysis.
Co-reporter:Dr. W. Hill Harman;Dr. Tzu-Pin Lin;Dr. Jonas C. Peters
Angewandte Chemie International Edition 2014 Volume 53( Issue 4) pp:1081-1086
Publication Date(Web):
DOI:10.1002/anie.201308175
Abstract
Bifunctional EH activation offers a promising approach for the design of two-electron-reduction catalysts with late first-row metals, such as Ni. To this end, we have been pursuing H2 activation reactions at late-metal boratranes and herein describe a diphosphine–borane-supported Ni—(H2) complex, [(PhDPBiPr)Ni(H2)], which has been characterized in solution. 1H NMR spectroscopy confirms the presence of an intact H2 ligand. A range of data, including electronic-structure calculations, suggests a d10 configuration for [(PhDPBiPr)Ni(H2)] as most appropriate. Such a configuration is highly unusual among transition-metal H2 adducts. The nonclassical H2 adduct is an intermediate in the complete activation of H2 across the NiB interaction. Reaction-coordinate analysis suggests synergistic activation of the H2 ligand by both the Ni and B centers of the nickel boratrane subunit, thus highlighting an important role of the borane ligand both in stabilizing the d10 Ni—(H2) interaction and in the H—H cleavage step.
Co-reporter:John S. Anderson ; Jonas C. Peters
Angewandte Chemie International Edition 2014 Volume 53( Issue 23) pp:5978-5981
Publication Date(Web):
DOI:10.1002/anie.201401018
Abstract
FeI centers in iron–sulfide complexes have little precedent in synthetic chemistry despite a growing interest in the possible role of unusually low valent iron in metalloenzymes that feature iron–sulfur clusters. A series of three diiron [(L3Fe)2(μ-S)] complexes that were isolated and characterized in the low-valent oxidation states FeIISFeII, FeIISFeI, and FeISFeI is described. This family of iron sulfides constitutes a unique redox series comprising three nearly isostructural but electronically distinct Fe2(μ-S) species. Combined structural, magnetic, and spectroscopic studies provided strong evidence that the pseudotetrahedral iron centers undergo a transition to low-spin S=1/2 states upon reduction from FeII to FeI. The possibility of accessing low-spin, pseudotetrahedral FeI sites compatible with S2− as a ligand was previously unknown.
Co-reporter:Dr. Gaël Ung;Jonathan Rittle;Dr. Michele Soleilhavoup; Guy Bertr; Jonas C. Peters
Angewandte Chemie International Edition 2014 Volume 53( Issue 32) pp:8427-8431
Publication Date(Web):
DOI:10.1002/anie.201404078
Abstract
The CAAC [CAAC=cyclic (alkyl)(amino)carbene] family of carbene ligands have shown promise in stabilizing unusually low-coordination number transition-metal complexes in low formal oxidation states. Here we extend this narrative by demonstrating their utility in affording access to the first examples of two-coordinate formal Fe0 and Co0 [(CAAC)2M] complexes, prepared by reduction of their corresponding two-coordinate cationic FeI and CoI precursors. The stability of these species arises from the strong σ-donating and π-accepting properties of the supporting CAAC ligands, in addition to steric protection.
Co-reporter:Dr. W. Hill Harman;Dr. Tzu-Pin Lin;Dr. Jonas C. Peters
Angewandte Chemie 2014 Volume 126( Issue 4) pp:1099-1104
Publication Date(Web):
DOI:10.1002/ange.201308175
Abstract
Bifunctional EH activation offers a promising approach for the design of two-electron-reduction catalysts with late first-row metals, such as Ni. To this end, we have been pursuing H2 activation reactions at late-metal boratranes and herein describe a diphosphine–borane-supported Ni—(H2) complex, [(PhDPBiPr)Ni(H2)], which has been characterized in solution. 1H NMR spectroscopy confirms the presence of an intact H2 ligand. A range of data, including electronic-structure calculations, suggests a d10 configuration for [(PhDPBiPr)Ni(H2)] as most appropriate. Such a configuration is highly unusual among transition-metal H2 adducts. The nonclassical H2 adduct is an intermediate in the complete activation of H2 across the NiB interaction. Reaction-coordinate analysis suggests synergistic activation of the H2 ligand by both the Ni and B centers of the nickel boratrane subunit, thus highlighting an important role of the borane ligand both in stabilizing the d10 Ni—(H2) interaction and in the H—H cleavage step.
Co-reporter:John S. Anderson ; Jonas C. Peters
Angewandte Chemie 2014 Volume 126( Issue 23) pp:6088-6091
Publication Date(Web):
DOI:10.1002/ange.201401018
Abstract
FeI centers in iron–sulfide complexes have little precedent in synthetic chemistry despite a growing interest in the possible role of unusually low valent iron in metalloenzymes that feature iron–sulfur clusters. A series of three diiron [(L3Fe)2(μ-S)] complexes that were isolated and characterized in the low-valent oxidation states FeIISFeII, FeIISFeI, and FeISFeI is described. This family of iron sulfides constitutes a unique redox series comprising three nearly isostructural but electronically distinct Fe2(μ-S) species. Combined structural, magnetic, and spectroscopic studies provided strong evidence that the pseudotetrahedral iron centers undergo a transition to low-spin S=1/2 states upon reduction from FeII to FeI. The possibility of accessing low-spin, pseudotetrahedral FeI sites compatible with S2− as a ligand was previously unknown.
Co-reporter:Christopher Uyeda ; Yichen Tan ; Gregory C. Fu
Journal of the American Chemical Society 2013 Volume 135(Issue 25) pp:9548-9552
Publication Date(Web):May 23, 2013
DOI:10.1021/ja404050f
Building on the known photophysical properties of well-defined copper–carbazolide complexes, we have recently described photoinduced, copper-catalyzed N-arylations and N-alkylations of carbazoles. Until now, there have been no examples of the use of other families of heteroatom nucleophiles in such photoinduced processes. Herein, we report a versatile photoinduced, copper-catalyzed method for coupling aryl thiols with aryl halides, wherein a single set of reaction conditions, using inexpensive CuI as a precatalyst without the need for an added ligand, is effective for a wide range of coupling partners. As far as we are aware, copper-catalyzed C–S cross-couplings at 0 °C have not previously been achieved, which renders our observation of efficient reaction of an unactivated aryl iodide at −40 °C especially striking. Mechanistic investigations are consistent with these photoinduced C–S cross-couplings following a SET/radical pathway for C–X bond cleavage (via a Cu(I)–thiolate), which contrasts with nonphotoinduced, copper-catalyzed processes wherein a concerted mechanism is believed to occur.
Co-reporter:Daniel L. M. Suess
Journal of the American Chemical Society 2013 Volume 135(Issue 34) pp:12580-12583
Publication Date(Web):August 9, 2013
DOI:10.1021/ja406874k
An iron diphosphineborane platform that was previously reported to facilitate a high degree of N2 functionalization is herein shown to effect reductive CO coupling. Disilylation of an iron dicarbonyl precursor furnishes a structurally unprecedented iron dicarbyne complex. Several complexes related to this process are also characterized which allows for a comparative analysis of their respective Fe–B and Fe–C bonding. Facile hydrogenation of the iron dicarbyne at ambient temperature and 1 atm H2 results in release of a CO-derived olefin.
Co-reporter:Daniel T. Ziegler ; Junwon Choi ; José María Muñoz-Molina ; Alex C. Bissember ; Jonas C. Peters ;Gregory C. Fu
Journal of the American Chemical Society 2013 Volume 135(Issue 35) pp:13107-13112
Publication Date(Web):August 22, 2013
DOI:10.1021/ja4060806
The use of light to facilitate copper-catalyzed cross-couplings of nitrogen nucleophiles can enable C–N bond formation to occur under unusually mild conditions. In this study, we substantially expand the scope of such processes, establishing that this approach is not limited to reactions of carbazoles with iodobenzene and alkyl halides. Specifically, we demonstrate for the first time that other nitrogen nucleophiles (e.g., common pharmacophores such as indoles, benzimidazoles, and imidazoles) as well as other electrophiles (e.g., hindered/deactivated/heterocyclic aryl iodides, an aryl bromide, an activated aryl chloride, alkenyl halides, and an alkynyl bromide) serve as suitable partners. Photoinduced C–N bond formation can be achieved at room temperature using a common procedure with an inexpensive catalyst (CuI) that does not require a ligand coadditive and is tolerant of moisture and a variety of functional groups.
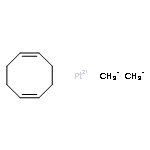
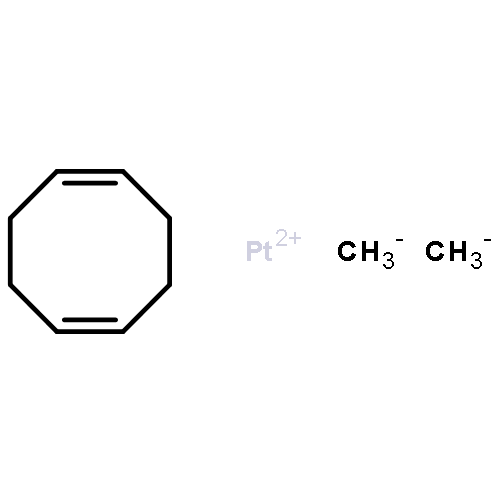
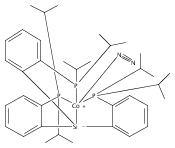
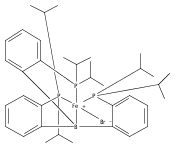
Co-reporter:Tzu-Pin Lin
Journal of the American Chemical Society 2013 Volume 135(Issue 41) pp:15310-15313
Publication Date(Web):September 30, 2013
DOI:10.1021/ja408397v
We describe the synthesis of a cobalt(I)–N2 complex (2) supported by a meridional bis-phosphino-boryl (PBP) ligand. Complex 2 undergoes a clean reaction with 2 equiv of dihydrogen to afford a dihydridoboratocobalt dihydride (3). The ability of boron to switch between a boryl and a dihydridoborate conformation makes possible the reversible conversion of 2 and 3. Complex 3 reacts with HMe2N–BH3 to give a hydridoborane cobalt tetrahydridoborate complex. We explore this boryl–cobalt system in the context of catalytic olefin hydrogenation as well as amine–borane dehydrogenation/transfer hydrogenation.
Co-reporter:Sidney E. Creutz
Journal of the American Chemical Society 2013 Volume 136(Issue 3) pp:1105-1115
Publication Date(Web):December 18, 2013
