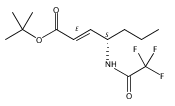
Co-reporter:Wei Zhao, Ryan P. Wurz, Jonas C. Peters, and Gregory C. Fu
Journal of the American Chemical Society September 6, 2017 Volume 139(Issue 35) pp:12153-12153
Publication Date(Web):August 25, 2017
DOI:10.1021/jacs.7b07546
The Curtius rearrangement is a classic, powerful method for converting carboxylic acids into protected amines, but its widespread use is impeded by safety issues (the need to handle azides). We have developed an alternative to the Curtius rearrangement that employs a copper catalyst in combination with blue-LED irradiation to achieve the decarboxylative coupling of aliphatic carboxylic acid derivatives (specifically, readily available N-hydroxyphthalimide esters) to afford protected amines under mild conditions. This C–N bond-forming process is compatible with a wide array of functional groups, including an alcohol, aldehyde, epoxide, indole, nitroalkane, and sulfide. Control reactions and mechanistic studies are consistent with the hypothesis that copper species are engaged in both the photochemistry and the key bond-forming step, which occurs through out-of-cage coupling of an alkyl radical.
Co-reporter:Jun Myun Ahn, Tanvi S. Ratani, Kareem I. Hannoun, Gregory C. Fu, and Jonas C. Peters
Journal of the American Chemical Society September 13, 2017 Volume 139(Issue 36) pp:12716-12716
Publication Date(Web):August 17, 2017
DOI:10.1021/jacs.7b07052
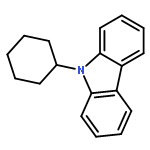
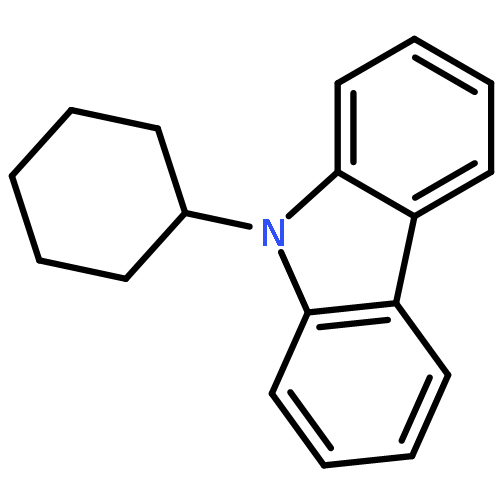
We have recently reported that a variety of couplings of nitrogen, sulfur, oxygen, and carbon nucleophiles with organic halides can be achieved under mild conditions (−40 to 30 °C) through the use of light and a copper catalyst. Insight into the various mechanisms by which these reactions proceed may enhance our understanding of chemical reactivity and facilitate the development of new methods. In this report, we apply an array of tools (EPR, NMR, transient absorption, and UV–vis spectroscopy; ESI–MS; X-ray crystallography; DFT calculations; reactivity, stereochemical, and product studies) to investigate the photoinduced, copper-catalyzed coupling of carbazole with alkyl bromides. Our observations are consistent with pathways wherein both an excited state of the copper(I) carbazolide complex ([CuI(carb)2]−) and an excited state of the nucleophile (Li(carb)) can serve as photoreductants of the alkyl bromide. The catalytically dominant pathway proceeds from the excited state of Li(carb), generating a carbazyl radical and an alkyl radical. The cross-coupling of these radicals is catalyzed by copper via an out-of-cage mechanism in which [CuI(carb)2]− and [CuII(carb)3]− (carb = carbazolide), both of which have been identified under coupling conditions, are key intermediates, and [CuII(carb)3]− serves as the persistent radical that is responsible for predominant cross-coupling. This study underscores the versatility of copper(II) complexes in engaging with radical intermediates that are generated by disparate pathways, en route to targeted bond constructions.
Co-reporter:Marcin Kalek and Gregory C. Fu
Journal of the American Chemical Society March 22, 2017 Volume 139(Issue 11) pp:4225-4225
Publication Date(Web):March 9, 2017
DOI:10.1021/jacs.7b01826
Investigation of the dependence of product enantiometric excess (ee) on catalyst ee is a widely used tool to probe the mechanism of an enantioselective reaction; in particular, the observation of a nonlinear relationship is usually interpreted as an indication of the presence of one or more species that contain at least two units of the chiral entity. In this report, we demonstrate that catalytic enantioconvergent reactions can display an intrinsic negative nonlinear effect that originates purely from the kinetic characteristics of certain enantioconvergent processes and is independent of possible aggregation of the chiral entity. Specifically, this intrinsic negative nonlinear effect can arise when there is a kinetic resolution of the racemic starting material, and its magnitude is correlated with the selectivity factor and the conversion; the dependence on conversion provides a ready means to distinguish it from a more conventional nonlinear effect. We support our analysis with experimental data for two distinct enantioconvergent processes, one catalyzed by a chiral phosphine and the other by a chiral Pd/phosphine complex.
Co-reporter:Sarah Yunmi Lee, Stefan Neufeind, and Gregory C. Fu
Journal of the American Chemical Society June 25, 2014 Volume 136(Issue 25) pp:8899-8902
Publication Date(Web):June 12, 2014
DOI:10.1021/ja5044209
The catalytic asymmetric synthesis of alkyl fluorides, particularly α-fluorocarbonyl compounds, has been the focus of substantial effort in recent years. While significant progress has been described in the formation of enantioenriched secondary alkyl fluorides, advances in the generation of tertiary alkyl fluorides have been more limited. Here, we describe a method for the catalytic asymmetric coupling of aryl alkyl ketenes with commercially available N-fluorodibenzenesulfonimide (NFSI) and C6F5ONa to furnish tertiary α-fluoroesters. Mechanistic studies are consistent with the hypothesis that the addition of an external nucleophile (C6F5ONa) is critical for turnover, releasing the catalyst (PPY*) from an N-acylated intermediate. The available data can be explained by a reaction pathway wherein the enantioselectivity is determined in the turnover-limiting transfer of fluorine from NFSI to a chiral enolate derived from the addition of PPY* to the ketene. The structure and the reactivity of the product of this proposed elementary step, an α-fluoro-N-acylpyridinium salt, have been examined.
Co-reporter:Junwon Choi
Science 2017 Volume 356(Issue 6334) pp:
Publication Date(Web):
DOI:10.1126/science.aaf7230
Stitching one alkyl group to another
Chemical reactions such as Heck and Suzuki coupling facilitate access to an enormous range of relatively flat molecules. This geometrical constraint is associated with the comparative ease of linking together aryl and alkenyl carbons. Choi and Fu review recent developments in forming bonds between the more abundant alkyl carbon centers that underlie diverse molecules with complex three-dimensional structures. Nickel catalysis in particular has emerged as a powerful method to access individual mirror-image isomers selectively and thereby tune the biological properties of the targeted products.
Science, this issue p. eaaf7230
Co-reporter:Xin Mu;Yu Shibata;Yusuke Makida; Gregory C. Fu
Angewandte Chemie International Edition 2017 Volume 56(Issue 21) pp:5821-5824
Publication Date(Web):2017/05/15
DOI:10.1002/anie.201702402
AbstractVicinal stereocenters are found in many natural and unnatural compounds. Although metal-catalyzed cross-coupling reactions of unactivated alkyl electrophiles are emerging as a powerful tool in organic synthesis, there have been virtually no reports of processes that generate, much less control, vicinal stereocenters. In this investigation, we establish that a chiral nickel catalyst can mediate doubly stereoconvergent alkyl–alkyl cross-coupling, specifically, reactions of a racemic pyrrolidine-derived nucleophile with cyclic alkyl halides (as mixtures of stereoisomers) to produce vicinal stereocenters with very good stereoselectivity.
Co-reporter:Xin Mu;Yu Shibata;Yusuke Makida; Gregory C. Fu
Angewandte Chemie 2017 Volume 129(Issue 21) pp:5915-5918
Publication Date(Web):2017/05/15
DOI:10.1002/ange.201702402
AbstractVicinal stereocenters are found in many natural and unnatural compounds. Although metal-catalyzed cross-coupling reactions of unactivated alkyl electrophiles are emerging as a powerful tool in organic synthesis, there have been virtually no reports of processes that generate, much less control, vicinal stereocenters. In this investigation, we establish that a chiral nickel catalyst can mediate doubly stereoconvergent alkyl–alkyl cross-coupling, specifically, reactions of a racemic pyrrolidine-derived nucleophile with cyclic alkyl halides (as mixtures of stereoisomers) to produce vicinal stereocenters with very good stereoselectivity.
Co-reporter:Daniel T. Ziegler and Gregory C. Fu
Journal of the American Chemical Society 2016 Volume 138(Issue 37) pp:12069-12072
Publication Date(Web):September 12, 2016
DOI:10.1021/jacs.6b08486
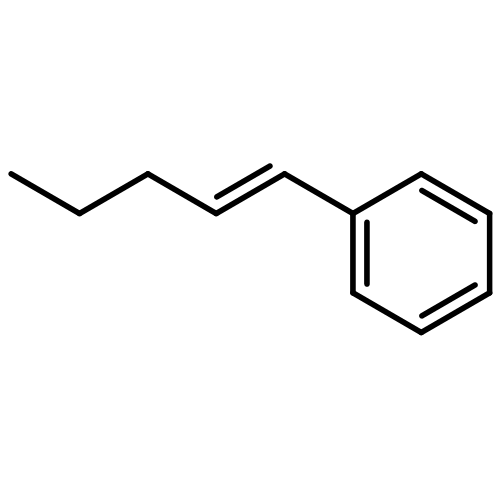
Benzylic alcohols and ethers are common subunits in bioactive molecules, as well as useful intermediates in organic chemistry. In this Communication, we describe a new approach to the enantioselective synthesis of benzylic ethers through the chiral phosphine-catalyzed coupling of two readily available partners, γ-aryl-substituted alkynoates and alcohols, under mild conditions. In this process, the alkynoate partner undergoes an internal redox reaction. Specifically, the benzylic position is oxidized with good enantioselectivity, and the alkyne is reduced to the alkene.
Co-reporter:Crystal K. Chu; Yufan Liang
Journal of the American Chemical Society 2016 Volume 138(Issue 20) pp:6404-6407
Publication Date(Web):May 17, 2016
DOI:10.1021/jacs.6b03465
A wide array of cross-coupling methods for the formation of C–C bonds from unactivated alkyl electrophiles have been described in recent years. In contrast, progress in the development of methods for the construction of C–heteroatom bonds has lagged; for example, there have been no reports of metal-catalyzed cross-couplings of unactivated secondary or tertiary alkyl halides with silicon nucleophiles to form C–Si bonds. In this study, we address this challenge, establishing that a simple, commercially available nickel catalyst (NiBr2·diglyme) can achieve couplings of alkyl bromides with nucleophilic silicon reagents under unusually mild conditions (e.g., −20 °C); especially noteworthy is our ability to employ unactivated tertiary alkyl halides as electrophilic coupling partners, which is still relatively uncommon in the field of cross-coupling chemistry. Stereochemical, relative reactivity, and radical-trap studies are consistent with a homolytic pathway for C–X bond cleavage.
Co-reporter:Zhiwei Zuo; Huan Cong; Wei Li; Junwon Choi; Gregory C. Fu;David W. C. MacMillan
Journal of the American Chemical Society 2016 Volume 138(Issue 6) pp:1832-1835
Publication Date(Web):February 5, 2016
DOI:10.1021/jacs.5b13211
An asymmetric decarboxylative Csp3–Csp2 cross-coupling has been achieved via the synergistic merger of photoredox and nickel catalysis. This mild, operationally simple protocol transforms a wide variety of naturally abundant α-amino acids and readily available aryl halides into valuable chiral benzylic amines in high enantiomeric excess, thereby producing motifs found in pharmacologically active agents.
