Co-reporter:Long Jiang, Ruijuan Yin, Xueting Wang, Jiajia Dai, Jing Li, Tao Jiang, Rilei Yu
European Journal of Medicinal Chemistry 2017 Volume 135(Volume 135) pp:
Publication Date(Web):28 July 2017
DOI:10.1016/j.ejmech.2017.04.019
•A novel family of neolamellarin A derivatives showed high inhibitory activity toward heat shock protein 90 (Hsp90).•3,4-Bis(catechol)pyrrole scaffold and methoxy groups were essential to the Hsp90 inhibitory activity of these compounds.•The hydrophobic interactions were the main driving force for the binding affinity of these compounds.In this study, we designed and synthesized a novel family of neolamellarin A derivatives that showed high inhibitory activity toward heat shock protein 90 (Hsp90), a kinase associated with cell proliferation. The 3,4-bis(catechol)pyrrole scaffold and the benzyl group with methoxy modification at N position of pyrrole are essential to the Hsp90 inhibitory activity and cytotoxicity of these compounds. Western blot analysis demonstrated that these compounds induced dramatic depletion of the examined client proteins of Hsp90, and accelerated cancer cell apoptosis. Docking simulations suggested that the binding mode of 9p was similar to that of the VER49009, a potent inhibitor of Hsp90. Further molecular dynamics simulation indicated that the hydrophobic interactions as well as the hydrogen bonds contributed to the high affinity of 9p to Hsp90.Download high-res image (328KB)Download full-size image
Co-reporter:Yunyan Zhao, Zhiming Zhang, Liangmin Yu, Tao Jiang
Synthetic Metals 2017 Volume 234(Volume 234) pp:
Publication Date(Web):1 December 2017
DOI:10.1016/j.synthmet.2017.11.005
•Hydrophobic polystyrene/electro-spun polyaniline coatings were prepared through a superposition combination method.•Electrochemical measurements showed that the bi-layer coating system has the outstanding corrosion protection property.•The coating could give full play to the shielding performance of topcoat and anodic protection performance of primer.Herein, bi-layer coatings were prepared through a superposition combination method, which contains polystyrene (PS) topcoat and polyaniline (PANI)/polymethyl methacrylate (PMMA) primer. PS topcoats prepared by different preparation methods show diverse microstructures, hydrophobicity, anti-medium permeability and corrosion resistance. In order to study the anti-corrosion mechanism of (PANI/PMMA primer − PS topcoat) coating system, Q235 carbon steel covered with bi-layer coatings were immersed in 3% NaCl solution for 720 h. And the anti-corrosion properties of samples were studied by open circuit potential, visual observations, electrochemical impedance spectroscopy and Tafel polarization test. Electrochemical techniques confirm that collocation of PS topcoat and PANI/PMMA primer could give full play to the shielding performance of topcoat and anodic protection performance of PANI/PMMA primer. Observed results show (PANI/PMMA primer − PS topcoat) coating system prepared by spray method has the best anti-medium permeability and outstanding corrosion protection property, which is attributed to the special microstructure.
Co-reporter:Sheng Tong, Meng Zhang, Shixi Wang, Ruijuan Yin, Rilei Yu, Shengbiao Wan, Tao Jiang, Lijuan Zhang
European Journal of Medicinal Chemistry 2016 Volume 123() pp:849-857
Publication Date(Web):10 November 2016
DOI:10.1016/j.ejmech.2016.07.071
•Novel isothiouronium-modified pyrimidine-substituted curcumin analogs were synthesized.•Isothiouronium modifications greatly enhanced anticancer activities of the analogs.•Only the fluorescence compound with isothiouronium modification showed unique Golgi localization.•The isothiouronium analogs were novel Golgi staining compounds.•The isothiouronium analogs were useful in studying the biological functions of Golgi.Most of protein post-translational modifications occur in the Golgi and many human diseases are associated with abnormal Golgi function or improper post translational modifications of proteins in the Golgi. In this study, we designed and synthesized 4 × 6 series of novel isothiouronium-modified (E,E)-4,6-bis(styryl)-pyrimidine analogs and found that they localized at the Golgi as visualized by the intrinsic fluorescence of the analogs. The isothiouronium-modified analogs had potent cytotoxicity in both normal (Chinese Hamster Ovary or CHO) and cancer cells. Furthermore, permethylated isothiouronium-modified analogs showed cancer cell-selective cytotoxicity. The molecular mechanisms underlying Golgi localization of isothiouronium-modified compounds were investigated using 7 CHO and 4 human cancer cell lines and the results indicated that the compounds had binding partners in the Golgi. Thus, isothiouronium-modified analogs might be promising anticancer agents, novel Golgi staining reagents, and useful research tools for studying Golgi functions in normal or cancer cells and in Golgi-related human diseases.
