Co-reporter:Mariko Kikuchi, Reiko Kurotani, Hiroyuki Konno
Tetrahedron Letters 2017 Volume 58, Issue 43(Issue 43) pp:
Publication Date(Web):25 October 2017
DOI:10.1016/j.tetlet.2017.09.058
•Native chemical ligation by Dawson’s linker.•Solid phase peptide synthesis.•Secretoglobin 3A2 as the diagnostics of pulmonary tumors.A secretoglobin 3A2 type C (98–139) peptide was synthesized by native chemical ligation between 115Ile and 116Cys residues using Dawson’s linker. The peptide-N-acyl-benzimidazolinone-glycine amide, a C-terminal thioesters precursor, was provided from 3-amino-4-(methylamino)benzoic acid. In addition, an N-terminal cysteine fragment, the (116–139) peptide, was prepared by ordinary Fmoc-solid phase peptide synthesis. Native chemical ligation of the (98–115) fragment with the Dawson’s linker and the (116–139) peptide smoothly proceeded to give SCGB3A2 type C (98–139) peptide.Download high-res image (88KB)Download full-size image
Co-reporter:Yoshinori Tokairin, Hiroyuki Konno
Tetrahedron 2017 Volume 73(Issue 1) pp:39-45
Publication Date(Web):5 January 2017
DOI:10.1016/j.tet.2016.11.050
Synthesis of a unique fatty acyl unit to build the N-terminus of callipeltin A and homophymine B is described. Our approach to access (2R, 3R, 4R)-3-hydroxy-2,4,6-trimethylheptanoic acid uses enzymatic hydrolysis for the desymmetrization of achiral acetate, followed by diastereoselective Roush crotylboration and Wittig olefination for the backbone construction.
Co-reporter:Hiroyuki Konno, Takumi Onuma, Ikumi Nitanai, Masaki Wakabayashi, Shigekazu Yano, Kenta Teruya, Kenichi Akaji
Bioorganic & Medicinal Chemistry Letters 2017 Volume 27, Issue 12(Issue 12) pp:
Publication Date(Web):15 June 2017
DOI:10.1016/j.bmcl.2017.04.056
Synthesis and evaluation of new scaffold phenylisoserine derivatives connected with the essential functional groups against SARS CoV 3CL protease are described. The phenylisoserine backbone was found by simulation on GOLD software and the structure activity relationship study of phenylisoserine derivatives gave SK80 with an IC50 value of 43 μM against SARS CoV 3CL R188I mutant protease.Download high-res image (48KB)Download full-size image
Co-reporter:Ryo Yoshino, Yoshinori Tokairin, Hiroyuki Konno
Tetrahedron Letters 2017 Volume 58, Issue 16(Issue 16) pp:
Publication Date(Web):19 April 2017
DOI:10.1016/j.tetlet.2017.03.025
•The guanidination by the Mitsunobu condition.•An orthogonally protected Fmoc-AGDHE derivative.•Osmium-catalyzed dihydroxylation of the corresponding Z-olefin.(2R,3R,4S)-4-Amino-7-guanidino-2,3-dihydroxyheptanoic acid (AGDHE), a common constituent of biologically active marine peptides, callipeltin A (1) and neamphamide A, was synthesized as its orthogonally protected derivative from l-glutamic acid in 15 steps. Guanidination by the Mitsunobu condition and osmium-catalyzed dihydroxylation of the corresponding Z-olefin were employed as the key steps.Download high-res image (41KB)Download full-size image
Co-reporter:Ryo Yoshino, Yoshinori Tokairin, Mari Kikuchi, Hiroyuki Konno
Tetrahedron Letters 2017 Volume 58, Issue 16(Issue 16) pp:
Publication Date(Web):19 April 2017
DOI:10.1016/j.tetlet.2017.03.021
•A new Fmoc reagent to avoid the formation of Fmoc-βAla-OH.•Easy and cheap preparation of the reagent.•The reagent gives Fmoc-amino acid in good yields.Fmoc-OSu has been widely used for Fmoc protection of amino groups, especially amino acids, in solid phase peptide synthesis. However, it has been recognized that Fmoc-βAla-OH is formed as a by-product via the Lossen rearrangement during the reaction. Since we reconfirmed the formation of Fmoc-βAla-OH during the preparation of Fmoc-AA-OH by Fmoc-OSu, Fmoc-OPhth was designed and synthesized as a new Fmoc reagent to avoid the formation of Fmoc-βAla-OH. Furthermore, Fmoc protection by Fmoc-OPhth and Fmoc-SPPS were evaluated. The various Fmoc-amino acids prepared by Fmoc-OPhth were carried out in good yields and these are applicable in Fmoc-SPPS.Download high-res image (82KB)Download full-size image
Co-reporter:Hiroyuki Konno, Masaki Wakabayashi, Daiki Takanuma, Yota Saito, Kenichi Akaji
Bioorganic & Medicinal Chemistry 2016 Volume 24(Issue 6) pp:1241-1254
Publication Date(Web):15 March 2016
DOI:10.1016/j.bmc.2016.01.052
Synthesis of serine derivatives having the essential functional groups for the inhibitor of SARS 3CL protease and evaluation of their inhibitory activities using SARS 3CL R188I mutant protease are described. The lead compounds, functionalized serine derivatives, were designed based on the tetrapeptide aldehyde and Bai’s cinnamoly inhibitor, and additionally performed with simulation on GOLD softwear. Structure activity relationship studies of the candidate compounds were given reasonable inhibitors ent-3 and ent-7k against SARS 3CL R188I mutant protease. These inhibitors showed protease selectivity and no cytotoxicity.