Co-reporter:Miles W. Johnson, Kareem I. Hannoun, Yichen Tan, Gregory C. Fu and Jonas C. Peters
Chemical Science 2016 vol. 7(Issue 7) pp:4091-4100
Publication Date(Web):24 Feb 2016
DOI:10.1039/C5SC04709A
Photoinduced, copper-catalyzed cross-coupling can offer a complementary approach to thermal (non-photoinduced) methods for generating C–X (X = C, N, O, S, etc.) bonds. In this report, we describe the first detailed mechanistic investigation of one of the processes that we have developed, specifically, the (stoichiometric) coupling of a copper–thiolate with an aryl iodide. In particular, we focus on the chemistry of a discrete [CuI(SAr)2]− complex (Ar = 2,6-dimethylphenyl), applying a range of techniques, including ESI-MS, cyclic voltammetry, transient luminescence spectroscopy, optical spectroscopy, DFT calculations, Stern–Volmer analysis, EPR spectroscopy, actinometry, and reactivity studies. The available data are consistent with the viability of a pathway in which photoexcited [CuI(SAr)2]−* serves as an electron donor to an aryl iodide to afford an aryl radical, which then reacts in cage with the newly generated copper(II)–thiolate to furnish the cross-coupling product in a non-chain process.
Co-reporter:Quirin M. Kainz;Susan L. Zultanski;Agnieszka Bartoszewicz;Jonas C. Peters;Carson D. Matier
Science 2016 Volume 351(Issue 6274) pp:681-684
Publication Date(Web):12 Feb 2016
DOI:10.1126/science.aad8313
Copper's light touch forges C-N bonds
Organic photochemistry has traditionally relied on excitation in the ultraviolet, where carbon-based compounds tend to absorb. Over the past decade, the field has undergone a renaissance as compounds that absorb visible light have proven to be versatile catalysts for organic reactions. For the most part, however, these catalysts have contained rare metals such as ruthenium or iridium. Kainz et al. now report a blue light-driven C-N bond-forming reaction catalyzed by Earth-abundant copper (see the Perspective by Greaney). Through coordination to a chiral ligand, the copper center couples alkyl chlorides to indoles and carbazoles with a high degree of enantioselectivity.
Science, this issue p. 681; see also p. 666
Co-reporter:Jens Schmidt;Junwon Choi;Martin Slusarczyk;Albert Tianxiang Liu
Science 2016 Volume 354(Issue 6317) pp:
Publication Date(Web):
DOI:10.1126/science.aai8611
Crafting chiral boron building blocks
Carbon-boron bonds are easily transformed into a wide variety of C–C, C–N, and C–O bonds. With that flexibility in mind, Schmidt et al. show that nickel complexes can catalyze asymmetric alkylation of carbon centers adjacent to boron. This protocol creates chiral alkylboronates that function as stable precursors to numerous complex molecules. The reaction proceeds in stereo-convergent fashion—forming a single product from either mirror image of the α-haloboronate reagent. Successive reactions can also create chains of adjacent chiral alkyl centers with stereochemistry set by the configuration of the ligand bound to nickel.
Science, this issue p. 1265
Co-reporter:Søren Kramer
Journal of the American Chemical Society 2015 Volume 137(Issue 11) pp:3803-3806
Publication Date(Web):March 17, 2015
DOI:10.1021/jacs.5b01944
Due in part to the common occurrence of five-membered nitrogen heterocycles in bioactive molecules, the discovery of methods for the enantioselective synthesis of such structures is a useful endeavor. Building on a single example by Tong of a phosphine-catalyzed [4 + 1] annulation of an amine with an allene that furnished an achiral dihydropyrrole in 22% yield, we have developed, with the aid of a new chiral spirophosphine catalyst, a method with increased utility, specifically, improved yield, enhanced scope (the use of γ-substituted allenes), and good ee. The enantioenriched dihydropyrrole products can be transformed into other interesting families of compounds with very good stereoselectivity.
Co-reporter:Sarah Yunmi Lee; Yuji Fujiwara; Atsuko Nishiguchi; Marcin Kalek
Journal of the American Chemical Society 2015 Volume 137(Issue 13) pp:4587-4591
Publication Date(Web):March 27, 2015
DOI:10.1021/jacs.5b01985
Substantial progress has been described in the development of asymmetric variants of the phosphine-catalyzed intermolecular [3+2] annulation of allenes with alkenes; however, there have not been corresponding advances for the intramolecular process, which can generate a higher level of complexity (an additional ring and stereocenter(s)). In this study, we describe the application of chiral phosphepine catalysts to address this challenge, thereby providing access to useful scaffolds that are found in bioactive compounds, including diquinane and quinolin-2-one derivatives, with very good stereoselectivity. The products of the [3+2] annulation can be readily transformed into structures that are even more stereochemically rich. Mechanistic studies are consistent with β addition of the phosphepine to the allene being the turnover-limiting step of the catalytic cycle, followed by a concerted [3+2] cycloaddition to the pendant olefin.
Co-reporter:Tanvi S. Ratani; Shoshana Bachman; Gregory C. Fu;Jonas C. Peters
Journal of the American Chemical Society 2015 Volume 137(Issue 43) pp:13902-13907
Publication Date(Web):October 22, 2015
DOI:10.1021/jacs.5b08452
We have recently reported that, in the presence of light and a copper catalyst, nitrogen nucleophiles such as carbazoles and primary amides undergo C–N coupling with alkyl halides under mild conditions. In the present study, we establish that photoinduced, copper-catalyzed alkylation can also be applied to C–C bond formation, specifically, that the cyanation of unactivated secondary alkyl chlorides can be achieved at room temperature to afford nitriles, an important class of target molecules. Thus, in the presence of an inexpensive copper catalyst (CuI; no ligand coadditive) and a readily available light source (UVC compact fluorescent light bulb), a wide array of alkyl halides undergo cyanation in good yield. Our initial mechanistic studies are consistent with the hypothesis that an excited state of [Cu(CN)2]− may play a role, via single electron transfer, in this process. This investigation provides a rare example of a transition metal-catalyzed cyanation of an alkyl halide, as well as the first illustrations of photoinduced, copper-catalyzed alkylation with either a carbon nucleophile or a secondary alkyl chloride.
Co-reporter:Marcin Kalek
Journal of the American Chemical Society 2015 Volume 137(Issue 29) pp:9438-9442
Publication Date(Web):July 20, 2015
DOI:10.1021/jacs.5b05528
Methods have recently been developed for the phosphine-catalyzed asymmetric γ-addition of nucleophiles to readily available allenoates and alkynoates to generate useful α,β-unsaturated carbonyl compounds that bear a stereogenic center in either the γ or the δ position (but not both) with high stereoselectivity. The utility of this approach would be enhanced considerably if the stereochemistry at both termini of the new bond could be controlled effectively. In this report, we describe the achievement of this objective, specifically, that a chiral phosphepine can catalyze the stereoconvergent γ-addition of a racemic nucleophile to a racemic electrophile; through the choice of an appropriate heterocycle as the nucleophilic partner, this new method enables the synthesis of protected α,α-disubstituted α-amino acid derivatives in good yield, diastereoselectivity, and enantioselectivity.
Co-reporter:Yufan Liang
Journal of the American Chemical Society 2015 Volume 137(Issue 30) pp:9523-9526
Publication Date(Web):July 23, 2015
DOI:10.1021/jacs.5b04725
In this report, we establish that a readily available nickel/bis(oxazoline) catalyst accomplishes a wide array of enantioconvergent cross-couplings of arylzinc reagents with CF3-substituted racemic secondary alkyl halides, a process that necessitates that the chiral catalyst be able to effectively distinguish between a CF3 and an alkyl group in order to provide good ee. We further demonstrate that this method can be applied without modification to the catalytic asymmetric synthesis of other families of fluorinated organic compounds.
Co-reporter:Yufan Liang ; Gregory C. Fu
Angewandte Chemie 2015 Volume 127( Issue 31) pp:9175-9179
Publication Date(Web):
DOI:10.1002/ange.201503297
Abstract
Fluorinated organic molecules are of interest in fields ranging from medicinal chemistry to polymer science. Described herein is a mild, convenient, and versatile method for the synthesis of compounds bearing a perfluoroalkyl group attached to a tertiary carbon atom by using an alkyl–alkyl cross-coupling. A nickel catalyst derived from NiCl2⋅glyme and a pybox ligand achieves the coupling of a wide range of fluorinated alkyl halides with alkylzinc reagents at room temperature. A broad array of functional groups is compatible with the reaction conditions, and highly selective couplings can be achieved on the basis of differing levels of fluorination. A mechanistic investigation has established that the presence of 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) inhibits cross-coupling under these conditions and that a TEMPO–electrophile adduct can be isolated.
Co-reporter:Yufan Liang ; Gregory C. Fu
Angewandte Chemie International Edition 2015 Volume 54( Issue 31) pp:9047-9051
Publication Date(Web):
DOI:10.1002/anie.201503297
Abstract
Fluorinated organic molecules are of interest in fields ranging from medicinal chemistry to polymer science. Described herein is a mild, convenient, and versatile method for the synthesis of compounds bearing a perfluoroalkyl group attached to a tertiary carbon atom by using an alkyl–alkyl cross-coupling. A nickel catalyst derived from NiCl2⋅glyme and a pybox ligand achieves the coupling of a wide range of fluorinated alkyl halides with alkylzinc reagents at room temperature. A broad array of functional groups is compatible with the reaction conditions, and highly selective couplings can be achieved on the basis of differing levels of fluorination. A mechanistic investigation has established that the presence of 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) inhibits cross-coupling under these conditions and that a TEMPO–electrophile adduct can be isolated.
Co-reporter:Junwon Choi ; Pablo Martín-Gago
Journal of the American Chemical Society 2014 Volume 136(Issue 34) pp:12161-12165
Publication Date(Web):August 15, 2014
DOI:10.1021/ja506885s