Co-reporter:Zongjiang Yu; Weizhi Sun; Weibing Peng; Rilei Yu; Guoqiang Li
Molecular Pharmaceutics 2016 Volume 13(Issue 5) pp:1699-1710
Publication Date(Web):March 28, 2016
DOI:10.1021/acs.molpharmaceut.6b00129
Oleanolic acid (OA) is a well-known pentacyclic triterpenoid compound, which has been used as a dietary supplement and is supplied as an over-the-counter drug for the treatment of human liver diseases. These are reasons for the low bioavailability of OA which have restricted its wider application. In this study, two OA prodrugs (1,3-cyclic propanyl phosphate esters of OA) were designed and synthesized. The hepatoprotective effects of these prodrugs were evaluated against carbon tetrachloride (CCl4) induced liver injury in mice; the levels of alanine aminotransferase (ALT), lactic dehydrogenase (LDH), and aspartate aminotransferase (AST) were significantly increased, and the level of the hepatic malondialdehyde (MDA) was increased. The metabolism, in vitro, of the prodrugs was studied by incubation in rat liver microsome; the plasma pharmacokinetics and the biodistribution in vivo after intravenous (iv) injection to six rats were investigated, respectively. The prodrugs diminished gradually with time; most of the parent drugs were released within 30 min in vitro, and the presumed mechanism of the in vitro metabolism was confirmed. The plasma–concentration data in vivo was analyzed by a compartmental method: both the prodrugs and the corresponding released parent drugs existed at up to 48 h in rats. The t1/2 improved after intravenous administration in rats compared with direct injection of the parent drugs. All analyte concentrations were highest in the liver, and most of the prodrugs were excreted in feces (>47.11%). Therefore, 1,3-cyclic propanyl phosphate esters of OA can serve as a promising lead candidate for drugs.
Co-reporter:Renshuai Zhang, Shaopeng Chen, Xiaowei Zhang, Rilei Yu, Shengbiao Wan, Meiyu Geng and Tao Jiang
RSC Advances 2016 vol. 6(Issue 43) pp:36857-36862
Publication Date(Web):07 Apr 2016
DOI:10.1039/C6RA06818A
A series of novel quinazoline glycoside derivatives were designed, synthesized, and evaluated for their inhibition activities against EGFR-WT, EGFR/L858R/T790M, and skin epidermoid carcinoma cell line (A431). Several L-rhamnose derivatives were found to be as efficient as the irreversible inhibitor HKI-272 or BIBW2992 in inhibiting EGFR/T790M/L858R. Molecular dynamic simulations indicated that the saccharide group plays a significant role in stabilization of the quinazoline glycoside derivative through the hydrogen bonding with several polar residues at the edge of the ATP-binding cleft.
Co-reporter:Renshuai Zhang, Shaopeng Chen, Xueting Wang, Rilei Yu, Mingjing Li, Sumei Ren, Tao Jiang
Carbohydrate Research 2016 Volume 429() pp:48-53
Publication Date(Web):24 June 2016
DOI:10.1016/j.carres.2016.04.011
•Three DNA cyclo-intercalators containing short linker-bridged were synthesized.•The DNA binding properties of novel cyclo-intercalators were studied.•Docking simulations explained the mechanism of novel cyclo-intercalators.Novel DNA cyclo-intercalators, which incorporated two intercalator subunits linked by two bridges, were synthesized. Binding of the compounds to calf-thymus DNA was studied by fluorescence spectroscopy, and docking simulations were used to predict the binding modes of these cyclic compounds. The spectral data demonstrated that all of these compounds can interact with CT-DNA. The sugar moiety played an important role in the process of binding between the intercalators containing glucuronic acid and DNA. The length and flexibility of the connecting bridges affected the binding affinity of the resultant cyclo-intercalators. Docking simulations showed that compounds 7 and 8 interact with DNA as mono-intercalators.