DOI:10.1021/ja4114962
While recent spectroscopic studies have established the presence of an interstitial carbon atom at the center of the iron–molybdenum cofactor (FeMoco) of MoFe-nitrogenase, its role is unknown. We have pursued Fe–N2 model chemistry to explore a hypothesis whereby this C-atom (previously denoted as a light X-atom) may provide a flexible trans interaction with an Fe center to expose an Fe–N2 binding site. In this context, we now report on Fe complexes of a new tris(phosphino)alkyl (CPiPr3) ligand featuring an axial carbon donor. It is established that the iron center in this scaffold binds dinitrogen trans to the Calkyl-atom anchor in three distinct and structurally characterized oxidation states. Fe–Calkyl lengthening is observed upon reduction, reflective of significant ionic character in the Fe–Calkyl interaction. The anionic (CPiPr3)FeN2– species can be functionalized by a silyl electrophile to generate (CPiPr3)Fe–N2SiR3. (CPiPr3)FeN2– also functions as a modest catalyst for the reduction of N2 to NH3 when supplied with electrons and protons at −78 °C under 1 atm N2 (4.6 equiv NH3/Fe).
Co-reporter:Marc-Etienne Moret ; Limei Zhang
Journal of the American Chemical Society 2013 Volume 135(Issue 10) pp:3792-3795
Publication Date(Web):February 18, 2013
DOI:10.1021/ja4006578
Virtually all chemical bonds consist of one or several pairs of electrons shared by two atoms. Examples of σ-bonds made of a single electron delocalized over two neighboring atoms were until recently found only in gas-phase cations such as H2+ and Li2+ and in highly unstable species generated in solid matrices. Only in the past decade was bona fide one-electron bonding observed for molecules in fluid solution. Here we report the isolation and structural characterization of a thermally stable compound featuring a Cu–B one-electron bond, as well as its oxidized (nonbonded) and reduced (two-electrons-bonded) congeners. This triad provides an excellent opportunity to study the degree of σ-bonding in a metalloboratrane as a function of electron count.
Co-reporter:Daniel L. M. Suess
Journal of the American Chemical Society 2013 Volume 135(Issue 13) pp:4938-4941
Publication Date(Web):March 11, 2013
DOI:10.1021/ja400836u
The synthesis and characterization of Fe–diphosphineborane complexes are described in the context of N2 functionalization chemistry. Iron aminoimides can be generated at room temperature under 1 atm N2 and are shown to react with E–H bonds from PhSiH3 and H2. The resulting products derive from delivery of the E fragment to Nα and the H atom to B. The flexibility and lability of the Fe–BPh interactions in these complexes engender this reactivity.
Co-reporter:Christopher Uyeda and Jonas C. Peters
Journal of the American Chemical Society 2013 Volume 135(Issue 32) pp:12023-12031
Publication Date(Web):July 18, 2013
DOI:10.1021/ja4053653
Heme-containing nitrite reductases bind and activate nitrite by a mechanism that is proposed to involve interactions with Brønsted acidic residues in the secondary coordination sphere. To model this functionality using synthetic platforms that incorporate a Lewis acidic site, heterobimetallic CoMg complexes supported by diimine–dioxime ligands are described. The neutral (μ-NO2)CoMg species 3 is synthesized from the [(μ-OAc)(Br)CoMg]+ complex 1 by a sequence of one-electron reduction and ligand substitution reactions. Data are presented for a redox series of nitrite adducts, featuring a conserved μ-(η1-N:η1-O)-NO2 motif, derived from this synthon. Conditions are identified for the proton-induced N–O bond heterolysis of bound NO2– in the most reduced member of this series, affording the [(NO)(Cl)CoMg(H2O)]+ complex 6. Reduction of this complex followed by protonation leads to the evolution of free N2O. On the basis of these stoichiometric reactivity studies, the competence of complex 1 as a NO2– reduction catalyst is evaluated using electrochemical methods. In bulk electrolysis experiments, conducted at −1.2 V vs SCE using Et3NHCl as a proton source, N2O is produced selectively without the competing formation of NH3, NH2OH, or H2.
Co-reporter:Caroline T. Saouma, Connie C. Lu, Michael W. Day and Jonas C. Peters
Chemical Science 2013 vol. 4(Issue 10) pp:4042-4051
Publication Date(Web):12 Jul 2013
DOI:10.1039/C3SC51262B
This manuscript explores the product distribution of the reaction of carbon dioxide with reactive iron(I) complexes supported by tris(phosphino)borate ligands, [PhBPR3]− ([PhBPR3]− = [PhB(CH2PR2)3]−; R = CH2Cy, Ph, iPr, mter; mter = 3,5-meta-terphenyl). Our studies reveal an interesting and unexpected role for the solvent medium with respect to the course of the CO2 activation reaction. For instance, exposure of methylcyclohexane (MeCy) solutions of to CO2 yields the partial decarbonylation product . When the reaction is instead carried out in benzene or THF, reductive coupling of CO2 occurs to give the bridging oxalate species . Reaction studies aimed at understanding this solvent effect are presented, and suggest that the product profile is ultimately determined by the ability of the solvent to coordinate the iron center. When more sterically encumbering auxiliary ligands are employed to support the iron(I) center (i.e., [PhBPPh3]− and [PhBPiPr3]−), complete decarbonylation is observed to afford structurally unusual diiron(II) products of the type {[PhBPR3]Fe}2(μ-O). A mechanistic hypothesis that is consistent with the collection of results described is offered, and suggests that reductive coupling of CO2 likely occurs from an electronically saturated “FeII–CO2˙−” species.
Co-reporter:Christopher Uyeda and Jonas C. Peters
Chemical Science 2013 vol. 4(Issue 1) pp:157-163
Publication Date(Web):12 Sep 2012
DOI:10.1039/C2SC21231E
Heterobimetallic NiZn complexes featuring metal centers in distinct coordination environments have been synthesized using diimine–dioxime ligands as binucleating scaffolds. A tetramethylfuran-containing ligand derivative enables a stable one-electron-reduced S = 1/2 species to be accessed using Cp2Co as a chemical reductant. The resulting pseudo-square planar complex exhibits spectroscopic and crystallographic characteristics of a ligand-centered radical bound to a Ni(II) center. Upon coordination of a π-acidic ligand such as PPh3, however, a five-coordinate Ni(I) metalloradical is formed. The electronic structures of these reduced species provide insight into the subtle effects of ligand structure on the potential and reversibility of the NiII/I couple for complexes of redox-active tetraazamacrocycles.
Co-reporter:Dr. Alex C. Bissember;Dr. Rylan J. Lundgren;Sidney E. Creutz; Jonas C. Peters; Gregory C. Fu
Angewandte Chemie 2013 Volume 125( Issue 19) pp:5233-5237
Publication Date(Web):
DOI:10.1002/ange.201301202
Co-reporter:Jonathan Rittle;
Proceedings of the National Academy of Sciences 2013 110(40) pp:15898-15903
Publication Date(Web):September 16, 2013
DOI:10.1073/pnas.1310153110
We report here a series of four- and five-coordinate Fe model complexes that feature an axial tri(silyl)methyl ligand positioned
trans to a substrate-binding site. This arrangement is used to crudely model a single-belt Fe site of the FeMo-cofactor that might
bind N2 at a position trans to the interstitial C atom. Reduction of a trigonal pyramidal Fe(I) complex leads to uptake of N2 and subsequent functionalization furnishes an open-shell Fe–diazenido complex. A related series of five-coordinate Fe–CO
complexes stable across three redox states is also described. Spectroscopic, crystallographic, and Density Functional Theory
(DFT) studies of these complexes suggest that a decrease in the covalency of the Fe–Calkyl interaction occurs upon reduction and substrate binding. This leads to unusually long Fe–Calkyl bond distances that reflect an ionic Fe–C bond. The data presented are contextualized in support of a hypothesis wherein
modulation of a belt Fe–C interaction in the FeMo-cofactor facilitates substrate binding and reduction.
Co-reporter:Dr. Alex C. Bissember;Dr. Rylan J. Lundgren;Sidney E. Creutz; Jonas C. Peters; Gregory C. Fu
Angewandte Chemie International Edition 2013 Volume 52( Issue 19) pp:5129-5133
Publication Date(Web):
DOI:10.1002/anie.201301202
Co-reporter:Henry Fong, Marc-Etienne Moret, Yunho Lee, and Jonas C. Peters
Organometallics 2013 Volume 32(Issue 10) pp:3053-3062
Publication Date(Web):May 6, 2013
DOI:10.1021/om400281v
Reversible, heterolytic addition of H2 across an iron–boron bond in a ferraboratrane with formal hydride transfer to the boron gives iron-borohydrido-hydride complexes. These compounds catalyze the hydrogenation of alkenes and alkynes to the respective alkanes. Notably, the boron is capable of acting as a shuttle for hydride transfer to substrates. The results are interesting in the context of heterolytic substrate addition across metal–boron bonds in metallaboratranes and related systems, as well as metal–ligand bifunctional catalysis.
Co-reporter:Daniel L. M. Suess ; Charlene Tsay
Journal of the American Chemical Society 2012 Volume 134(Issue 34) pp:14158-14164
Publication Date(Web):August 14, 2012
DOI:10.1021/ja305248f