The development of efficient methods for the generation of enantioenriched sulfonamides and sulfones is an important objective for fields such as organic synthesis and medicinal chemistry; however, there have been relatively few reports of direct catalytic asymmetric approaches to controlling the stereochemistry of the sulfur-bearing carbon of such targets. In this report, we describe nickel-catalyzed stereoconvergent Negishi arylations and alkenylations of racemic α-bromosulfonamides and -sulfones that furnish the desired cross-coupling product in very good ee and yield for an array of reaction partners. Mechanistic studies are consistent with the generation of a radical intermediate that has a sufficient lifetime to diffuse out of the solvent cage and to cyclize onto a pendant olefin.
Co-reporter:Yufan Liang
Journal of the American Chemical Society 2014 Volume 136(Issue 14) pp:5520-5524
Publication Date(Web):March 31, 2014
DOI:10.1021/ja501815p
The development of new approaches to the construction of fluorine-containing target molecules is important for a variety of scientific disciplines, including medicinal chemistry. In this Article, we describe a method for the catalytic enantioselective synthesis of tertiary alkyl fluorides through Negishi reactions of racemic α-halo-α-fluoroketones, which represents the first catalytic asymmetric cross-coupling that employs geminal dihalides as electrophiles. Thus, selective reaction of a C–Br (or C–Cl) bond in the presence of a C–F bond can be achieved with the aid of a nickel/bis(oxazoline) catalyst. The products of the stereoconvergent cross-couplings, enantioenriched tertiary α-fluoroketones, can be converted into an array of interesting organofluorine compounds.
Co-reporter:Hien-Quang Do ; Shoshana Bachman ; Alex C. Bissember ; Jonas C. Peters
Journal of the American Chemical Society 2014 Volume 136(Issue 5) pp:2162-2167
Publication Date(Web):January 21, 2014
DOI:10.1021/ja4126609
The development of a mild and general method for the alkylation of amides with relatively unreactive alkyl halides (i.e., poor substrates for SN2 reactions) is an ongoing challenge in organic synthesis. We describe herein a versatile transition-metal-catalyzed approach: in particular, a photoinduced, copper-catalyzed monoalkylation of primary amides. A broad array of alkyl and aryl amides (as well as a lactam and a 2-oxazolidinone) couple with unactivated secondary (and hindered primary) alkyl bromides and iodides using a single set of comparatively simple and mild conditions: inexpensive CuI as the catalyst, no separate added ligand, and C–N bond formation at room temperature. The method is compatible with a variety of functional groups, such as an olefin, a carbamate, a thiophene, and a pyridine, and it has been applied to the synthesis of an opioid receptor antagonist. A range of mechanistic observations, including reactivity and stereochemical studies, are consistent with a coupling pathway that includes photoexcitation of a copper–amidate complex, followed by electron transfer to form an alkyl radical.
Co-reporter:Huan Cong
Journal of the American Chemical Society 2014 Volume 136(Issue 10) pp:3788-3791
Publication Date(Web):February 27, 2014
DOI:10.1021/ja500706v
As part of our ongoing effort to expand the scope of cross-coupling reactions of alkyl electrophiles, we have pursued a strategy wherein the nucleophilic coupling partner includes a pendant olefin; after transmetalation by such a substrate, if β-migratory insertion proceeds faster than direct cross-coupling, an additional carbon–carbon bond and stereocenter can be formed. With the aid of a nickel/diamine catalyst (both components are commercially available), we have established the viability of this approach for the catalytic asymmetric synthesis of 2,3-dihydrobenzofurans and indanes. Furthermore, we have applied this new method to the construction of the dihydrobenzofuran core of fasiglifam, as well as to a cross-coupling with a racemic alkyl electrophile; in the latter process, the chiral catalyst controls two stereocenters, one that is newly generated in a β-migratory insertion and one that begins as a mixture of enantiomers.
Co-reporter:Sarah Yunmi Lee, Stefan Neufeind, and Gregory C. Fu
Journal of the American Chemical Society 2014 136(25) pp: 8899-8902
Publication Date(Web):June 12, 2014
DOI:10.1021/ja5044209
The catalytic asymmetric synthesis of alkyl fluorides, particularly α-fluorocarbonyl compounds, has been the focus of substantial effort in recent years. While significant progress has been described in the formation of enantioenriched secondary alkyl fluorides, advances in the generation of tertiary alkyl fluorides have been more limited. Here, we describe a method for the catalytic asymmetric coupling of aryl alkyl ketenes with commercially available N-fluorodibenzenesulfonimide (NFSI) and C6F5ONa to furnish tertiary α-fluoroesters. Mechanistic studies are consistent with the hypothesis that the addition of an external nucleophile (C6F5ONa) is critical for turnover, releasing the catalyst (PPY*) from an N-acylated intermediate. The available data can be explained by a reaction pathway wherein the enantioselectivity is determined in the turnover-limiting transfer of fluorine from NFSI to a chiral enolate derived from the addition of PPY* to the ketene. The structure and the reactivity of the product of this proposed elementary step, an α-fluoro-N-acylpyridinium salt, have been examined.
Co-reporter:Nathan D. Schley
Journal of the American Chemical Society 2014 Volume 136(Issue 47) pp:16588-16593
Publication Date(Web):November 17, 2014
DOI:10.1021/ja508718m
Although nickel-catalyzed stereoconvergent couplings of racemic alkyl electrophiles are emerging as a powerful tool in organic chemistry, to date there have been no systematic mechanistic studies of such processes. Herein, we examine the pathway for enantioselective Negishi arylations of secondary propargylic bromides, and we provide evidence for an unanticipated radical chain pathway wherein oxidative addition of the C–Br bond occurs through a bimetallic mechanism. In particular, we have crystallographically characterized a diamagnetic arylnickel(II) complex, [(i-Pr-pybox)NiIIPh]BArF4, and furnished support for [(i-Pr-pybox)NiIIPh]+ being the predominant nickel-containing species formed under the catalyzed conditions as well as a key player in the cross-coupling mechanism. On the other hand, our observations do not require a role for an organonickel(I) intermediate (e.g., (i-Pr-pybox)NiIPh), which has previously been suggested to be an intermediate in nickel-catalyzed cross-couplings, oxidatively adding alkyl electrophiles through a monometallic pathway.
Co-reporter:Yichen Tan, José María Muñoz-Molina, Gregory C. Fu and Jonas C. Peters
Chemical Science 2014 vol. 5(Issue 7) pp:2831-2835
Publication Date(Web):22 Apr 2014
DOI:10.1039/C4SC00368C
Most copper-catalyzed cross-couplings require an elevated reaction temperature. Recently, a photoinduced variant has been developed that enables C–X bond-forming reactions of certain nitrogen and sulfur nucleophiles to proceed under unusually mild conditions (−40 °C to room temperature). In view of the importance of carbon–oxygen bond construction in organic chemistry, the expansion of this photochemical approach to oxygen nucleophiles is an important objective. In this report, we establish that, in the presence of light and an inexpensive copper pre-catalyst (CuI), a wide array of phenols and aryl iodides can be coupled to generate diaryl ethers under mild conditions (room temperature) in the presence of a variety of functional groups. Our studies indicate that a Cu(I)–phenoxide complex is a viable intermediate in photoinduced C–O bond-formation.
Co-reporter:Daniel T. Ziegler;Lorena Riesgo;Takuya Ikeda;Yuji Fujiwara; Gregory C. Fu
Angewandte Chemie 2014 Volume 126( Issue 48) pp:13399-13403
Publication Date(Web):
DOI:10.1002/ange.201405854
Abstract
Because of the frequent occurrence of cyclopentane subunits in bioactive compounds, the development of efficient catalytic asymmetric methods for their synthesis is an important objective. Introduced herein is a new family of chiral nucleophilic catalysts, biphenyl-derived phosphepines, and we apply them to an enantioselective variant of a useful [4+1] annulation. A range of one-carbon coupling partners can be employed, thereby generating cyclopentenes which bear a fully substituted stereocenter [either all-carbon or heteroatom-substituted (sulfur and phosphorus)]. Stereocenters at the other four positions of the cyclopentane ring can also be introduced with good stereoselectivity. An initial mechanistic study indicates that phosphine addition to the electrophilic four-carbon coupling partner is not the turnover-limiting step of the catalytic cycle.
Co-reporter:Daniel T. Ziegler;Lorena Riesgo;Takuya Ikeda;Yuji Fujiwara; Gregory C. Fu
Angewandte Chemie International Edition 2014 Volume 53( Issue 48) pp:13183-13187
Publication Date(Web):
DOI:10.1002/anie.201405854
Abstract
Because of the frequent occurrence of cyclopentane subunits in bioactive compounds, the development of efficient catalytic asymmetric methods for their synthesis is an important objective. Introduced herein is a new family of chiral nucleophilic catalysts, biphenyl-derived phosphepines, and we apply them to an enantioselective variant of a useful [4+1] annulation. A range of one-carbon coupling partners can be employed, thereby generating cyclopentenes which bear a fully substituted stereocenter [either all-carbon or heteroatom-substituted (sulfur and phosphorus)]. Stereocenters at the other four positions of the cyclopentane ring can also be introduced with good stereoselectivity. An initial mechanistic study indicates that phosphine addition to the electrophilic four-carbon coupling partner is not the turnover-limiting step of the catalytic cycle.
Co-reporter:Christopher J. Cordier ; Rylan J. Lundgren
Journal of the American Chemical Society 2013 Volume 135(Issue 30) pp:10946-10949
Publication Date(Web):July 19, 2013
DOI:10.1021/ja4054114
Although enantioconvergent alkyl–alkyl couplings of racemic electrophiles have been developed, there have been no reports of the corresponding reactions of racemic nucleophiles. Herein we describe Negishi cross-couplings of racemic α-zincated N-Boc-pyrrolidine with unactivated secondary halides, thus providing a one-pot, catalytic asymmetric method for the synthesis of a range of 2-alkylpyrrolidines (an important family of target molecules) from N-Boc-pyrrolidine, a commercially available precursor. Preliminary mechanistic studies indicated that two of the most straightforward mechanisms for enantioconvergence (dynamic kinetic resolution of the organometallic coupling partner and a simple β-hydride elimination/β-migratory insertion pathway) are unlikely to be operative.
Co-reporter:Daniel T. Ziegler ; Junwon Choi ; José María Muñoz-Molina ; Alex C. Bissember ; Jonas C. Peters
Journal of the American Chemical Society 2013 Volume 135(Issue 35) pp:13107-13112
Publication Date(Web):August 22, 2013
DOI:10.1021/ja4060806