Co-reporter:Shixu Liu;Junxiu Meng;Wei Zhang;Shengbiao Wan
Journal of Heterocyclic Chemistry 2016 Volume 53( Issue 4) pp:1194-1199
Publication Date(Web):
DOI:10.1002/jhet.2381
Six novel analogues of bioactive natural indolo[3,2-b]quinoline alkaloid glycoside jusbetonin were designed and synthesized, employing polyphosphoric acid mediated cyclization and two different amidation strategies on introduction of tetra-O-acetyl-β-d-glucosamine to tetracyclic carboxylic acids.
Co-reporter:Qurat-ul-ain Shaikh, Meiting Yang, Khadim Hussain Memon, Mehreen Lateef, Du Na, Shengbiao Wan, Deslandes Eric, Lijuan Zhang, Tao Jiang
Carbohydrate Research 2016 430() pp: 72-81
Publication Date(Web):22 July 2016
DOI:10.1016/j.carres.2016.04.021
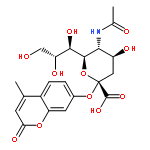
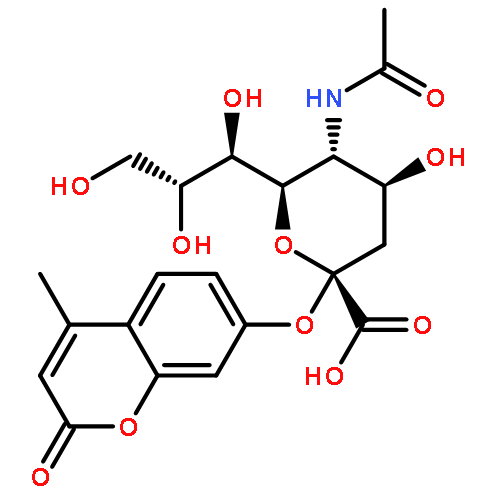
•A series of new benzoic and cinnamic acid analogs of PGG were synthesized.•The SAR of PGG analogs for antitumor and antioxidant activity has been studied.•PGG had highest cytotoxicities in HCT116 and A549 cells.•Pentavanilloylglucose was most effective at killing HT29 and H1299 cells.•Different molecular targets might be involved for antitumor and antioxidant activities.1,2,3,4,6-Pentakis[-O-(3,4,5-trihydroxybenzoyl)]-α,β-D-glucopyranose (PGG) 12 has been reported for its antioxidant activities, where the free OH groups in PGG seem to be critical for activities. To explore PGG-based compounds as chemotherapeutic agents and to analyze the contribution of specific OH groups in PGG for anti-cancer activities, we designed and synthesized a series of 27 benzoic and cinnamic acid analogs of PGG. These analogs were tested for cytotoxicities against two human lung (A549 and H1299) and two human colon (HCT116 and HT29) cancer cell lines. Compound 12 (PGG) had highest cytotoxicities against HCT116 and A549 cells with IC50 of 1.61 µM and 3.02 µM, respectively. In contrast, the compound 16 (1,2,3,4,6-pentakis[-O-(4-hydroxy-3-methoxybenzoyl)]-α,β-D-glucopyranose, PVG) was most effective at killing HT29 and H1299 cells with IC50 of 1.76 µM and 3.65 µM, respectively, indicating the mutual contribution of m-methoxy and p-hydroxy groups to the observed cytotoxicities. Moreover, cinnamic acid analogs were less active than the benzoic acid analogs evidenced by higher IC50 values. Furthermore, in cinnamic acid analogs the hydrogenation of double bond to saturated 2-C side chain enhance the cytotoxicities in all four cell lines. Compounds also possess good anti-oxidant and reducing activities. Compound 12 and 26 show the highest antioxidant and reducing activities.
Co-reporter:Jiachi Chiou, Shengbiao Wan, Kin-Fai Chan, Pui-Kin So, Dandan He, Edward Wai-chi Chan, Tak-hang Chan, Kwok-yin Wong, Jiang Tao and Sheng Chen
Chemical Communications 2015 vol. 51(Issue 46) pp:9543-9546
Publication Date(Web):20 Apr 2015
DOI:10.1039/C5CC02594J
We report the discovery of a promising NDM-1 inhibitor, ebselen, through a cell-based screening approach. Enzymatic kinetic study and ESI-MS analysis suggested that ebselen could bind to NDM-1 by forming a S–Se bond with the Cys221 residue at the active site, thereby exhibiting a new inhibition mechanism with broad spectrum inhibitory potential.