Two isostructural, nonclassical Co(H2) complexes are prepared from their Co(N2) precursors using tris(phosphino)silyl and tris(phosphino)borane ancillary ligands. Comproportionation of CoBr2 and Co metal in the presence of TPB (tris-(o-diisopropylphophinophenyl)borane) gives (TPB)CoBr (4). One-electron reduction of 4 triggers N2 binding to give (TPB)Co(N2) (2-N2) which is isostructural to previously reported [SiP3]Co(N2) (1-N2) ([SiP3] = tris-(o-diisopropylphosphinophenyl)silyl). Both 1-N2 and 2-N2 react with 1 atm H2 to generate thermally stable H2 complexes 1-H2 and 2-H2, respectively. Both complexes are characterized by a suite of spectroscopic techniques in solution and by X-ray crystallography. The H2 and N2 ligands in 2-H2 and 2-N2 are labile under ambient conditions and the binding equilibria are observable by temperature-dependent UV/vis. A van’t Hoff analysis allows for the ligand binding energetics to be determined (H2: ΔHo = −12.5(3) kcal mol–1 and ΔSo = −26(3) cal K–1 mol–1; N2: ΔHo = −13.9(7) kcal mol–1 and ΔSo = −32(5) cal K–1 mol–1).
Co-reporter:John S. Anderson ; Marc-Etienne Moret
Journal of the American Chemical Society 2012 Volume 135(Issue 2) pp:534-537
Publication Date(Web):December 21, 2012
DOI:10.1021/ja307714m
Tris(phosphine)borane ligated Fe(I) centers featuring N2H4, NH3, NH2, and OH ligands are described. Conversion of Fe–NH2 to Fe–NH3+ by the addition of acid, and subsequent reductive release of NH3 to generate Fe–N2, is demonstrated. This sequence models the final steps of proposed Fe–mediated nitrogen fixation pathways. The five-coordinate trigonal bipyramidal complexes described are unusual in that they adopt S = 3/2 ground states and are prepared from a four-coordinate, S = 3/2 trigonal pyramidal precursor.
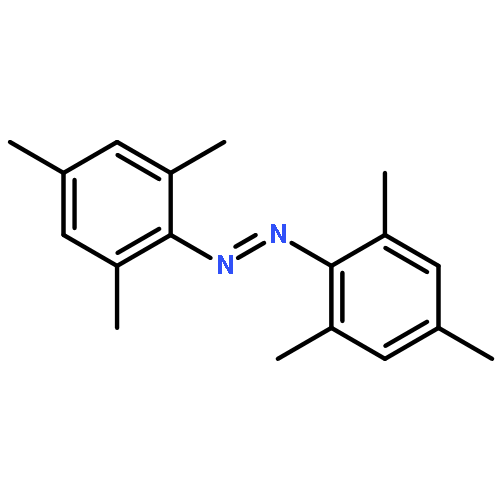
Co-reporter:Ayumi Takaoka ; Marc-Etienne Moret
Journal of the American Chemical Society 2012 Volume 134(Issue 15) pp:6695-6706
Publication Date(Web):March 1, 2012
DOI:10.1021/ja211603f
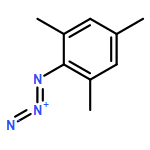
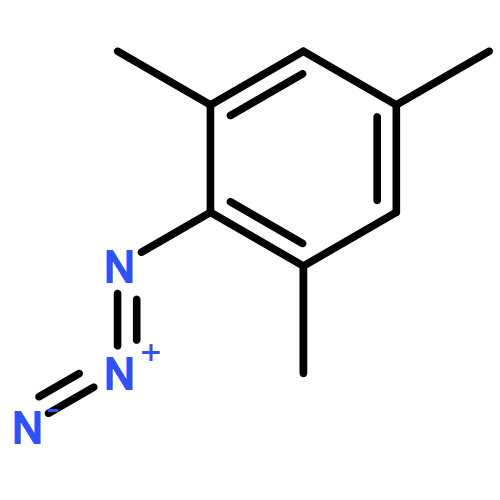
Unusual N–N coupling of aryl azides to yield azoarenes is demonstrated by the Ru(I) metalloradical, [SiPiPr3]Ru(N2) (4) ([SiPiPr3] = (2-iPr2PC6H4)3Si–). The yield of the azoarene is dependent on the substituent on the aryl azide, and the reaction is catalytic for p-methoxy and p-ethoxy phenyl azides, while no azoarene is observed for p-trifluoromethylphenyl azide. Studies aimed at probing the viability of a bimolecular coupling mechanism of metal imide species, as shown in the related [SiPiPr3]Fe system, have led to the isolation of several structurally unusual complexes including the ruthenium(IV) imide, 7-OMe, as well as the Ru(II) azide adduct 8-OMe. One electron reduction of 7-OMe complex led to the isolation of the formally Ru(III) imide complex, [SiPiPr3]Ru(NAr) (Ar = p-MeOC6H4, 5-OMe). EPR spectroscopy on 5-OMe suggests that the complex is electronically similar to the previously reported imide complex, [SiPiPr3]Ru(NAr) (Ar = p-CF3C6H4,5-CF3), and features radical character on the NAr moiety, but to a greater degree. The stability of 5-OMe establishes that bimolecular coupling of 5-OMe is kinetically inconsistent with the reaction. Further studies rule out mechanisms in which 5-OMe reacts directly with free aryl azide or a transient Ru(I) azide adduct. Together, these studies show that 5-OMe is likely uninvolved in the catalytic cycle and demonstrates the influence of the metal center on the mechanism of reaction. Instead, we favor a mechanism in which free aryl nitrene is released during the catalytic cycle and combines with itself or with free aryl azide to yield the azoarene.
Co-reporter:Charles C. L. McCrory ; Christopher Uyeda
Journal of the American Chemical Society 2012 Volume 134(Issue 6) pp:3164-3170
Publication Date(Web):January 12, 2012
DOI:10.1021/ja210661k
A series of water-soluble molecular cobalt complexes of tetraazamacrocyclic ligands are reported for the electrocatalytic production of H2 from pH 2.2 aqueous solutions. The comparative data reported for this family of complexes shed light on their relative efficiencies for hydrogen evolution in water. Rotating disk electrode voltammetry data are presented for each of the complexes discussed, as are data concerning their respective pH-dependent electrocatalytic activity. In particular, two diimine–dioxime complexes were identified as exhibiting catalytic onset at comparatively low overpotentials relative to other reported homogeneous cobalt and nickel electrocatalysts in aqueous solution. These complexes are stable at pH 2.2 and produce hydrogen with high Faradaic efficiency in bulk electrolysis experiments over time intervals ranging from 2 to 24 h.
Co-reporter:W. Hill Harman
Journal of the American Chemical Society 2012 Volume 134(Issue 11) pp:5080-5082
Publication Date(Web):March 1, 2012
DOI:10.1021/ja211419t
We report the synthesis and characterization of a series of nickel complexes of the chelating diphosphine-borane ligands ArB(o-Ph2PC6H4)2 ([ArDPBPh]; Ar = Ph, Mes). The [ArDPBPh] framework supports pseudo-tetrahedral nickel complexes featuring η2-B,C coordination from the ligand backbone. For the B-phenyl derivative, the THF adduct [PhDPBPh]Ni(THF) has been characterized by X-ray diffraction and features a very short interaction between nickel and the η2-B,C ligand. For the B-mesityl derivative, the reduced nickel complex [MesDPBPh]Ni is isolated as a pseudo-three-coordinate “naked” species that undergoes reversible, nearly thermoneutral oxidative addition of dihydrogen to give a borohydrido-hydride complex of nickel(II) which has been characterized in solution by multinuclear NMR. Furthermore, [MesDPBPh]Ni is an efficient catalyst for the hydrogenation of olefin substrates under mild conditions.
Co-reporter:Charlene Tsay and Jonas C. Peters
Chemical Science 2012 vol. 3(Issue 4) pp:1313-1318
Publication Date(Web):19 Jan 2012
DOI:10.1039/C2SC01033J
The first examples of thermally stable molecular dihydrogen adducts of nickel were synthesized from their dinitrogen adduct precursors, which are themselves among the first examples of Ni(II)-N2 complexes. The minimal activation of the bound N2 moieties suggests that these adducts are stabilized predominantly through σ-donation from the adduct to the electrophilic metal center. We further show that the bound H2 ligand can undergo heterolytic cleavage to deliver hydride to the nickel center. The H2 adducts are of particular interest in the context of hypotheses suggesting that Ni can serve as the site for H2 binding and heterolytic activation in [NiFe] hydrogenases.
Co-reporter:Ayumi Takaoka and Jonas C. Peters
Inorganic Chemistry 2012 Volume 51(Issue 1) pp:16-18
Publication Date(Web):December 1, 2011
DOI:10.1021/ic202079r
We herein present a series of d7 trimethylphosphine complexes of group 9 metals that are chelated by the tripodal tetradentate tris(phosphino)silyl ligand [SiPiPr3]H ([SiPiPr3] = (2-iPr2PC6H4)3Si–). Both electron paramagnetic resonance (EPR) simulations and density functional theory (DFT) calculations indicate largely metalloradical character. These complexes provide a rare opportunity to compare the properties between the low-valent metalloradicals of the second- and third-row transition metals with the corresponding first-row analogues.
Co-reporter:Caroline T. Saouma, Connie C. Lu, and Jonas C. Peters
Inorganic Chemistry 2012 Volume 51(Issue 18) pp:10043-10054
Publication Date(Web):September 5, 2012
DOI:10.1021/ic301704f
This article describes the synthesis and characterization of several low-spin iron(II) complexes that coordinate hydrazine (N2H4), hydrazido (N2H3–), and ammonia. The sterically encumbered tris(di-meta-terphenylphosphino)borate ligand, [PhBPmter3]−, is introduced to provide access to species that cannot be stabilized with the [PhBPPh3]− ligand ([PhBPR3]− = PhB(CH2PR2)3–). Treatment of [PhBPmter3]FeMe with hydrazine generates the unusual 5-coordinate hydrazido complex [PhBPmter3]Fe(η2-N2H3) (1), in which the hydrazido serves as an L2X-type ligand. Upon coordination of an L-type ligand, the hydrazido shifts to an LX-type ligand, generating [PhBPmter3]Fe(L)(η2-N2H3) (L = N2H4 (2) or NH3 (3)). In contrast, treatment of [PhBPPh3]FeMe with hydrazine forms the adduct [PhBPPh3]Fe(Me)(η2-N2H4) (5). Complex 5 is thermally unstable to methane loss, generating intermediate [PhBPPh3]Fe(η2-N2H3), which undergoes bimolecular coupling to produce {[PhBPPh3]Fe}2(μ-η1:η1-N2H4)(μ-η2:η2-N2H2). The oxidation of these and related hydrazine and hydrazido species is also presented. For example, oxidation of 1 or 5 with Pb(OAc)4 results in disproportionation of the N2Hx ligand (x = 3, 4), and formation of [PhBPR3]Fe(NH3)(OAc) (R = Ph (9) and mter (11)).
Co-reporter:Daniel L. M. Suess
Organometallics 2012 Volume 31(Issue 15) pp:5213-5222
Publication Date(Web):May 31, 2012
DOI:10.1021/om3001229
The preparation of a new tridentate diphosphinosulfinyl ligand is described as well as the synthesis and properties of some of its Rh, Ir, Ni, Pd, and Pt complexes. The ligand binds in a κ3-PS(O)P fashion in all cases. The M–S lengths in (SOP2)RhCl and (SOP2)IrCl (2.1340(8) and 2.1341(5) Å, respectively) are the shortest of all crystallographically characterized Rh– and Ir–S(O)R2 complexes, which illustrates the significant M–S(O) π-backbonding for these metals. Akin to Vaska’s complex, (SOP2)IrCl binds O2 to yield a peroxide complex with an O–O length of 1.465(3) Å and ν(O–O) = 847 cm–1. The C–O stretches of (SOP2)M(CO) (M = Ni, Pd, Pt) are ∼30–40 cm–1 higher than those of the analogous (PPh3)3M(CO). A number of divalent group 10 complexes of the form [(SOP2)MX]+ for Ni (X = Cl), Pd (X = Cl, Me), and Pt (X = Cl, Me) are reported, as well as the highly electrophilic complex [(SOP2)Pd(NCCH3)]2+. An analysis of the S–O length as a function of the M–S length for all d8 SOP2 Rh, Ir, Pd, and Pt complexes reveals that the electron-withdrawing capabilities of the sulfinyl group are mediated largely through π-backbonding for Rh and Ir and mostly through poor σ-donation for Pd and Pt.