The use of light to facilitate copper-catalyzed cross-couplings of nitrogen nucleophiles can enable C–N bond formation to occur under unusually mild conditions. In this study, we substantially expand the scope of such processes, establishing that this approach is not limited to reactions of carbazoles with iodobenzene and alkyl halides. Specifically, we demonstrate for the first time that other nitrogen nucleophiles (e.g., common pharmacophores such as indoles, benzimidazoles, and imidazoles) as well as other electrophiles (e.g., hindered/deactivated/heterocyclic aryl iodides, an aryl bromide, an activated aryl chloride, alkenyl halides, and an alkynyl bromide) serve as suitable partners. Photoinduced C–N bond formation can be achieved at room temperature using a common procedure with an inexpensive catalyst (CuI) that does not require a ligand coadditive and is tolerant of moisture and a variety of functional groups.
Co-reporter:Hien-Quang Do ; E. R. R. Chandrashekar
Journal of the American Chemical Society 2013 Volume 135(Issue 44) pp:16288-16291
Publication Date(Web):October 28, 2013
DOI:10.1021/ja408561b
A tertiary stereogenic center that bears two different aryl substituents is found in a variety of bioactive compounds, including medicines such as Zoloft and Detrol. We have developed an efficient method for the synthesis of enantioenriched 1,1-diarylalkanes from readily available racemic benzylic alcohols. Formation of a benzylic mesylate (which is not isolated), followed by treatment with an arylzinc reagent, LiI, and a chiral nickel/bis(oxazoline) catalyst, furnishes the Negishi cross-coupling product in high ee and good yield. A wide array of functional groups (e.g., an aryl iodide, a thiophene, and an N-Boc-indole) are compatible with the mild reaction conditions. This method has been applied to a gram-scale synthesis of a precursor to Zoloft.
Co-reporter:Susan L. Zultanski
Journal of the American Chemical Society 2013 Volume 135(Issue 2) pp:624-627
Publication Date(Web):January 2, 2013
DOI:10.1021/ja311669p
The first Suzuki cross-couplings of unactivated tertiary alkyl electrophiles are described. The method employs a readily accessible catalyst (NiBr2·diglyme/4,4′-di-tert-butyl-2,2′-bipyridine, both commercially available) and represents the initial example of the use of a group 10 catalyst to cross-couple unactivated tertiary electrophiles to form C–C bonds. This approach to the synthesis of all-carbon quaternary carbon centers does not suffer from isomerization of the alkyl group, in contrast with the umpolung strategy for this bond construction (cross-coupling of a tertiary alkylmetal with an aryl electrophile). Preliminary mechanistic studies are consistent with the generation of a radical intermediate along the reaction pathway.
Co-reporter:Christopher Uyeda ; Yichen Tan ; Gregory C. Fu ;Jonas C. Peters
Journal of the American Chemical Society 2013 Volume 135(Issue 25) pp:9548-9552
Publication Date(Web):May 23, 2013
DOI:10.1021/ja404050f
Building on the known photophysical properties of well-defined copper–carbazolide complexes, we have recently described photoinduced, copper-catalyzed N-arylations and N-alkylations of carbazoles. Until now, there have been no examples of the use of other families of heteroatom nucleophiles in such photoinduced processes. Herein, we report a versatile photoinduced, copper-catalyzed method for coupling aryl thiols with aryl halides, wherein a single set of reaction conditions, using inexpensive CuI as a precatalyst without the need for an added ligand, is effective for a wide range of coupling partners. As far as we are aware, copper-catalyzed C–S cross-couplings at 0 °C have not previously been achieved, which renders our observation of efficient reaction of an unactivated aryl iodide at −40 °C especially striking. Mechanistic investigations are consistent with these photoinduced C–S cross-couplings following a SET/radical pathway for C–X bond cleavage (via a Cu(I)–thiolate), which contrasts with nonphotoinduced, copper-catalyzed processes wherein a concerted mechanism is believed to occur.
Co-reporter:Dr. Rylan J. Lundgren;Dr. Ashraf Wilsily;Dr. Nicolas Marion;Dr. Cong Ma;Dr. Ying Kit Chung; Gregory C. Fu
Angewandte Chemie International Edition 2013 Volume 52( Issue 9) pp:2525-2528
Publication Date(Web):
DOI:10.1002/anie.201208957
Co-reporter:Dr. Alex C. Bissember;Dr. Rylan J. Lundgren;Sidney E. Creutz; Jonas C. Peters; Gregory C. Fu
Angewandte Chemie International Edition 2013 Volume 52( Issue 19) pp:5129-5133
Publication Date(Web):
DOI:10.1002/anie.201301202
Co-reporter:Dr. Rylan J. Lundgren;Dr. Ashraf Wilsily;Dr. Nicolas Marion;Dr. Cong Ma;Dr. Ying Kit Chung; Gregory C. Fu
Angewandte Chemie 2013 Volume 125( Issue 9) pp:2585-2588
Publication Date(Web):
DOI:10.1002/ange.201208957
Co-reporter:Alex C. Bissember ; Anna Levina
Journal of the American Chemical Society 2012 Volume 134(Issue 34) pp:14232-14237
Publication Date(Web):August 20, 2012
DOI:10.1021/ja306323x
We have exploited a typically undesired elementary step in cross-coupling reactions, β-hydride elimination, to accomplish palladium-catalyzed dehydrohalogenations of alkyl bromides to form terminal olefins. We have applied this method, which proceeds in excellent yield at room temperature in the presence of a variety of functional groups, to a formal total synthesis of (R)-mevalonolactone. Our mechanistic studies have established that the rate-determining step can vary with the structure of the alkyl bromide and, most significantly, that L2PdHBr (L = phosphine), an intermediate that is often invoked in palladium-catalyzed processes such as the Heck reaction, is not an intermediate in the active catalytic cycle.
Co-reporter:Sarah Yunmi Lee ; Jaclyn M. Murphy ; Atsushi Ukai
Journal of the American Chemical Society 2012 Volume 134(Issue 36) pp:15149-15153
Publication Date(Web):August 30, 2012
DOI:10.1021/ja307425g
Because of the ubiquity of the secondary carbinol subunit, the development of new methods for its enantioselective synthesis remains an important ongoing challenge. In this report, we describe the first nonenzymatic method for the dynamic kinetic resolution (DKR) of secondary alcohols (specifically, aryl alkyl carbinols) through enantioselective acylation, and we substantially expand the scope of this approach, vis-à-vis enzymatic reactions. Simply combining an effective process for the kinetic resolution of alcohols with an active catalyst for the racemization of alcohols did not lead to DKR, due to the incompatibility of the ruthenium-based racemization catalyst with the acylating agent (Ac2O) used in the kinetic resolution. A mechanistic investigation revealed that the ruthenium catalyst is deactivated through the formation of a stable ruthenium–acetate complex; this deleterious pathway was circumvented through the appropriate choice of acylating agent (an acyl carbonate). Mechanistic studies of this new process point to reversible N-acylation of the nucleophilic catalyst, acyl transfer from the catalyst to the alcohol as the rate-determining step, and carbonate anion serving as the Brønsted base in that acyl-transfer step.
Co-reporter:Jörg T. Binder ; Christopher J. Cordier
Journal of the American Chemical Society 2012 Volume 134(Issue 41) pp:17003-17006
Publication Date(Web):October 5, 2012
DOI:10.1021/ja308460z
We have developed a nickel-catalyzed method for the asymmetric cross-coupling of secondary electrophiles with secondary nucleophiles, specifically, stereoconvergent Negishi reactions of racemic benzylic bromides with achiral cycloalkylzinc reagents. In contrast to most previous studies of enantioselective Negishi cross-couplings, tridentate pybox ligands are ineffective in this process; however, a new, readily available bidentate isoquinoline–oxazoline ligand furnishes excellent ee’s and good yields. The use of acyclic alkylzinc reagents as coupling partners led to the discovery of a highly unusual isomerization that generates a significant quantity of a branched cross-coupling product from an unbranched nucleophile.
Co-reporter:Sidney E. Creutz;Kenneth J. Lotito;Jonas C. Peters
Science 2012 Volume 338(Issue 6107) pp:647-651
Publication Date(Web):02 Nov 2012
DOI:10.1126/science.1226458
Co-reporter:Yichen Tan, José María Muñoz-Molina, Gregory C. Fu and Jonas C. Peters
Chemical Science (2010-Present) 2014 - vol. 5(Issue 7) pp:NaN2835-2835
Publication Date(Web):2014/04/22
DOI:10.1039/C4SC00368C
Most copper-catalyzed cross-couplings require an elevated reaction temperature. Recently, a photoinduced variant has been developed that enables C–X bond-forming reactions of certain nitrogen and sulfur nucleophiles to proceed under unusually mild conditions (−40 °C to room temperature). In view of the importance of carbon–oxygen bond construction in organic chemistry, the expansion of this photochemical approach to oxygen nucleophiles is an important objective. In this report, we establish that, in the presence of light and an inexpensive copper pre-catalyst (CuI), a wide array of phenols and aryl iodides can be coupled to generate diaryl ethers under mild conditions (room temperature) in the presence of a variety of functional groups. Our studies indicate that a Cu(I)–phenoxide complex is a viable intermediate in photoinduced C–O bond-formation.
Co-reporter:Miles W. Johnson, Kareem I. Hannoun, Yichen Tan, Gregory C. Fu and Jonas C. Peters
Chemical Science (2010-Present) 2016 - vol. 7(Issue 7) pp:NaN4100-4100
Publication Date(Web):2016/02/24
DOI:10.1039/C5SC04709A
Photoinduced, copper-catalyzed cross-coupling can offer a complementary approach to thermal (non-photoinduced) methods for generating C–X (X = C, N, O, S, etc.) bonds. In this report, we describe the first detailed mechanistic investigation of one of the processes that we have developed, specifically, the (stoichiometric) coupling of a copper–thiolate with an aryl iodide. In particular, we focus on the chemistry of a discrete [CuI(SAr)2]− complex (Ar = 2,6-dimethylphenyl), applying a range of techniques, including ESI-MS, cyclic voltammetry, transient luminescence spectroscopy, optical spectroscopy, DFT calculations, Stern–Volmer analysis, EPR spectroscopy, actinometry, and reactivity studies. The available data are consistent with the viability of a pathway in which photoexcited [CuI(SAr)2]−* serves as an electron donor to an aryl iodide to afford an aryl radical, which then reacts in cage with the newly generated copper(II)–thiolate to furnish the cross-coupling product in a non-chain process.
.jpg)
![L-ISOLEUCINE, N-[(6-BROMO-2,3-DIHYDRO-1-OXO-1H-INDEN-4-YL)CARBONYL]-](http://img.cochemist.com/ccimg/863500/863489-95-6.png)
![L-ISOLEUCINE, N-[(6-BROMO-2,3-DIHYDRO-1-OXO-1H-INDEN-4-YL)CARBONYL]-](http://img.cochemist.com/ccimg/863500/863489-95-6_b.png)