Co-reporter:Shixu Liu;Wei Wang;Long Jiang;Shengbiao Wan;Lijuan Zhang;Rilei Yu
Chemical Biology & Drug Design 2015 Volume 86( Issue 5) pp:1221-1225
Publication Date(Web):
DOI:10.1111/cbdd.12589
A series of 2-pyridinyl-3-substituted-4(3H)-quinazolinones were synthesized, and their anti-influenza A virus activities were determined using the cytopathic effect inhibition assay. Most of the compounds were potent with IC50 values ranging from 51.6 to 93.0 μm, which are better than that of the currently marketed drug ribavirin. The molecular mechanisms of the new compounds were investigated using neuraminidase inhibition assay, cellular NF-κB signaling pathway inhibition assay, and computational docking. Compound 4e, which is a N3 imidazol-1-ylpropyl-substituted derivative of 2-pyridinyl-4(3H)-quinazolinone, had the most potent anti-influenza A virus activity in vitro, and inhibited both virus neuraminidase and cellular NF-κB signaling pathway. In conclusion, 2-pyridinyl-4(3H)-quinazolinone is a new scaffold for the design of potent anti-influenza A virus compounds, offering an alternative approach to tackle influenza drug resistance.
Co-reporter:Shaikh Qurat-ul-ain, Wei Wang, Meiting Yang, Na Du, Shengbiao Wan, Lijuan Zhang, Tao Jiang
Carbohydrate Research 2015 Volume 402() pp:152-157
Publication Date(Web):30 January 2015
DOI:10.1016/j.carres.2014.10.011
•Anomeric selectivity in galloylation of d-glucose/d-mannose was explored.•The synthesized compounds were tested for anti-IAV activity on MDCK cell line.•The double bond in cinnamic PGG analogues contributes for anti-IAV activity.•Anomeric mixtures appeared two times more active than Ribavirin.•αβ-Anomeric mixture was more potent than α- or β-pure anomers.Anomeric selectivity in galloylation of d-glucose and d-mannose with carboxylic acid was explored under steglich conditions. Base catalyst 4-dimethylaminopyridine favored the formation of alpha-anomers, while adding an acid and carbodiimide favored the formation of beta-anomers. Steric hindrance between α,β-unsaturated acid and C-2 OH stereochemistry (adjacent carbon to anomeric) influenced anomeric selectivity for both d-glucose and d-mannose. The influenza A virus inhibition activities of the synthesized compounds were evaluated in Madin–Darby canine kidney cell line using the cytopathic effect inhibition assay. All the synthetic methoxylated analogues showed more considerable activity against influenza A virus than their corresponding acids, which indicated the sugar core as key functionality for anti-viral activity. The activities of trimethoxy-cinnamic acid Pentagalloylglucose analogues, 3α, 3β, 4α, and 4β (IC50, 109.1 μM, 134.4 μM, 119.5 μM, 111.1 μM, respectively) were better than those of trimethoxy-benzoic acid Pentagalloylglucose analogues, 1-αβ and 2α, 2β (IC50, 209.8 μM, 132.9 μM, 161.2 μM, respectively), which suggested that the double bond in cinnamic acid Pentagalloylglucose analogues makes the major contribution for influenza A virus inhibitory activity. Notably, several anomeric mixtures showed better activities than pure alpha or beta anomer and were almost two times more effective than Ribavirin, a clinically used anti-viral drug.