Co-reporter:Sidney E. Creutz;Kenneth J. Lotito;Gregory C. Fu
Science 2012 Volume 338(Issue 6107) pp:647-651
Publication Date(Web):02 Nov 2012
DOI:10.1126/science.1226458
Co-reporter:Yunho Lee
Journal of the American Chemical Society 2011 Volume 133(Issue 12) pp:4438-4446
Publication Date(Web):March 4, 2011
DOI:10.1021/ja109678y
A series of monocarbonyl iron complexes in the formal oxidation states 0, +1, and +2 are accessible when supported by a tetradentate tris(phosphino)silyl ligand (SiPiPr3 = [Si(o-C6H4PiPr2)3]−). X-ray diffraction (XRD) studies of these carbonyl complexes establish little geometrical change about the iron center as a function of oxidation state. It is possible to functionalize the terminal CO ligand of the most reduced carbonyl adduct by addition of SiMe3+ to afford a well-defined iron carbyne species, (SiPiPr3)Fe≡C—OSiMe3. Single-crystal XRD data of this iron carbyne derivative reveal an unusually short Fe≡C—OSiMe3 bond distance (1.671(2) Å) and a substantially elongated C−O distance (1.278(3) Å), consistent with Fe−C carbyne character. The overall trigonal bipyramidal geometry of (SiPiPr3)Fe≡C—OSiMe3 compares well with that of the corresponding carbonyls, (SiPiPr3)Fe(CO)−, (SiPiPr3)Fe(CO), and (SiPiPr3)Fe(CO)+. Details regarding the electronic structure of the carbyne complex have been explored via the collection of comparative Mössbauer data for all of the complexes featured and also via DFT calculations. In sum, these data point to a strongly π-accepting Fischer-type carbyne ligand that confers stability to a low-valent iron(0) rather than high-valent iron(IV) center.
Co-reporter:Yunho Lee ; R. Adam Kinney ; Brian M. Hoffman
Journal of the American Chemical Society 2011 Volume 133(Issue 41) pp:16366-16369
Publication Date(Web):September 28, 2011
DOI:10.1021/ja207003m
We have exploited the capacity of the “(SiPiPr3)Fe(I)” scaffold to accommodate additional axial ligands and characterized the mononuclear S = 1/2 H2 adduct complex (SiPiPr3)FeI(H2). EPR and ENDOR data, in the context of X-ray structural results, revealed that this complex provides a highly unusual example of an open-shell metal complex that binds dihydrogen as a ligand. The H2 ligand at 2 K dynamically reorients within the ligand-binding pocket, tunneling among the energy minima created by strong interactions with the three Fe–P bonds.
Co-reporter:Marc-Etienne Moret
Journal of the American Chemical Society 2011 Volume 133(Issue 45) pp:18118-18121
Publication Date(Web):October 18, 2011
DOI:10.1021/ja208675p
The reactivity of the anionic dinitrogen complex [(TPB)Fe(N2)]− (TPB = tris[2-(diisopropylphosphino)phenyl]borane) toward silicon electrophiles has been examined. [(TPB)Fe(N2)]− reacts with trimethylsilyl chloride to yield the silyldiazenido complex (TPB)Fe(NNSiMe3), which is reduced by Na/Hg in THF to yield the corresponding sodium-bound anion [(TPB)Fe(NNSiMe3)]Na(THF). The use of 1,2-bis(chlorodimethylsilyl)ethane in the presence of excess Na/Hg results in the disilylation of the bound N2 molecule to yield the disilylhydrazido(2−) complex (TPB)Fe≡NR (R = 2,2,5,5-tetramethyl-1-aza-2,5-disilacyclopentyl). One of the phosphine arms of TPB in (TPB)Fe≡NR can be substituted by CO or tBuNC to yield crystalline adducts (TPB)(L)Fe≡NR (L = CO, tBuNC). The N–N bond in (TPB)(tBuNC)Fe≡NR is cleaved upon standing at room temperature to yield a phosphoraniminato/disilylamido iron(II) complex. The flexibility of the Fe–B linkage is thought to play a key role in these transformations of Fe-bound dinitrogen.
Co-reporter:Bryan D. Stubbert ; Jonas C. Peters ;Harry B. Gray
Journal of the American Chemical Society 2011 Volume 133(Issue 45) pp:18070-18073
Publication Date(Web):October 24, 2011
DOI:10.1021/ja2078015
A cobalt bis(iminopyridine) complex is a highly active electrocatalyst for water reduction, with an estimated apparent second order rate constant kapp ≤ 107 M–1s–1 over a range of buffer/salt concentrations. Scan rate dependence data are consistent with freely diffusing electroactive species over pH 4–9 at room temperature for each of two catalytic reduction events, one of which is believed to be ligand based. Faradaic H2 yields up to 87 ± 10% measured in constant potential electrolyses (−1.4 V vs SCE) confirm high reactivity and high fidelity in a catalyst supported by the noninnocent bis(iminopyridine) ligand. A mechanism involving initial reduction of Co2+ and subsequent protonation is proposed.
Co-reporter:Ayumi Takaoka ; Neal P. Mankad
Journal of the American Chemical Society 2011 Volume 133(Issue 22) pp:8440-8443
Publication Date(Web):May 16, 2011
DOI:10.1021/ja2020907
We report a unique class of dinitrogen complexes of iron featuring sulfur donors in the ancillary ligand. The ligands utilized are related to the recently studied tris(phosphino)silyl ligands (2-R2PC6H4)3Si (R = Ph, iPr) but have one or two phosphine arms replaced with thioether donors. Depending on the number of phosphine arms replaced, both mononuclear and dinuclear iron complexes with dinitrogen are accessible. These complexes contribute to a desirable class of model complexes that possess both dinitrogen and sulfur ligands in the immediate iron coordination sphere.
Co-reporter:Caroline T. Saouma, Jonas C. Peters
Coordination Chemistry Reviews 2011 Volume 255(7–8) pp:920-937
Publication Date(Web):April 2011
DOI:10.1016/j.ccr.2011.01.009
Mid-to-late transition metal complexes that feature terminal, multiply bonded ligands such as oxos, imides, and nitrides have been invoked as intermediates in several catalytic transformations of synthetic and biological significance. Until about ten years ago, isolable examples of such species were virtually unknown. Over the past decade or so, numerous chemically well-defined examples of such species have been discovered. In this context, the present review summarizes the development of 4- and 5-coordinate Fe(E) and Co(E) species under local three-fold symmetry.Research highlights► Mid-to-late transition metal complexes that feature terminal, multiply bonded ligands. ► Review of 4- and 5-coordinate L3M(E) and L′L3M(E) species (E = oxo, imide, nitride, M = Fe or Co). ► FeNx triple bond across five oxidation states. ► Atom and group transfer reactions to install multiply bonded ligands.
Co-reporter:Caroline T. Saouma ; Curtis E. Moore ; Arnold L. Rheingold
Inorganic Chemistry 2011 Volume 50(Issue 22) pp:11285-11287
Publication Date(Web):October 17, 2011
DOI:10.1021/ic2016066
A family of iron(II) complexes that coordinate dinitrogen, diazene, hydrazine, and ammonia are presented. This series of complexes is unusual in that the complexes within it feature a common auxiliary ligand set and differ only by virtue of the nitrogenous NxHy ligand that occupies the sixth binding site. The ability of an iron center to bind N2, N2H2, N2H4, and NH3 is important to establish in the context of evaluating catalytic N2 reduction schemes that invoke these nitrogenous species. Such a scenario has been proposed as an iron-mediated, alternating reduction scheme within the cofactor of nitrogenase enzymes.
Co-reporter:Mina Mazzeo, Maria Strianese, Olaf Kühl and Jonas C. Peters
Dalton Transactions 2011 vol. 40(Issue 35) pp:9026-9033
Publication Date(Web):09 Aug 2011
DOI:10.1039/C1DT10825E
A series of platinum(II) complexes supported by the tridentate bis(phosphine)phosphido ligand bis(2-diisopropylphosphinophenyl)phosphide) [iPr–PPP] have been synthesized and characterized (1–4). X-Ray structural studies of [iPr–PPP]PtCl (1) and [iPr–PPP]PtCH3 (3) complexes show meridional [iPr–PPP] ligands around approximately square-planar platinum centers. Structural data and NMR analysis highlight a strong trans influence for the phosphido phosphorous donor, comparable to that of the anionic aryl carbon of the classic PCP pincer complexes. A series of thermally stable [PPP]Pt(IV) compounds, including [PPP]Pt(CH3)2X [X = I (5) and SbF6 (6)], were also synthesized. The study of the binding affinity of SO2 and NO to complex 1 has also been addressed.
Co-reporter:Dr. Marc-Etienne Moret ; Jonas C. Peters
Angewandte Chemie International Edition 2011 Volume 50( Issue 9) pp:2063-2067
Publication Date(Web):
DOI:10.1002/anie.201006918
Co-reporter:Caroline T. Saouma;R. Adam Kinney; Brian M. Hoffman; Jonas C. Peters
Angewandte Chemie International Edition 2011 Volume 50( Issue 15) pp:3446-3449
Publication Date(Web):
DOI:10.1002/anie.201006299
Co-reporter:Dr. Marc-Etienne Moret ; Jonas C. Peters
Angewandte Chemie 2011 Volume 123( Issue 9) pp:2111-2115
Publication Date(Web):
DOI:10.1002/ange.201006918
Co-reporter:Joseph C. Deaton ; Steven C. Switalski ; Denis Y. Kondakov ; Ralph H. Young ; Thomas D. Pawlik ; David J. Giesen ; Seth B. Harkins ; Alex J. M. Miller ; Seth F. Mickenberg
Journal of the American Chemical Society 2010 Volume 132(Issue 27) pp:9499-9508
Publication Date(Web):June 17, 2010
DOI:10.1021/ja1004575
A highly emissive bis(phosphine)diarylamido dinuclear copper(I) complex (quantum yield = 57%) was shown to exhibit E-type delayed fluorescence by variable temperature emission spectroscopy and photoluminescence decay measurement of doped vapor-deposited films. The lowest energy singlet and triplet excited states were assigned as charge transfer states on the basis of theoretical calculations and the small observed S1−T1 energy gap. Vapor-deposited OLEDs doped with the complex in the emissive layer gave a maximum external quantum efficiency of 16.1%, demonstrating that triplet excitons can be harvested very efficiently through the delayed fluorescence channel. The function of the emissive dopant in OLEDs was further probed by several physical methods, including electrically detected EPR, cyclic voltammetry, and photoluminescence in the presence of applied current.