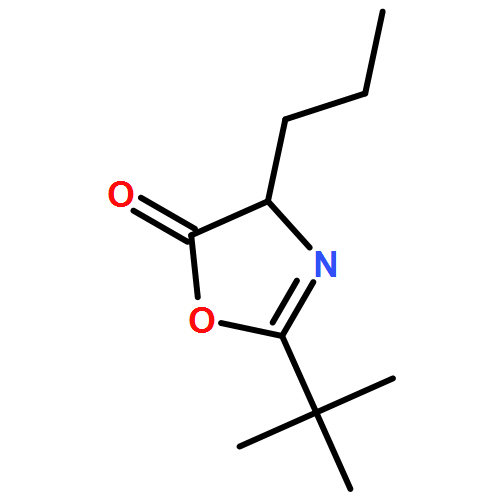
![5(4H)-Oxazolone, 2-(1,1-dimethylethyl)-4-[2-(methylthio)ethyl]-](/data/chemimg/3725200/1245935-92-5.png)
![5(4H)-Oxazolone, 2-(1,1-dimethylethyl)-4-[2-(methylthio)ethyl]-](/data/chemimg/3725200/1245935-92-5_b.png)
![Zinc, [1,1'-biphenyl]-4-ylchloro-](http://img.cochemist.com/ccimg/917100/917086-24-9.png)
![Zinc, [1,1'-biphenyl]-4-ylchloro-](http://img.cochemist.com/ccimg/917100/917086-24-9_b.png)