Co-reporter:Hongxia Liu;Libo Li;Shaikh Qurat-ul-ain
Journal of Heterocyclic Chemistry 2015 Volume 52( Issue 2) pp:473-477
Publication Date(Web):
DOI:10.1002/jhet.1959
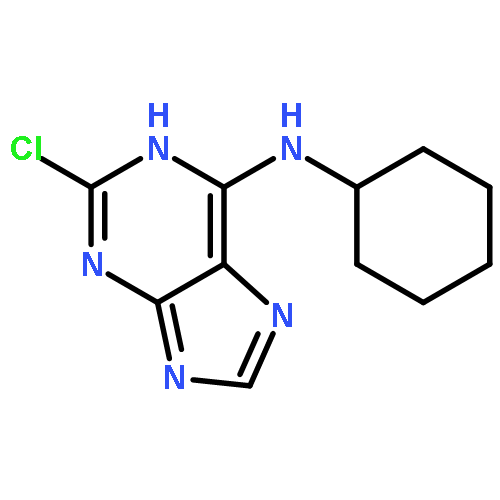
A series of novel 2,6-disubstituted purine derivatives were designed and synthesized from 2,6-dichloropurine. The structures of target compounds were determined by 1H-NMR, 13C-NMR, and HRMS. The synthesized compounds were evaluated for their inhibitory activities against lung cancer cell lines of A549 and liver cancer cell lines of Bel-7402. 2-(4-Benzyloxy-phenylamino)-6-(cyclohexylamino)purine(3), 2-(4-chloro-phenylamino)-6-(n-butylamino)purine (5), 2-(4-morpholinoamino)-6-(4-hydroxy-phenylamino)purine (9), and 2-(4-O-galactosyl-phenylamino)-6-(cyclohexylamino)purine (12) exhibited moderate inhibitory activity.
Co-reporter:Jiuyang Zhao, Xiang Ling, Shousong Cao, Xiaojun Liu, Shengbiao Wan, Tao Jiang, and Fengzhi Li
Molecular Pharmaceutics 2014 Volume 11(Issue 2) pp:457-467
Publication Date(Web):December 15, 2013
DOI:10.1021/mp4004282
We recently reported the identification and characterization of a novel small chemical molecule designated FL118. FL118 selectively inhibits multiple cancer survival and proliferation-associated antiapoptotic proteins (survivin, Mcl-1, XIAP, cIAP2) and eliminates small and large human tumor xenografts in animal models (Ling et al., PLoS One 2012, 7, e45571). Here, we report a follow-up study on the structure–activity relationship (SAR) of the hydroxyl group in the lactone ring of FL118. We found that the superior antitumor efficacy of FL118 heavily depends on its steric configuration through comparing the antitumor activity of FL118 with FL113 (the racemic mixture of FL118). Consistently, FL118 proved much more effective in inhibiting the expression of survivin, Mcl-1, and cIAP2, both in vitro and in vivo, compared to FL113. Additionally, Tet-on controlled induction of survivin or forced expression of Mcl-1 protects cancer cells from FL118-mediated growth inhibition and cell death. To further explore the SAR, we synthesized seven position 20-esterifiable FL118 and FL113 derivatives. Studies on these seven new compounds revealed that keeping a free hydroxyl group of FL118 is also important for high antitumor efficacy. Together, these studies confirm the superior anticancer activity of FL118 and narrow the window for further SAR studies to generate novel analogues based on FL118 core structure on its other potential chemical positions.Keywords: camptothecin analogue; cancer cells; FL118; human tumor animal models; IAP inhibitor; SAR analysis;
Co-reporter:Dacheng Fan, Weizhi Sun, Peiju Qiu, Zhiyong Wu, Yantuan Li, Shengbiao Wan, Tao Jiang, Lijuan Zhang
European Journal of Medicinal Chemistry 2014 Volume 74() pp:533-540
Publication Date(Web):3 March 2014
DOI:10.1016/j.ejmech.2013.08.012
•The TFA catalyzed 3-vinylindoles cyclodimerization was investigated.•Benzyl and phenyl substituted in 1,2-C of compounds gave highest cytotoxic effect.•F-benzyl and F-phenyl substitution in 1,2-C of compound altered EGFR expression.We synthesized a series of novel 3-indolyl cyclopent[b]indoles by trifluoroacetic acid mediated cyclodimerizations. The reaction showed high stereoselectivity and moderate to good yields. The influencing factors for stereoselectivity were systematically analyzed and a stepwise reaction mechanism was proposed. The cell viability tests in two colon and two lung cancer cell lines indicated the 1-benzyl-2-phenyl-group in 3-indolyl cyclopent[b]indoles was critical for the observed lower IC50s in these compounds. Western blot analysis demonstrated that the compound inhibited the expression and phosphorylation of EGFR through altered HSP90 expression. Further cell cycle and cell cycle check point protein analyses showed expected anti-cellular proliferation and cell cycle arresting properties associated with suppressed EGFR expression and phosphorylation. These data revealed a novel molecular mechanism explaining the observed cytotoxicities for these compounds.Compound 8 and 9 inhibited cancer cell proliferation. Western blot analysis demonstrated that compound 9 altered the expression and phosphorylation of EGFR.