Co-reporter:Ayumi Takaoka;LauraC.H. Gerber;JonasC. Peters
Angewandte Chemie 2010 Volume 122( Issue 24) pp:4182-4185
Publication Date(Web):
DOI:10.1002/ange.201001199
Co-reporter:Ayumi Takaoka;LauraC.H. Gerber;JonasC. Peters
Angewandte Chemie International Edition 2010 Volume 49( Issue 24) pp:4088-4091
Publication Date(Web):
DOI:10.1002/anie.201001199
Co-reporter:Neal P. Mankad;Matthew T. Whited;Jonas C. Peters
Angewandte Chemie 2007 Volume 119(Issue 30) pp:
Publication Date(Web):28 JUN 2007
DOI:10.1002/ange.200701188
Eine Stütze für FeI: Monoanionische Tris(phosphanyl)silyl-Liganden ([SiPR3]=[(2-R2PC6H4)3Si]−) stabilisieren fünffach-koordinierte Eisen(I)-Komplexe. Das Silan [SiPPh3]H reagiert mit Mesityleisen(II) zu [(SiPPh3)FeIIMes], in dem eine agostische Wechselwirkung mit einer C-H-Bindung vorliegt. Nach Umsetzung mit HCl und Reduktion entsteht der terminale FeI-N2-Komplex [(SiPPh3)FeN2] (siehe Schema), der protolytisch zu Hydrazin gespalten wird.
Co-reporter:Neal P. Mankad;Matthew T. Whited;Jonas C. Peters
Angewandte Chemie International Edition 2007 Volume 46(Issue 30) pp:
Publication Date(Web):28 JUN 2007
DOI:10.1002/anie.200701188
Taking hold: Monoanionic tris(phosphino)silyl ligands ([SiPR3]=[(2-R2PC6H4)3Si]−) stabilize unusual five-coordinate FeI complexes. The silane [SiPPh3]H reacts with mesityliron(II) to give [(SiPPh3)FeIIMes], which displays a CH agostic interaction. Treatment with HCl and reduction affords the terminally bonded FeIN2 complex [(SiPPh3)FeN2] (see scheme; structures show only donor atoms of [SiPPh3]), which yields hydrazine under protolytic conditions.
Co-reporter:Matthew T. Whited, Eric Rivard and Jonas C. Peters
Chemical Communications 2006 (Issue 15) pp:1613-1615
Publication Date(Web):10 Mar 2006
DOI:10.1039/B516046D
Divalent complexes of iron and cobalt with new, monoanionic tripodal amido-polyphosphine ligands have been thoroughly characterized, and XRD analysis reveals geometries that are distinct for this class of ligand.
Co-reporter:Mark P. Mehn, Steven D. Brown, Tapan K. Paine, William W. Brennessel, Christopher J. Cramer, Jonas C. Peters and Lawrence Que, Jr.
Dalton Transactions 2006 (Issue 10) pp:1347-1351
Publication Date(Web):17 Nov 2005
DOI:10.1039/B509580H
Rare examples of monometallic high-spin and low-spin L3Fe(H3BH) complexes have been characterized, where the two L3 ligands are [TpPh2] and [PhBP3] ([TpPh2] = [HB(3,5-Ph2pz)3]− and [PhBP3] = [PhB(CH2PPh2)3]−). The structures are reported wherein the borohydride ligand is facially coordinated to the iron center in each complex. Density functional methods have been employed to explain the bonding in these unusual iron(II) centers. Despite the differences in spin states, short Fe–B distances are observed in both complexes and there is significant theoretical evidence to support a substantial bonding interaction between the iron and boron nuclei. In light of this interaction, we suggest that these complexes can be described as (L3)Fe(η4-H3BH) complexes.
Co-reporter:Christine M. Thomas Professor
Angewandte Chemie 2006 Volume 118(Issue 5) pp:
Publication Date(Web):21 DEC 2005
DOI:10.1002/ange.200502527
Die Reaktion des High-Spin-Eisen(II)-Alkylkomplexes [{PhB(CH2PiPr2)3}FeMe] mit Phenyl- oder Mesitylsilan ergibt ungewöhnliche diamagnetische Silan-Addukte des Eisen(II)-hydrids [{PhB(CH2PiPr2)3}FeH]. Die Ergebnisse von NMR-Spektroskopie, Röntgenkristallographie und DFT-Rechnungen sprechen für einen η3-Bindungsmodus. Für den schnellen Austausch zwischen den Hydridpositionen in Lösung wurde ein Silylen-Intermediat vorgeschlagen.
Co-reporter:Christine M. Thomas,Jonas C. Peters
Angewandte Chemie International Edition 2006 45(5) pp:776-780
Publication Date(Web):
DOI:10.1002/anie.200502527
Co-reporter:Xile Hu, Brandi M. Cossairt, Bruce S. Brunschwig, Nathan S. Lewis and Jonas C. Peters
Chemical Communications 2005 (Issue 37) pp:4723-4725
Publication Date(Web):23 Aug 2005
DOI:10.1039/B509188H
In the presence of moderately strong acids in CH3CN, cobalt complexes with BF2-bridged diglyoxime ligands are active catalysts for the reduction of protons to H2 at potentials as positive as −0.28 V vs. SCE.
Co-reporter:Theodore A. Betley
Angewandte Chemie International Edition 2003 Volume 42(Issue 21) pp:
Publication Date(Web):28 MAY 2003
DOI:10.1002/anie.200250378
Formally zwitterionic bis(phosphanyl)- and bis(amino)borate rhodium(I) complexes (see picture) can catalytically mediate the hydrogenation, hydroacylation, hydroboration, and hydrosilation of double bonds. These neutral systems are shown to be highly active, even under conditions incompatible with their isostructural, but formally cationic, relatives.
Co-reporter:Theodore A. Betley
Angewandte Chemie 2003 Volume 115(Issue 21) pp:
Publication Date(Web):28 MAY 2003
DOI:10.1002/ange.200250378
Zwitterionische Bis(phosphanyl)- und Bis(amino)borat-Rhodium(I)-Komplexe (siehe Bild) katalysieren Hydrierungen, Hydroacylierungen, Hydroborierungen und Hydrosilylierungen von Doppelbindungen. Die Neutralverbindungen arbeiten hocheffizient und reagieren auch unter Bedingungen, bei denen ihre isostrukturellen kationischen Analoga unwirksam sind.
Co-reporter:Ian R. Shapiro, David M. Jenkins, J. Christopher Thomas, Michael W. Day and Jonas C. Peters
Chemical Communications 2001 (Issue 20) pp:2152-2153
Publication Date(Web):27 Sep 2001
DOI:10.1039/B104447H
A homoleptic phosphine adduct of thallium(I) supported by a tris(phosphino)borate ligand has been isolated and structurally characterized.
Co-reporter:Tzu-Pin Lin
Journal of the American Chemical Society () pp:
Publication Date(Web):September 2, 2014
DOI:10.1021/ja504667f
New approaches toward the generation of late first-row metal catalysts that efficiently facilitate two-electron reductive transformations (e.g., hydrogenation) more typical of noble-metal catalysts is an important goal. Herein we describe the synthesis of a structurally unusual S = 1 bimetallic Co complex, [(CyPBP)CoH]2 (1), supported by bis(phosphino)boryl and bis(phosphino)hydridoborane ligands. This complex reacts reversibly with a second equivalent of H2 (1 atm) and serves as an olefin hydrogenation catalyst under mild conditions (room temperature, 1 atm H2). A bimetallic Co species is invoked in the rate-determining step of the catalysis according to kinetic studies. A structurally related NiINiI dimer, [(PhPBP)Ni]2 (3), has also been prepared. Like Co catalyst 1, Ni complex 3 displays reversible reactivity toward H2, affording the bimetallic complex [(PhPBHP)NiH]2 (4). This reversible behavior is unprecedented for NiI species and is attributed to the presence of a boryl–Ni bond. Lastly, a series of monomeric (tBuPBP)NiX complexes (X = Cl (5), OTf (6), H (7), OC(H)O (8)) have been prepared. The complex (tBuPBP)NiH (7) shows enhanced catalytic olefin hydrogenation activity when directly compared with its isoelectronic/isostructural analogues where the boryl unit is substituted by a phenyl or amine donor, a phenomenon that we posit is related to the strong trans influence exerted by the boryl ligand.
Co-reporter:Charlene Tsay ; Neal P. Mankad
Journal of the American Chemical Society () pp:
Publication Date(Web):
DOI:10.1021/ja105284p
We report herein the characterization of electrophilic, trigonal bipyramidal {[SiP3R]Pt(L)}+ cations ([SiP3R] = [(2-R2PC6H4)3Si]; R = Ph, iPr) that feature weakly coordinated ligands including CH2Cl2, Et2O, toluene, and H2. A cationic toluene adduct that shows a close platinum aryl C−H σ-contact is perhaps most noteworthy in this context. For the isopropyl-substituted ligand, [SiP3iPr], it has proven possible to exclude the fifth axial donor to afford the rigorously four-coordinate, trigonal pyramidal (TP) complex {[SiP3iPr]Pt}+. An isostructural TP palladium complex {[SiP3iPr]Pd}+ is also accessible. Prototypical four-coordinate d8 platinum and palladium complexes are square planar. The TP d8 cations described herein are hence geometrically distinct.
Co-reporter:Trevor J. Del Castillo; Niklas B. Thompson; Daniel L. M. Suess; Gaël Ung
Inorganic Chemistry () pp:
Publication Date(Web):
DOI:10.1021/acs.inorgchem.5b00645
Well-defined molecular catalysts for the reduction of N2 to NH3 with protons and electrons remain very rare despite decades of interest and are currently limited to systems featuring molybdenum or iron. This report details the synthesis of a molecular cobalt complex that generates superstoichiometric yields of NH3 (>200% NH3 per Co–N2 precursor) via the direct reduction of N2 with protons and electrons. While the NH3 yields reported herein are modest by comparison to those of previously described iron and molybdenum systems, they intimate that other metals are likely to be viable as molecular N2 reduction catalysts. Additionally, a comparison of the featured tris(phosphine)borane Co–N2 complex with structurally related Co–N2 and Fe–N2 species shows how remarkably sensitive the N2 reduction performance of potential precatalysts is. These studies enable consideration of the structural and electronic effects that are likely relevant to N2 conversion activity, including the π basicity, charge state, and geometric flexibility.
Co-reporter:Miles W. Johnson, Kareem I. Hannoun, Yichen Tan, Gregory C. Fu and Jonas C. Peters
Chemical Science (2010-Present) 2016 - vol. 7(Issue 7) pp:NaN4100-4100
Publication Date(Web):2016/02/24
DOI:10.1039/C5SC04709A