![Pentanal, 5-[[tris(1-methylethyl)silyl]oxy]-](http://img.cochemist.com/ccimg/245100/245039-02-5.png)
![Pentanal, 5-[[tris(1-methylethyl)silyl]oxy]-](http://img.cochemist.com/ccimg/245100/245039-02-5_b.png)


![Zinc, chloro[3-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/154500/154407-12-2.png)
![Zinc, chloro[3-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/154500/154407-12-2_b.png)

![(+)-2,2'-Isopropylidenebis[(4R)-4-phenyl-2-oxazoline]](/data/chemimg/116500/150529-93-4.png)
![(+)-2,2'-Isopropylidenebis[(4R)-4-phenyl-2-oxazoline]](/data/chemimg/116500/150529-93-4_b.png)


![Hexanoyl chloride, 6-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/134200/134163-12-5.png)
![Hexanoyl chloride, 6-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/134200/134163-12-5_b.png)
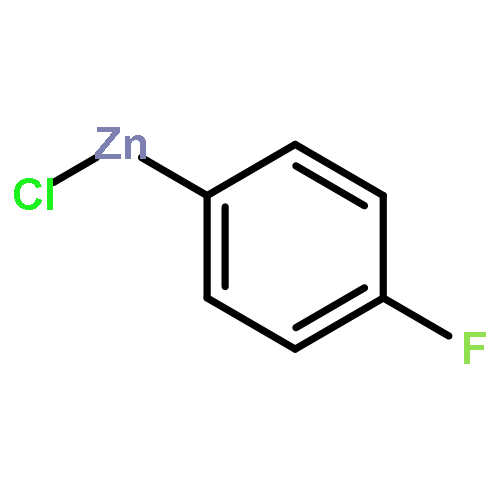