Co-reporter:Nan Zhang, Zhaohui Zhang, Iris L.K. Wong, Shengbiao Wan, Larry M.C. Chow, Tao Jiang
European Journal of Medicinal Chemistry 2014 Volume 83() pp:74-83
Publication Date(Web):18 August 2014
DOI:10.1016/j.ejmech.2014.06.016
•Flexible Y or X-shaped scaffold was used for P-gp modulators design.•Two Naamidine analogues exhibited notably potent P-gp modulating activity.•Compound lipophilicity is a key factor for P-gp modulating activity.•p-Diethylamino introduced to 5-benzyl of imidazole significantly improved activity.Over-expression of P-glycoprotein (P-gp), a primary multidrug transporter which is located in plasma membranes, plays a major role in the multidrug resistance (MDR) of cytotoxic chemotherapy. Naamidines are a class of marine imidazole alkaloids isolated from Leucetta and Clathrina sponges, possessing a Y-shaped scaffold. Based on the results previously obtained from the third-generation MDR modulator ONT-093 and other modulators developed in our group, we designed and synthesized a series of novel 4,5-di-substituted benzyl-1-methyl-1H-imidazol-2-substituted amines using the Naamidine scaffold as the structure template. Subsequently, their reversing activity for Taxol resistance has been evaluated in P-gp-mediated multidrug resistance breast cancer cell line MDA435/LCC6MDR. Compounds 12c with a Y-shaped scaffold, and compound 17c which is ‘X-shaped’ scaffold and possesses a 4-diethylamino group at aryl ring B, turned out to be the most potent P-gp modulators. It appears that compounds 12c and 17c at 1 μM concentration can sensitize LCC6MDR cells toward Taxol by 26.4 and 24.5 folds, with an EC50 212.5 and 210.5 nM, respectively. These two compounds are about 5–6 folds more potent than verapamil (RF = 4.5). Moreover, compounds 12c and 17c did not exhibit obvious cytotoxicity in either cancer cell lines or normal mouse fibroblast cell lines. This study has demonstrated that the synthetic Naamidine analogues can be potentially employed as effective, safe modulators for the P-gp-mediated drug resistance cancer cells.Marine alkaloid Naamidines analogues, 12c and 17c at 1 μM can sensitize LCC6MDR cells toward taxol by 26.4 and 24.5 folds, with an EC50 212.5 and 210.5 nM respectively.
Co-reporter:Ruijuan Yin, Meng Zhang, Cui Hao, Wei Wang, Peiju Qiu, Shengbiao Wan, Lijuan Zhang and Tao Jiang
Chemical Communications 2013 vol. 49(Issue 76) pp:8516-8518
Publication Date(Web):25 Jul 2013
DOI:10.1039/C3CC45203D
The synthesis of a 4 × 4 series of novel quindoline derivatives with or without boronic acid modifications and their cytotoxicities, cellular localizations, and implications on cancer cells are presented and discussed.
Co-reporter:Krikor Bijian, Zhongwei Zhang, Bin Xu, Su Jie, Bo Chen, Shengbiao Wan, JianHui Wu, Tao Jiang, Moulay A. Alaoui-Jamali
European Journal of Medicinal Chemistry 2012 Volume 48() pp:143-152
Publication Date(Web):February 2012
DOI:10.1016/j.ejmech.2011.12.006
The present study reports synthesis and biological activity of novel benzoisoselenazolone compounds derived from ebselen and conjugated to a sugar molecule. Cell proliferation assay using cancer cells combined with in vitro biochemical assays revealed that benzoisoselenazolone 2d, 5a, and 6a exerted anti-proliferative activity, which correlated with selective in vitro inhibition of focal adhesion kinase, AKT-1, and protein kinase C-α. Active molecules were able to significantly inhibit cell migration and invasion in vitro compared to cells treated with the vehicle alone or ebselen. Moreover, in vivo anticancer activity focusing on lead compound 2d and using an invasive human breast cancer orthotopic mouse model revealed a potent anti-metastatic activity at well-tolerated doses. In summary, these novel benzoisoselenazolones we report herein target multiple kinases with established roles in cancer progression and possess anti-invasive and anti-metastatic activity in preclinical models supporting a potential for therapeutic application for human disease.Novel benzisoselenazolone derivatives that possess significant anticancer activity in vitro and in vivo.Highlights► Novel sugar conjugated benzisoselenazolone derivatives have been synthesized. ► Possess significant anti-invasive and anti-metastatic breast cancer activity. ► Selectively inhibits FAK, AKT-1 and PKC-α kinases.