Photoinduced, copper-catalyzed cross-coupling can offer a complementary approach to thermal (non-photoinduced) methods for generating C–X (X = C, N, O, S, etc.) bonds. In this report, we describe the first detailed mechanistic investigation of one of the processes that we have developed, specifically, the (stoichiometric) coupling of a copper–thiolate with an aryl iodide. In particular, we focus on the chemistry of a discrete [CuI(SAr)2]− complex (Ar = 2,6-dimethylphenyl), applying a range of techniques, including ESI-MS, cyclic voltammetry, transient luminescence spectroscopy, optical spectroscopy, DFT calculations, Stern–Volmer analysis, EPR spectroscopy, actinometry, and reactivity studies. The available data are consistent with the viability of a pathway in which photoexcited [CuI(SAr)2]−* serves as an electron donor to an aryl iodide to afford an aryl radical, which then reacts in cage with the newly generated copper(II)–thiolate to furnish the cross-coupling product in a non-chain process.
Co-reporter:Yichen Tan, José María Muñoz-Molina, Gregory C. Fu and Jonas C. Peters
Chemical Science (2010-Present) 2014 - vol. 5(Issue 7) pp:NaN2835-2835
Publication Date(Web):2014/04/22
DOI:10.1039/C4SC00368C
Most copper-catalyzed cross-couplings require an elevated reaction temperature. Recently, a photoinduced variant has been developed that enables C–X bond-forming reactions of certain nitrogen and sulfur nucleophiles to proceed under unusually mild conditions (−40 °C to room temperature). In view of the importance of carbon–oxygen bond construction in organic chemistry, the expansion of this photochemical approach to oxygen nucleophiles is an important objective. In this report, we establish that, in the presence of light and an inexpensive copper pre-catalyst (CuI), a wide array of phenols and aryl iodides can be coupled to generate diaryl ethers under mild conditions (room temperature) in the presence of a variety of functional groups. Our studies indicate that a Cu(I)–phenoxide complex is a viable intermediate in photoinduced C–O bond-formation.
Co-reporter:Sidney E. Creutz and Jonas C. Peters
Chemical Science (2010-Present) 2017 - vol. 8(Issue 3) pp:NaN2328-2328
Publication Date(Web):2016/12/08
DOI:10.1039/C6SC04805F
Hydrogen bonding and other types of secondary-sphere interactions are ubiquitous in metalloenzyme active sites and are critical to the transformations they mediate. Exploiting secondary sphere interactions in synthetic catalysts to study the role(s) they might play in biological systems, and to develop increasingly efficient catalysts, is an important challenge. Whereas model studies in this broad context are increasingly abundant, as yet there has been relatively little progress in the area of synthetic catalysts for nitrogen fixation that incorporate secondary sphere design elements. Herein we present our first study of Fe–NxHy complexes supported by new tris(phosphine)silyl ligands, abbreviated as [SiPNMe3] and [SiPiPr2PNMe], that incorporate remote tertiary amine hydrogen-bond acceptors within a tertiary phosphine/amine 6-membered ring. These remote amine sites facilitate hydrogen-bonding interactions via a boat conformation of the 6-membered ring when certain nitrogenous substrates (e.g., NH3 and N2H4) are coordinated to the apical site of a trigonal bipyramidal iron complex, and adopt a chair conformation when no H-bonding is possible (e.g., N2). Countercation binding at the cyclic amine is also observed for anionic {Fe–N2}− complexes. Reactivity studies in the presence of proton/electron sources show that the incorporated amine functionality leads to rapid generation of catalytically inactive Fe–H species, thereby substantiating a hydride termination pathway that we have previously proposed deactivates catalysts of the type [EPR3]FeN2 (E = Si, C).
Co-reporter:Mina Mazzeo, Maria Strianese, Olaf Kühl and Jonas C. Peters
Dalton Transactions 2011 - vol. 40(Issue 35) pp:NaN9033-9033
Publication Date(Web):2011/08/09
DOI:10.1039/C1DT10825E
A series of platinum(II) complexes supported by the tridentate bis(phosphine)phosphido ligand bis(2-diisopropylphosphinophenyl)phosphide) [iPr–PPP] have been synthesized and characterized (1–4). X-Ray structural studies of [iPr–PPP]PtCl (1) and [iPr–PPP]PtCH3 (3) complexes show meridional [iPr–PPP] ligands around approximately square-planar platinum centers. Structural data and NMR analysis highlight a strong trans influence for the phosphido phosphorous donor, comparable to that of the anionic aryl carbon of the classic PCP pincer complexes. A series of thermally stable [PPP]Pt(IV) compounds, including [PPP]Pt(CH3)2X [X = I (5) and SbF6 (6)], were also synthesized. The study of the binding affinity of SO2 and NO to complex 1 has also been addressed.
Co-reporter:Charlene Tsay and Jonas C. Peters
Chemical Science (2010-Present) 2012 - vol. 3(Issue 4) pp:NaN1318-1318
Publication Date(Web):2012/01/19
DOI:10.1039/C2SC01033J
The first examples of thermally stable molecular dihydrogen adducts of nickel were synthesized from their dinitrogen adduct precursors, which are themselves among the first examples of Ni(II)-N2 complexes. The minimal activation of the bound N2 moieties suggests that these adducts are stabilized predominantly through σ-donation from the adduct to the electrophilic metal center. We further show that the bound H2 ligand can undergo heterolytic cleavage to deliver hydride to the nickel center. The H2 adducts are of particular interest in the context of hypotheses suggesting that Ni can serve as the site for H2 binding and heterolytic activation in [NiFe] hydrogenases.
Co-reporter:Caroline T. Saouma, Connie C. Lu, Michael W. Day and Jonas C. Peters
Chemical Science (2010-Present) 2013 - vol. 4(Issue 10) pp:NaN4051-4051
Publication Date(Web):2013/07/12
DOI:10.1039/C3SC51262B
This manuscript explores the product distribution of the reaction of carbon dioxide with reactive iron(I) complexes supported by tris(phosphino)borate ligands, [PhBPR3]− ([PhBPR3]− = [PhB(CH2PR2)3]−; R = CH2Cy, Ph, iPr, mter; mter = 3,5-meta-terphenyl). Our studies reveal an interesting and unexpected role for the solvent medium with respect to the course of the CO2 activation reaction. For instance, exposure of methylcyclohexane (MeCy) solutions of to CO2 yields the partial decarbonylation product . When the reaction is instead carried out in benzene or THF, reductive coupling of CO2 occurs to give the bridging oxalate species . Reaction studies aimed at understanding this solvent effect are presented, and suggest that the product profile is ultimately determined by the ability of the solvent to coordinate the iron center. When more sterically encumbering auxiliary ligands are employed to support the iron(I) center (i.e., [PhBPPh3]− and [PhBPiPr3]−), complete decarbonylation is observed to afford structurally unusual diiron(II) products of the type {[PhBPR3]Fe}2(μ-O). A mechanistic hypothesis that is consistent with the collection of results described is offered, and suggests that reductive coupling of CO2 likely occurs from an electronically saturated “FeII–CO2˙−” species.
Co-reporter:Christopher Uyeda and Jonas C. Peters
Chemical Science (2010-Present) 2013 - vol. 4(Issue 1) pp:NaN163-163
Publication Date(Web):2012/09/12
DOI:10.1039/C2SC21231E
Heterobimetallic NiZn complexes featuring metal centers in distinct coordination environments have been synthesized using diimine–dioxime ligands as binucleating scaffolds. A tetramethylfuran-containing ligand derivative enables a stable one-electron-reduced S = 1/2 species to be accessed using Cp2Co as a chemical reductant. The resulting pseudo-square planar complex exhibits spectroscopic and crystallographic characteristics of a ligand-centered radical bound to a Ni(II) center. Upon coordination of a π-acidic ligand such as PPh3, however, a five-coordinate Ni(I) metalloradical is formed. The electronic structures of these reduced species provide insight into the subtle effects of ligand structure on the potential and reversibility of the NiII/I couple for complexes of redox-active tetraazamacrocycles.
Co-reporter:Samantha N. MacMillan, W. Hill Harman and Jonas C. Peters
Chemical Science (2010-Present) 2014 - vol. 5(Issue 2) pp:NaN597-597
Publication Date(Web):2013/10/22
DOI:10.1039/C3SC52626G
Metal–borane complexes are emerging as promising systems for study in the context of bifunctional catalysis. Herein we describe diphosphineborane nickel complexes that activate Si–H bonds and catalyze the hydrosilylation of aldehydes. Treatment of [MesDPBPh]Ni (1) ([MesDPBPh] = MesB(o-Ph2PC6H4)2) with organosilanes affords the complexes [MesDPBPh](μ-H)NiE (E = SiH2Ph (3), SiHPh2 (4)). Complex 4 is in solution equilibrium with 1 and the thermodynamic and kinetic parameters of their exchange have been characterized by NMR spectroscopy. Complex 1 is a catalyst for the hydrosilylation of a range of para-substituted benzaldehydes. Mechanistic studies on this reaction via multinuclear NMR spectroscopy are consistent with the intermediacy of a borohydrido-Ni-siloxyalkyl species.
.jpg)
![4H-Diindeno[7,1-cd:1',7'-ef]phosphocin,5,6,10,11,12,13-hexahydro-5-phenyl-, (11aR)- (9CI)](http://img.cochemist.com/ccimg/856500/856407-37-9.png)
![4H-Diindeno[7,1-cd:1',7'-ef]phosphocin,5,6,10,11,12,13-hexahydro-5-phenyl-, (11aR)- (9CI)](http://img.cochemist.com/ccimg/856500/856407-37-9_b.png)
![L-ISOLEUCINE, N-[(6-BROMO-2,3-DIHYDRO-1-OXO-1H-INDEN-4-YL)CARBONYL]-](http://img.cochemist.com/ccimg/863500/863489-95-6.png)
![L-ISOLEUCINE, N-[(6-BROMO-2,3-DIHYDRO-1-OXO-1H-INDEN-4-YL)CARBONYL]-](http://img.cochemist.com/ccimg/863500/863489-95-6_b.png)