Co-reporter:Hiroyuki Konno, Kanako Abumi, Yasuhiro Sasaki, Shigekazu Yano, Kazuto Nosaka
Bioorganic & Medicinal Chemistry Letters 2015 Volume 25(Issue 16) pp:3199-3202
Publication Date(Web):15 August 2015
DOI:10.1016/j.bmcl.2015.05.088
Cyclic and linear lipopeptides, burkholdine analogues, were synthesized by conventional Fmoc-SPPS and cyclisation with DIPC/HOBt in the solution phase. Synthesized peptides were evaluated for antifungal activities with MIC values against Saccharomyces cerevisiae and Aspergillus oryzae. As a result, the stereochemistry of the amino acid residues and sequences of burkholdine analogues exerted a significant influence on antifungal activities. In addition, we found a linear burkholdine analogue with moderate antifungal activities.
Co-reporter:Hiroyuki Konno, Taki Sato, Yugo Saito, Iori Sakamoto, Kenichi Akaji
Bioorganic & Medicinal Chemistry Letters 2015 25(22) pp: 5127-5132
Publication Date(Web):
DOI:10.1016/j.bmcl.2015.10.007
Co-reporter:Hiroyuki Konno, Yoshihiro Sema, Yoshinori Tokairin
Tetrahedron 2015 Volume 71(Issue 21) pp:3433-3438
Publication Date(Web):27 May 2015
DOI:10.1016/j.tet.2015.03.080
The preparation of peptide aldehydes via efficient transformation of acetal/thioacetal structures followed by treatment of N-bromo succinimide is described. Peptide acetals derived from the corresponding Fmoc-amino acids and hexane-1,2,6-triol were prepared by the conventional Fmoc-SPPS. Subsequently, the peptide acetals were efficiently converted to thioacetal structures followed by treatment of N-bromo succinimide to give peptide aldehydes. Since alkyl and phenyl-containing peptides were applied to give reasonable results in a previous report, we attempted the synthesis of three variant peptide aldehydes in the present study. A C-terminal Phe-containing peptide aldehyde, Ac-Leu-Ala-Phe-H (3), to compare the reactivity of C-terminal amino acids was designed and synthesized. In this case, the reactivity was guided by steric hindrance between acetal structures and side chains of selected amino acids. In contrast, the Lys-containing peptide aldehyde, Ac-Lys-Ala-Leu-H (4), and the Ser-containing peptide, Ac-Ser-Phe-Leu-H (5), were applicable. These results suggest that this methodology has good potential.
Co-reporter:Mari Kikuchi and Hiroyuki Konno
Organic Letters 2014 Volume 16(Issue 16) pp:4324-4327
Publication Date(Web):July 31, 2014
DOI:10.1021/ol5020619
Total synthesis of callipeltins B and M, peptidyl cytotoxic agents isolated from marine sponges, by the combination of Fmoc solid-phase peptide synthesis and cyclization and global deprotection in the solution phase is described. Eight amino acids, including several unusual amino acids, were assembled on a solid support, and effective TFA-mediated deprotection was employed to reach callipeltin M. Callipeltin B was accomplished via the macrolactonization between the side chain of d-aThr and the C-terminus carboxylic acid of protected callipeltin M.