Co-reporter:Fu Long Liu, Sheng Biao Wan, Tao Jiang
Chinese Chemical Letters 2012 Volume 23(Issue 9) pp:993-995
Publication Date(Web):September 2012
DOI:10.1016/j.cclet.2012.06.018
Co-reporter:Puyong Zhang, Xiaofei Sun, Bin Xu, Krikor Bijian, Shengbiao Wan, Guigen Li, Moulay Alaoui-Jamali, Tao Jiang
European Journal of Medicinal Chemistry 2011 Volume 46(Issue 12) pp:6089-6097
Publication Date(Web):December 2011
DOI:10.1016/j.ejmech.2011.10.036
We report herein the chemical synthesis and biological evaluation of β-carboline alkaloid pityriacitrin and some of its new derivatives. Using tryptophan or 5-hydroxytryptophan and 5-substituted indole-3-glyoxals as the starting materials, pityriacitrin and some of its derivatives were synthesized via the acid-catalyzed Pictet–Spengler reaction and fully characterized by 1H and 13C NMR, mass spectroscopy and IR determinations. Biological studies revealed that pityriacitrin has a weak antiproliferative activity against a panel of breast and prostate cancer cell lines, whereas some of its derivatives exhibited stronger and potent activity, which was associated with induction of both cell apoptosis and necrosis.Marine alkaloids Pityriacitrin and Pityriacitrin B were totally synthesized. New series of derivatives were obtained by modification. Some of the derivatives exhibited stronger anticancer activity comparing with Pityriacitrin.Highlights► synthesis of β-carboline alkaloid pityriacitrin and its new derivatives. ► Acid-catalyzed Pictet–Spengler reaction was introduced during the reaction. ► Some derivatives compounds were found to have stronger antiproliferative activity.
Co-reporter:Weizhi Sun;Weibing Peng;Guoqiang Li
Chemical Biology & Drug Design 2011 Volume 77( Issue 3) pp:206-211
Publication Date(Web):
DOI:10.1111/j.1747-0285.2010.01077.x
A new class of potential prodrugs, 1,3-cyclic propanyl phosphate esters of 18β-glycyrrhetic acid, was designed and synthesized through the key reaction of 18β-glycyrrhetic acid with 1,3-cyclic propanyl phosphate ester catalysed by lithium diisopropylamide. The sustained-release properties of 1,3-cyclic propanyl phosphate esters of 18β-glycyrrhetic acid in vivo were also investigated. The animal experiments showed that 18β-glycyrrhetic acid was released from 1,3-cyclic propanyl phosphate esters of 18β-glycyrrhetic acid at a steady rate in rats and the plasma concentrations of 18β-glycyrrhetic acid were nearly stable. The result indicated that 1,3-cyclic propanyl phosphate esters of 18β-glycyrrhetic acid have sustained-release properties to avoid the quick metabolism of 18β-glycyrrhetic acid. These prodrugs are highlighted as a promising new strategy to improve 18β-glycyrrhetic acid metabolism.
Co-reporter:Junxiu Meng;Shaoqing Yu;Shengbiao Wan;Sumei Ren
Chemical Biology & Drug Design 2011 Volume 78( Issue 5) pp:816-825
Publication Date(Web):
DOI:10.1111/j.1747-0285.2011.01196.x
A facile synthesis of a series of saccharide-binding arylboronic acid derivatives of indoloquinoline was described. The key synthetic steps were polyphosphoric acid-mediated cyclization, chlorinative aromatization, and amidation. Mass spectrometry experiments revealed these synthetic arylboronic acid derivatives of indoquinolines could bind to biologically important carbohydrates (sialic acid, fucose, glucose, and galactose) by forming boronate di-esters in alkaline aqueous solution. Most of the arylboronic acid derivatives of indoquinolines inhibited human breast cancer cell (MDA–231) proliferation at a concentration of 5 μm, whereas the compound 17 exhibited highest percentages (76.74%) of the cancer cell proliferation inhibition.