![Tert-butyl-[2-(1h-indol-3-yl)ethoxy]-dimethylsilane](http://img.cochemist.com/ccimg/101100/101079-48-5.png)
![Tert-butyl-[2-(1h-indol-3-yl)ethoxy]-dimethylsilane](http://img.cochemist.com/ccimg/101100/101079-48-5_b.png)




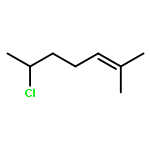



![ETHYL {3-[(DIETHYLCARBAMOYL)SULFANYL]PHENYL}ACETATE](http://img.cochemist.com/ccimg/74000/73900-07-9.png)
![ETHYL {3-[(DIETHYLCARBAMOYL)SULFANYL]PHENYL}ACETATE](http://img.cochemist.com/ccimg/74000/73900-07-9_b.png)




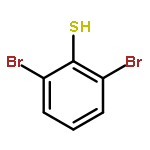
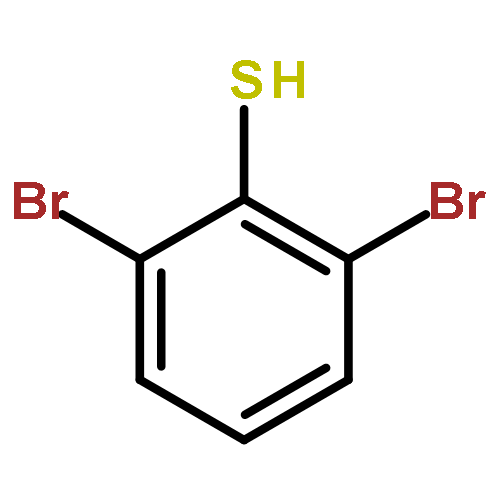



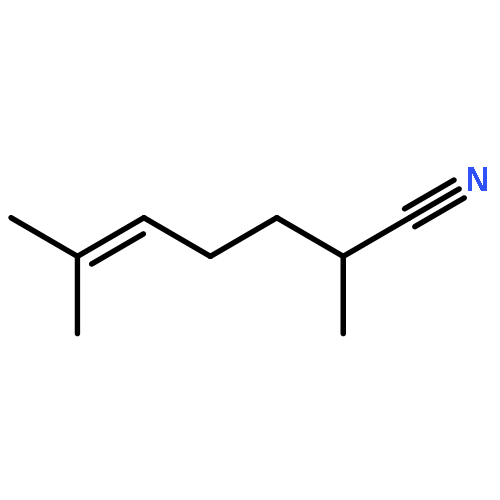





![Tricyclo[3.3.1.13,7]decane, 1-azido-](/data/chemimg/409500/24886-73-5.png)
![Tricyclo[3.3.1.13,7]decane, 1-azido-](/data/chemimg/409500/24886-73-5_b.png)







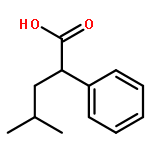







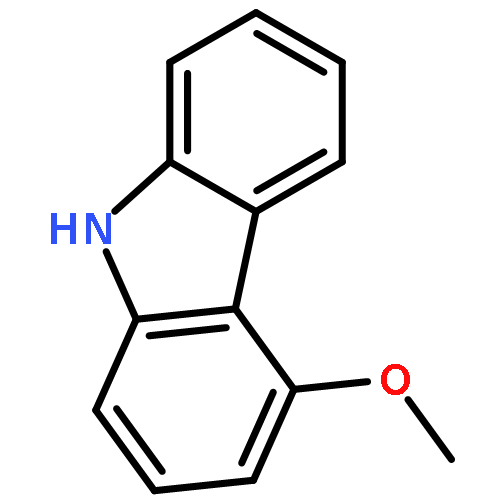





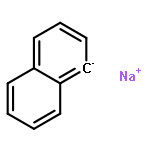
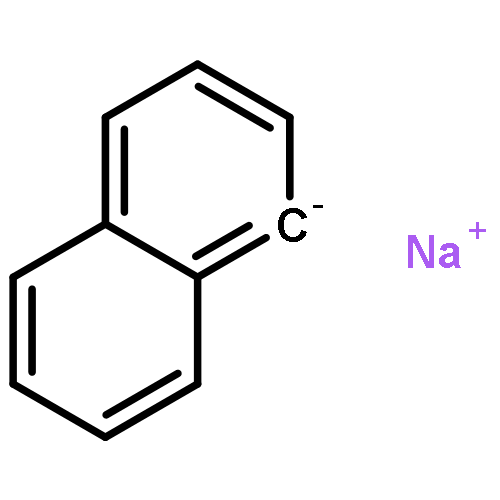
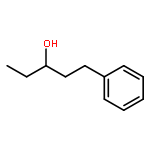
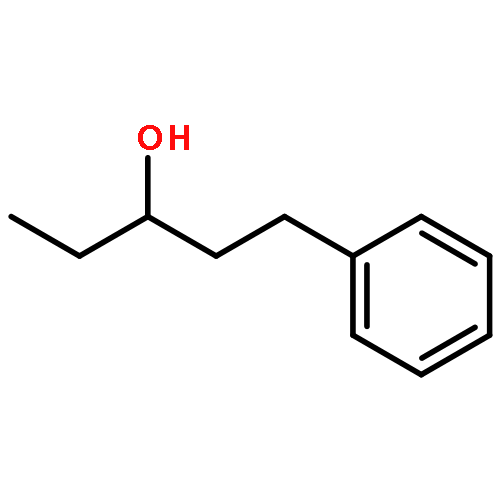


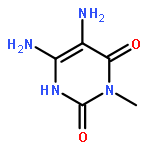

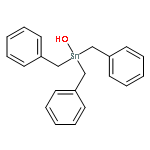
![Butanamide, 3-hydroxy-N-[2-(2-hydroxypropyl)-5-methylphenyl]-](http://img.cochemist.com/ccimg/61600/61564-08-7.png)
![Butanamide, 3-hydroxy-N-[2-(2-hydroxypropyl)-5-methylphenyl]-](http://img.cochemist.com/ccimg/61600/61564-08-7_b.png)





![L-Aspartic acid,N-[[[(4S)-4-amino-4-carboxybutyl]amino]iminomethyl]-](http://img.cochemist.com/ccimg/2400/2387-71-5.png)
![L-Aspartic acid,N-[[[(4S)-4-amino-4-carboxybutyl]amino]iminomethyl]-](http://img.cochemist.com/ccimg/2400/2387-71-5_b.png)
![1-[3-[4-[3-(trifluoromethyl)phenyl]piperazin-1-yl]azetidin-1-yl]ethanone](http://img.cochemist.com/ccimg/223400/223381-97-3.png)
![1-[3-[4-[3-(trifluoromethyl)phenyl]piperazin-1-yl]azetidin-1-yl]ethanone](http://img.cochemist.com/ccimg/223400/223381-97-3_b.png)








![Anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetrone, 2,9-bis(2,6-dimethylphenyl)-](/data/chemimg/673600/76372-76-4.png)
![Anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetrone, 2,9-bis(2,6-dimethylphenyl)-](/data/chemimg/673600/76372-76-4_b.png)











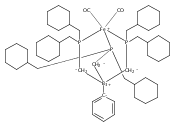




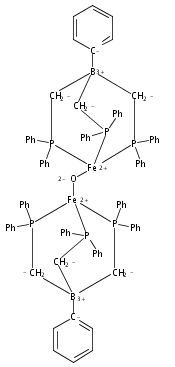

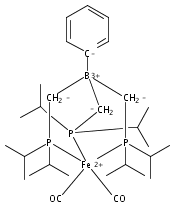
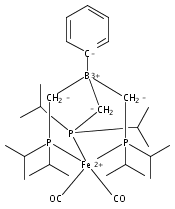
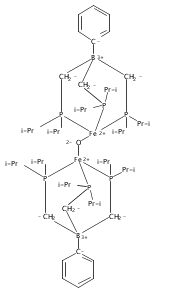
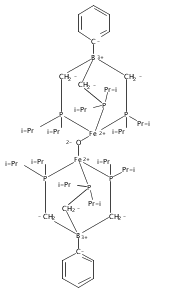

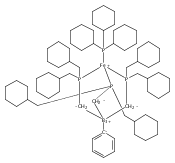
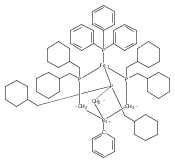








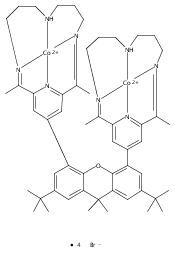
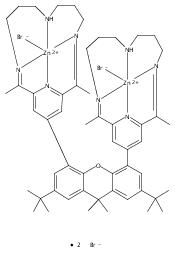
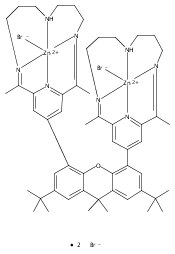


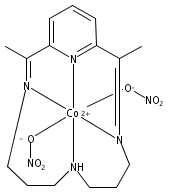
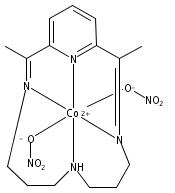


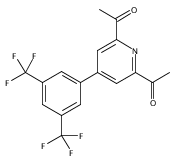












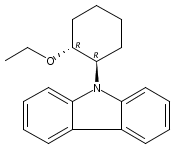














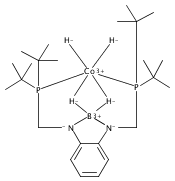
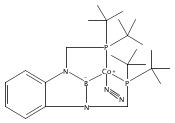























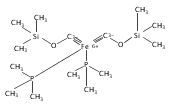
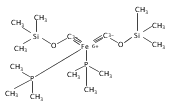

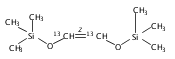


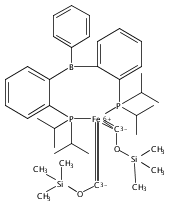
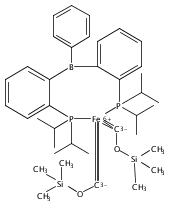




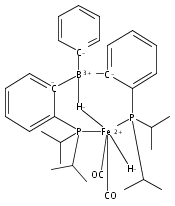

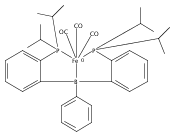
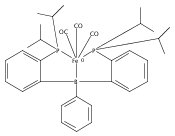
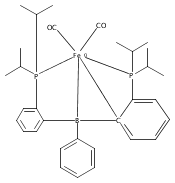
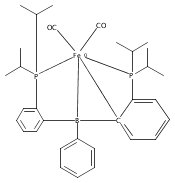









































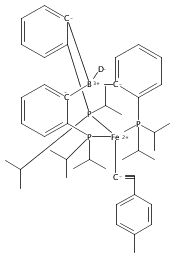
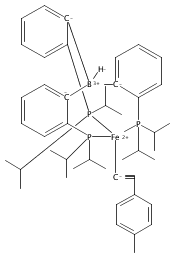



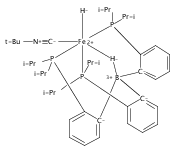
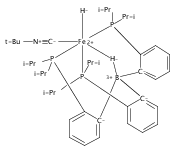
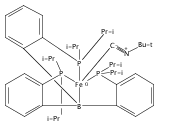
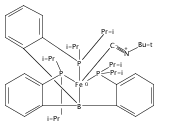