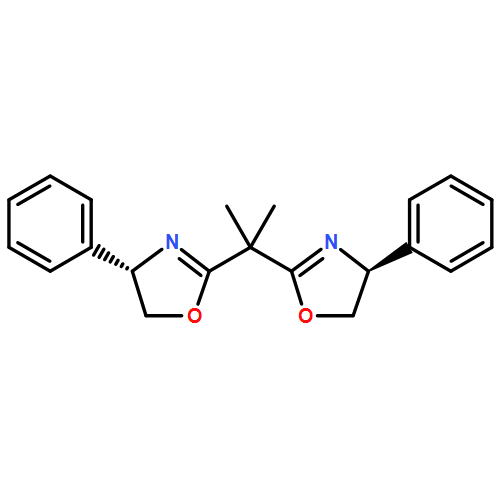


![1H-Indole, 5-(chloromethyl)-1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/119200/119159-26-1.png)
![1H-Indole, 5-(chloromethyl)-1-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/119200/119159-26-1_b.png)
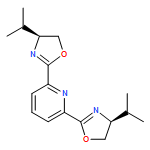

![Tert-butyl-[2-(1h-indol-3-yl)ethoxy]-dimethylsilane](http://img.cochemist.com/ccimg/101100/101079-48-5.png)
![Tert-butyl-[2-(1h-indol-3-yl)ethoxy]-dimethylsilane](http://img.cochemist.com/ccimg/101100/101079-48-5_b.png)
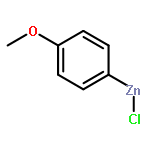
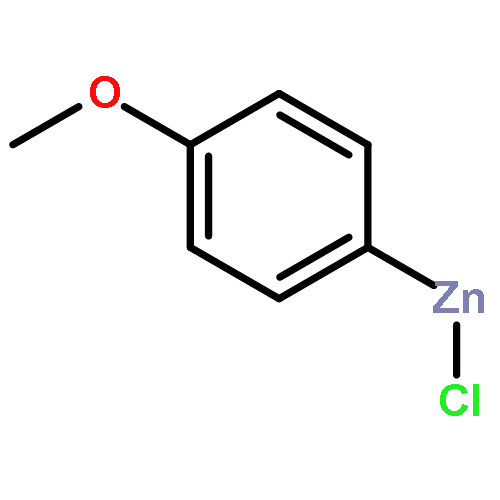
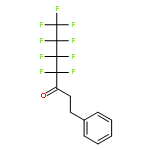











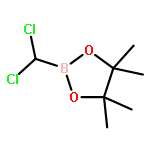
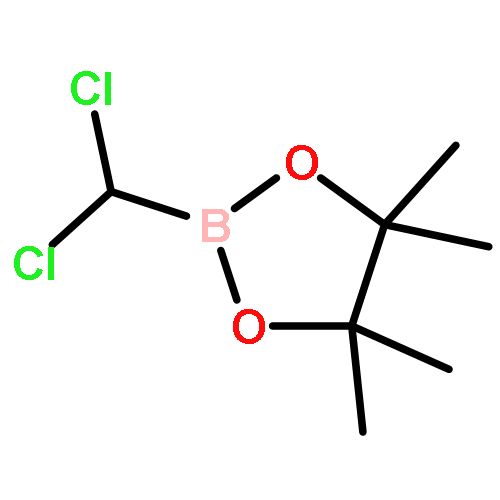









































![Bicyclo[2.2.1]heptane, 2-bromo-, (1R,2S,4S)-rel- (9CI)](http://img.cochemist.com/ccimg/13300/13237-87-1.png)
![Bicyclo[2.2.1]heptane, 2-bromo-, (1R,2S,4S)-rel- (9CI)](http://img.cochemist.com/ccimg/13300/13237-87-1_b.png)