Co-reporter:Hiroyuki Konno, Hitoshi Endo, Satomi Ise, Keiki Miyazaki, Hideo Aoki, Akira Sanjoh, Kazuya Kobayashi, Yasunao Hattori, Kenichi Akaji
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 2) pp:685-690
Publication Date(Web):15 January 2014
DOI:10.1016/j.bmcl.2013.11.039
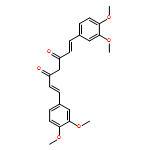
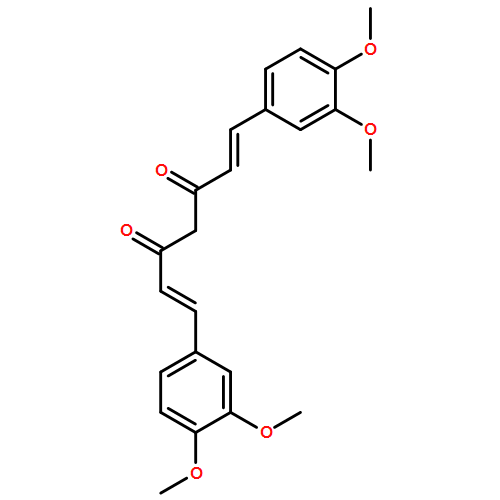
To research a new non-peptidyl inhibitor of beta-site amyloid precursor protein cleaving enzyme 1, we focused on the curcumin framework, two phenolic groups combined with an sp2 carbon spacer for low-molecular and high lipophilicity. The structure–activity relationship study of curcumin derivatives is described. Our results indicate that phenolic hydroxy groups and an alkenyl spacer are important structural factors for the inhibition of beta-site amyloid precursor protein cleaving enzyme 1 and, furthermore, non-competitive inhibition of enzyme activity is anticipated from an inhibitory kinetics experiment and docking simulation.
Co-reporter:Hitoshi Endo, Yuri Nikaido, Mamiko Nakadate, Satomi Ise, Hiroyuki Konno
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 24) pp:5621-5626
Publication Date(Web):15 December 2014
DOI:10.1016/j.bmcl.2014.10.076
Co-reporter:Hiroyuki Konno, Yoshihiro Sema, Manabu Ishii, Yasunao Hattori, Kazuto Nosaka, Kenichi Akaji
Tetrahedron Letters 2013 Volume 54(Issue 36) pp:4848-4850
Publication Date(Web):4 September 2013
DOI:10.1016/j.tetlet.2013.06.103
We have investigated practical synthetic routes for the preparation of peptide aldehyde on a solid support. Peptide aldehyde was synthesized via efficient transformation of acetal/thioacetal structures.
Co-reporter:Hiroyuki Konno, Yusuke Otsuki, Kenta Matsuzaki, Kazuto Nosaka
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 14) pp:4244-4247
Publication Date(Web):15 July 2013
DOI:10.1016/j.bmcl.2013.04.091
Synthesis and antifungal activity of cyclic octapeptide derivatives of burkholdines are described. To construct cyclic octapeptides, the combination of Fmoc-SPPS and cyclization with DIC/HOBt in the solution phase was employed. Synthesized peptides were evaluated for antifungal activity with MIC values against Saccharomyces cerevisiae, Aspergillus oryzae, and Candida viswanathii. As a result, the lipid side chain and the stereochemistry of each amino acid of Bk-1097 analogues significantly affected antifungal activity.
Co-reporter:Mari Kikuchi, Hiroyuki Konno
Tetrahedron 2013 69(34) pp: 7098-7101
Publication Date(Web):
DOI:10.1016/j.tet.2013.06.027
Co-reporter:Mari Kikuchi, Yoshihisa Watanabe, Masaki Tanaka, Kenichi Akaji, Hiroyuki Konno
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 16) pp:4865-4868
Publication Date(Web):15 August 2011
DOI:10.1016/j.bmcl.2011.06.026
Solid phase synthesis of the cyclic depsipeptides of callipeltin B analogues and evaluation of cytotoxicity of synthetic peptides are described. We attempted to synthesize cyclic depsipeptides via the esterification or the amide bond formation for cyclization steps. In the assay of synthetic peptides, the linear peptide (10) exhibited modest cytotoxicity against HeLa cells.
Co-reporter:Mari Kikuchi, Kazuto Nosaka, Kenichi Akaji, Hiroyuki Konno
Tetrahedron Letters 2011 Volume 52(Issue 30) pp:3872-3875
Publication Date(Web):27 July 2011
DOI:10.1016/j.tetlet.2011.05.062
Solid phase total synthesis of callipeltin E (1), truncated linear peptide isolated from marine sponge, Latrunculia sp. was achieved. Our strategy based on traditional Fmoc-SPPS was in common use TFA-treatment final deprotection to reach callipeltin E (1) contained acid-sensitive βMeOTyr.