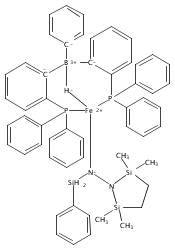

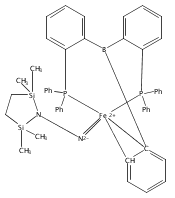
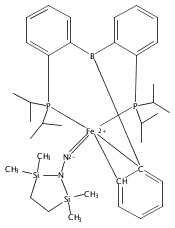

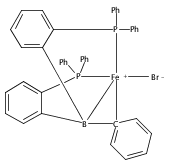

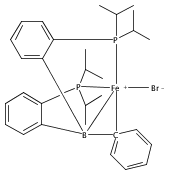
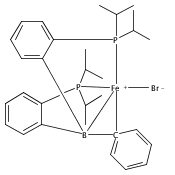


































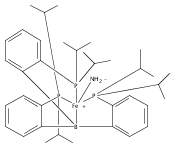



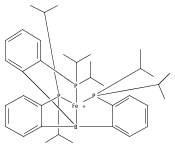
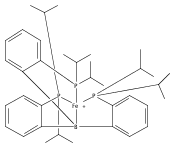
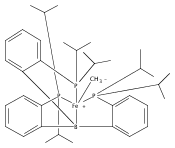



















































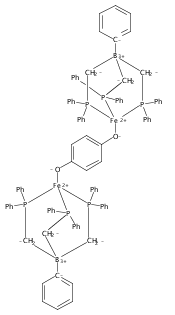
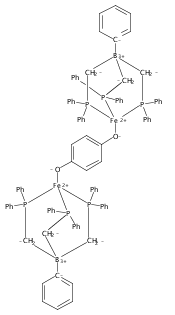
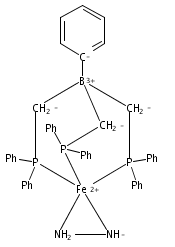
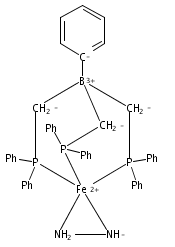












































































































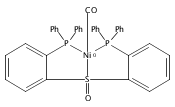
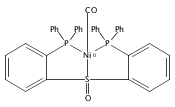


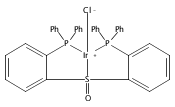
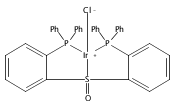






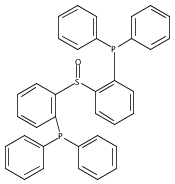
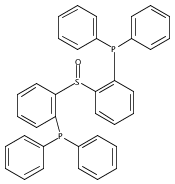




























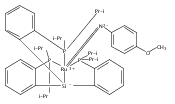














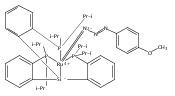








































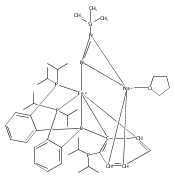



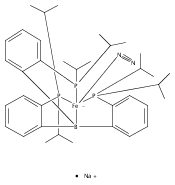








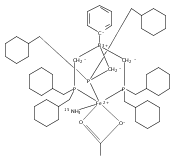







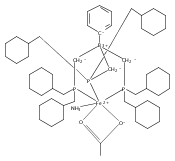
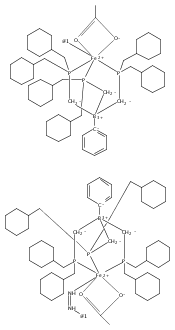







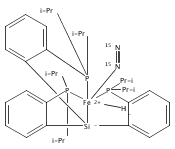
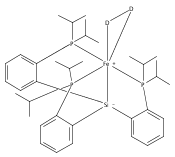

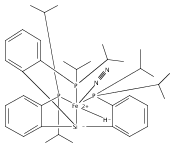
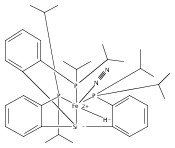


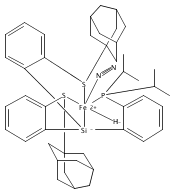
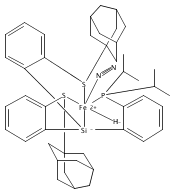


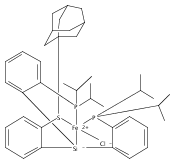
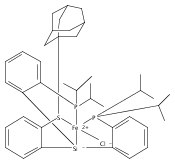
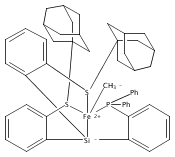
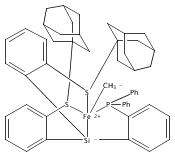
















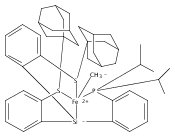
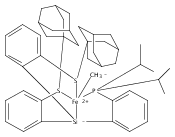
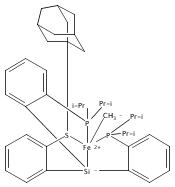




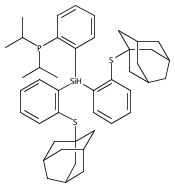
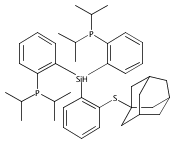

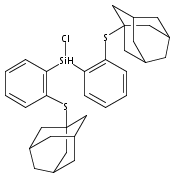
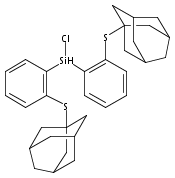
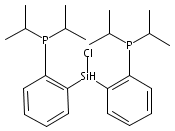











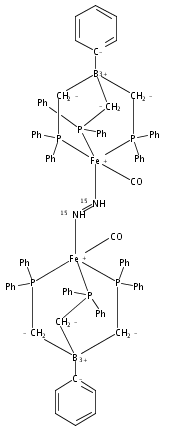

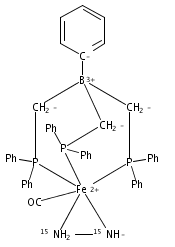



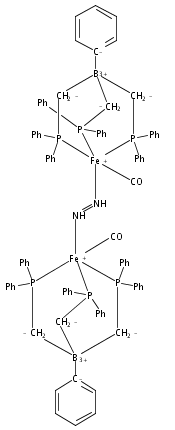

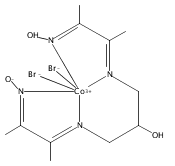
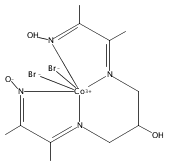
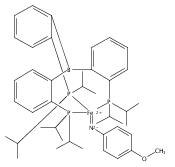
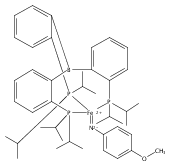


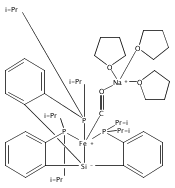



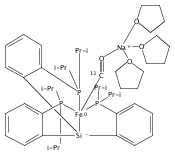


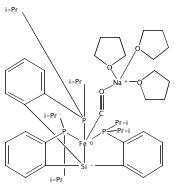
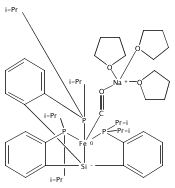
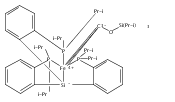
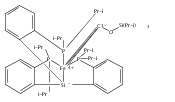
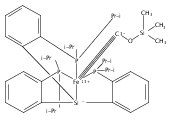
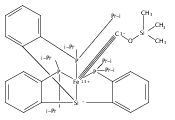

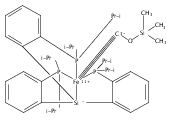

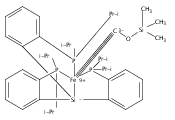






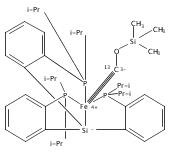


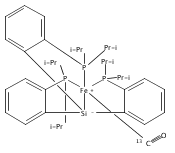


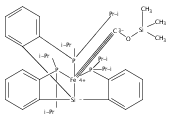

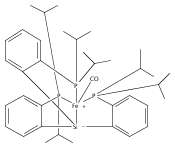


























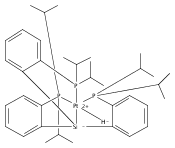










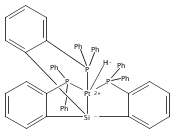

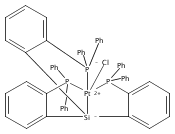

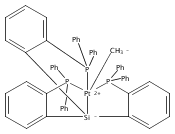



















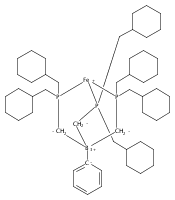
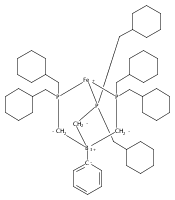
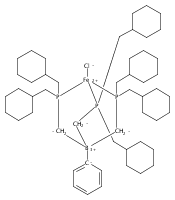
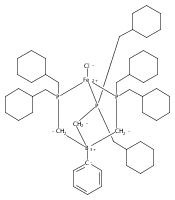







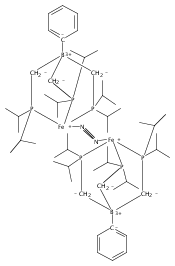
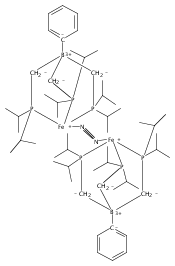


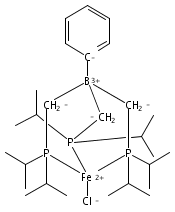
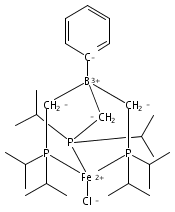




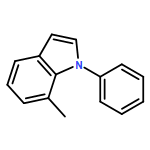





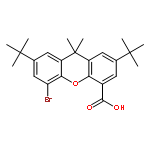




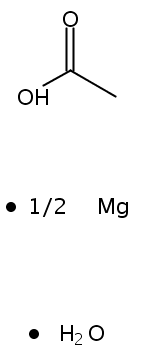
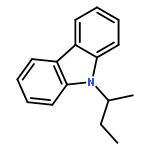









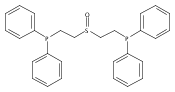










![Benzofuran, 2,3-dihydro-3-[(phenylthio)methyl]-](http://img.cochemist.com/ccimg/103400/103304-48-9.png)
![Benzofuran, 2,3-dihydro-3-[(phenylthio)methyl]-](http://img.cochemist.com/ccimg/103400/103304-48-9_b.png)