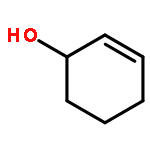

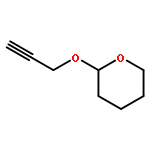
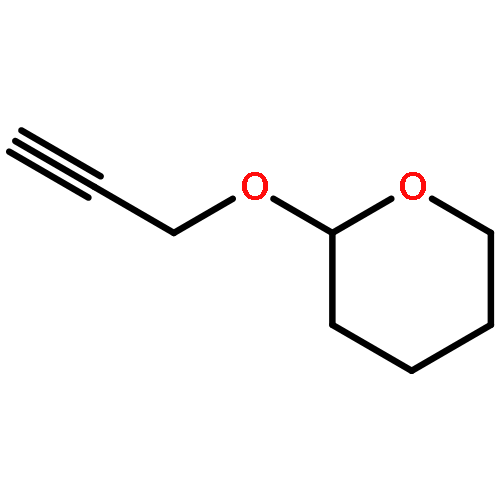
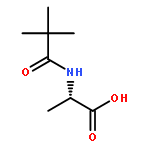
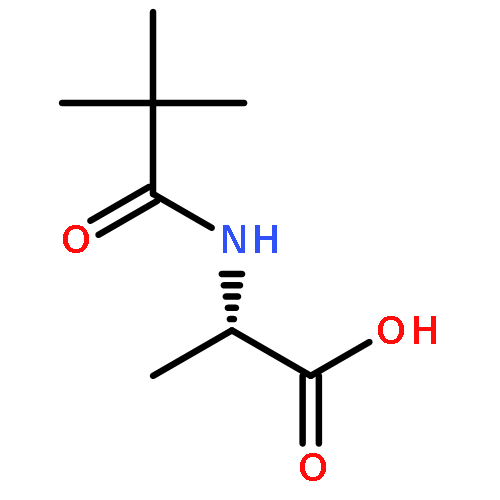

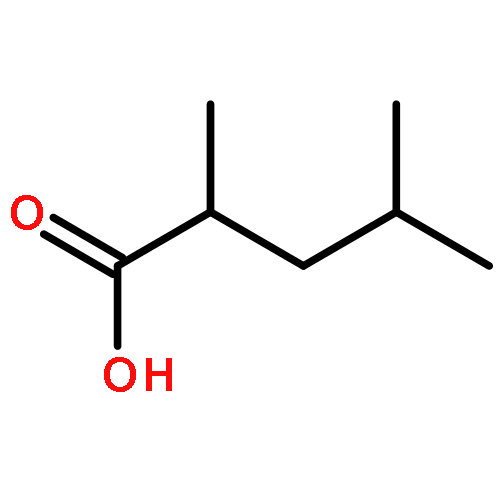
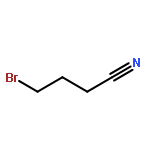
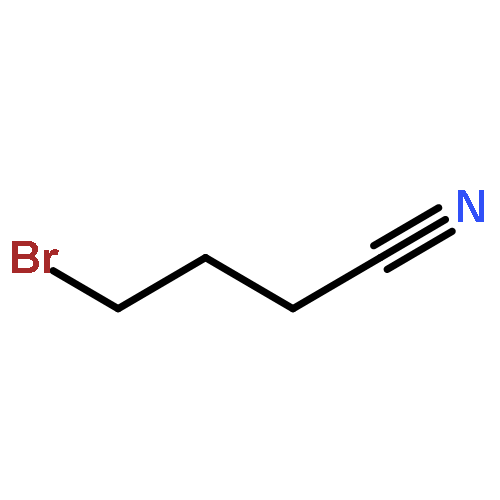






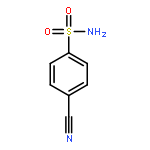







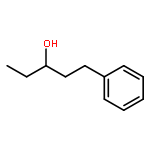
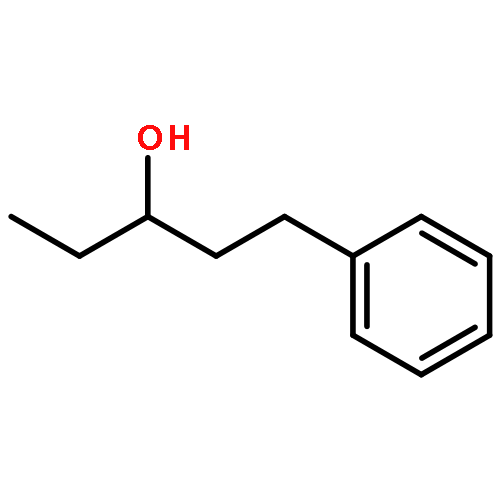


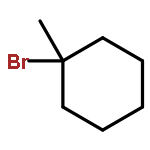

![(8R,9S,13S,14S)-13-methyl-6,7,8,9,11,12,13,14,15,16-decahydrospiro[cyclopenta[a]phenanthrene-17,2'-[1,3]dioxolan]-3-ol](http://img.cochemist.com/ccimg/1000/900-83-4.png)
![(8R,9S,13S,14S)-13-methyl-6,7,8,9,11,12,13,14,15,16-decahydrospiro[cyclopenta[a]phenanthrene-17,2'-[1,3]dioxolan]-3-ol](http://img.cochemist.com/ccimg/1000/900-83-4_b.png)












![1-Hexanol, 6-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-](http://img.cochemist.com/ccimg/121700/121671-77-0.png)
![1-Hexanol, 6-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-](http://img.cochemist.com/ccimg/121700/121671-77-0_b.png)
![6-[TERT-BUTYL(DIPHENYL)SILYL]OXYHEXANAL](/data/chemimg/393100/118794-70-0.png)
![6-[TERT-BUTYL(DIPHENYL)SILYL]OXYHEXANAL](/data/chemimg/393100/118794-70-0_b.png)
































































































































































































































































































































































![4H-Diindeno[7,1-cd:1',7'-ef]phosphocin,5,6,10,11,12,13-hexahydro-5-phenyl-, (11aS)- (9CI)](http://img.cochemist.com/ccimg/885800/885701-78-0.png)
![4H-Diindeno[7,1-cd:1',7'-ef]phosphocin,5,6,10,11,12,13-hexahydro-5-phenyl-, (11aS)- (9CI)](http://img.cochemist.com/ccimg/885800/885701-78-0_b.png)




![4H-Diindeno[7,1-cd:1',7'-ef]phosphocin,5,6,10,11,12,13-hexahydro-5-phenyl-, (11aR)- (9CI)](http://img.cochemist.com/ccimg/856500/856407-37-9.png)
![4H-Diindeno[7,1-cd:1',7'-ef]phosphocin,5,6,10,11,12,13-hexahydro-5-phenyl-, (11aR)- (9CI)](http://img.cochemist.com/ccimg/856500/856407-37-9_b.png)



















































![Benzofuran, 2,3-dihydro-3-[(phenylthio)methyl]-](http://img.cochemist.com/ccimg/103400/103304-48-9.png)
![Benzofuran, 2,3-dihydro-3-[(phenylthio)methyl]-](http://img.cochemist.com/ccimg/103400/103304-48-9_b.png)