Co-reporter:Hiroyuki Konno, Kazuto Nosaka, Kenichi Akaji
Tetrahedron 2011 67(47) pp: 9067-9071
Publication Date(Web):
DOI:10.1016/j.tet.2011.09.116
Co-reporter:Hiroyuki Konno, Hidefumi Makabe, Yasunao Hattori, Kazuto Nosaka, Kenichi Akaji
Tetrahedron 2010 66(40) pp: 7946-7953
Publication Date(Web):
DOI:10.1016/j.tet.2010.08.028













![Piperidine, 1-[(2S)-2-amino-3-hydroxy-1-oxopropyl]-](http://img.cochemist.com/ccimg/621000/620947-61-7.png)
![Piperidine, 1-[(2S)-2-amino-3-hydroxy-1-oxopropyl]-](http://img.cochemist.com/ccimg/621000/620947-61-7_b.png)












![Tert-butyl N-[(4-iodophenyl)methyl]carbamate](http://img.cochemist.com/ccimg/189200/189132-01-2.png)
![Tert-butyl N-[(4-iodophenyl)methyl]carbamate](http://img.cochemist.com/ccimg/189200/189132-01-2_b.png)










![Carbamic acid, [(1S)-1-formyl-2-phenylethyl]-, 9H-fluoren-9-ylmethylester](/data/chemimg/1729600/146803-43-2.png)
![Carbamic acid, [(1S)-1-formyl-2-phenylethyl]-, 9H-fluoren-9-ylmethylester](/data/chemimg/1729600/146803-43-2_b.png)
![Carbamic acid, [(1S)-1-formyl-3-methylbutyl]-, 9H-fluoren-9-ylmethylester](http://img.cochemist.com/ccimg/146900/146803-42-1.png)
![Carbamic acid, [(1S)-1-formyl-3-methylbutyl]-, 9H-fluoren-9-ylmethylester](http://img.cochemist.com/ccimg/146900/146803-42-1_b.png)
![3-Oxazolidinecarboxylic acid,4-[(1E)-3-ethoxy-3-oxo-1-propenyl]-2,2-dimethyl-, 1,1-dimethylethylester, (4S)-](http://img.cochemist.com/ccimg/134600/134525-18-1.png)
![3-Oxazolidinecarboxylic acid,4-[(1E)-3-ethoxy-3-oxo-1-propenyl]-2,2-dimethyl-, 1,1-dimethylethylester, (4S)-](http://img.cochemist.com/ccimg/134600/134525-18-1_b.png)
![Phenol, 4,4'-[(1E)-1H-pyrazole-3,5-diyldi-2,1-ethenediyl]bis[2-methoxy-](http://img.cochemist.com/ccimg/131100/131068-23-0.png)
![Phenol, 4,4'-[(1E)-1H-pyrazole-3,5-diyldi-2,1-ethenediyl]bis[2-methoxy-](http://img.cochemist.com/ccimg/131100/131068-23-0_b.png)
![D-Allothreonine,N-[(9H-fluoren-9-ylmethoxy)carbonyl]-](http://img.cochemist.com/ccimg/130700/130674-54-3.png)
![D-Allothreonine,N-[(9H-fluoren-9-ylmethoxy)carbonyl]-](http://img.cochemist.com/ccimg/130700/130674-54-3_b.png)






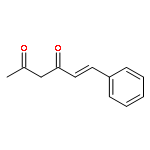
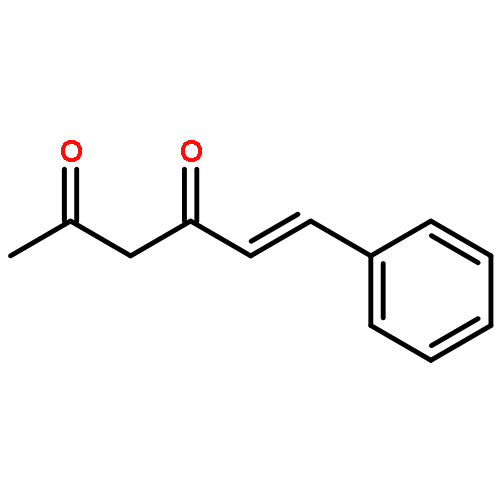














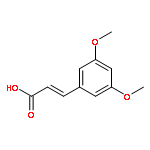



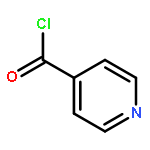
![(1S,4AR,5S,7AS)-1-(WEI -D-GLUCOPYRANOSYLOXY)-7-(HYDROXYMETHYL)-1,4A,<WBR />5,7A-TETRAHYDROCYCLOPENTA[C]PYRAN-5-YL 4-HYDROXYBENZOATE](http://img.cochemist.com/ccimg/7400/7362-37-0.png)
![(1S,4AR,5S,7AS)-1-(WEI -D-GLUCOPYRANOSYLOXY)-7-(HYDROXYMETHYL)-1,4A,<WBR />5,7A-TETRAHYDROCYCLOPENTA[C]PYRAN-5-YL 4-HYDROXYBENZOATE](http://img.cochemist.com/ccimg/7400/7362-37-0_b.png)