Co-reporter:Shaopeng Chen;Xiaowei Zhang;Junlei Wang;Shengbiao Wan;Meiyu Geng
Chemical Biology & Drug Design 2011 Volume 78( Issue 6) pp:1006-1013
Publication Date(Web):
DOI:10.1111/j.1747-0285.2011.01209.x
A new series of potential epidermal growth factor receptor inhibitors possessing bisquinazoline and saccharide moieties were designed and synthesized. The biological results demonstrated that the synthetic derivatives significantly inhibited epidermal growth factor receptor enzymatic activity in vitro. Of them, compound 14b showed the highest inhibitory rate toward epidermal growth factor receptor protein tyrosine kinase (81.36%) at a concentration of 1 μm. Further molecular simulation predicted that 14b offered its saccharide moieties hydrogen bonding to ATP-binding pocket.
Co-reporter:Shao Peng Chen, Yue Sun, Sheng Biao Wan, Tao Jiang
Chinese Chemical Letters 2011 Volume 22(Issue 9) pp:1033-1035
Publication Date(Web):September 2011
DOI:10.1016/j.cclet.2011.01.013
A novel 4-anilinoquinazoline dimer linked by a carbon–carbon bond in the C-7 position was synthesized via a one step Suzuki cross-coupling reaction. All structures of new compounds were characterized by 1H NMR, 13C NMR, and HRMS. The inhibition rate of the synthetic 4-anilinoquinazoline dimer 8 against epidermal growth factor receptor-tyrosine kinase enzymes (EGFR) in vitro was 44.4% at the concentration of 5.5 μmol/L.
Co-reporter:Pu Yong Zhang, Sheng Biao Wan, Su Mei Ren, Tao Jiang
Chinese Chemical Letters 2010 Volume 21(Issue 11) pp:1307-1309
Publication Date(Web):November 2010
DOI:10.1016/j.cclet.2010.05.012
Hyrtiosulawesine was isolated from Indonesian specimens of the marine sponges Hyrtios erectus and H. reticulatu in 2002. We report here the first total synthesis of hyrtiosulawesine using an efficient and convenient synthetic strategy which could be widely used in the synthesis of other β-caboline compounds. All structures of new compounds were confirmed by 1H NMR, 13C NMR and HRMS.
Co-reporter:Pu Yong Zhang, Jun Lei Wang, Sheng Biao Wan, Tao Jiang
Chinese Chemical Letters 2010 Volume 21(Issue 8) pp:889-891
Publication Date(Web):August 2010
DOI:10.1016/j.cclet.2010.03.017
Eudistalbin A was isolated from marine tunicate eudistoma album and possess cytotoxic activity (ED50 < 3.2 μg/mL) in vitro against the growth of KB human buccal carinoma cells. The synthetic eudistalbin A showed potent inhibitory activity against the breast carcinoma cell line MDA-231 with an IC50 value of 2.1 μmol/L using the metabolic assay MTT. All structures of new compounds were confirmed by 1H NMR, 13C NMR, HRMS and optical rotation.
Co-reporter:Wei Li;Zhong-Wei Zhang;Su-Mei Ren;Yann Sibiril;Dominique Parent Massin
Chemical Biology & Drug Design 2009 Volume 73( Issue 6) pp:682-686
Publication Date(Web):
DOI:10.1111/j.1747-0285.2009.00822.x
In this study, di-n-butyltin(IV) oxide was reacted with the amino glucose analog, cis-4-[N-(1′,3′,4′,6′-tetra-O-benzoyl-2-deoxy-glucopyranosyl)imido]-4-oxo-2-butenoic acid (1a) and o-[N-(1′,3′,4′,6′-tetra-O-benzoyl-2-deoxy-glucopyranosyl) carbamoyl] benzoic acid (2a) to give the complexes bis-{cis-4-[N-(1′,3′,4′,6′-tetra-O-benzoyl-2-deoxy-glucopyranosyl)imido-4-oxo-2-butenoic acid]-di-n-butyltin} carboxylate (1) and bis-{o-[N-(1′,3′,4′,6′-tetra-O-benzoyl-2-deoxy-glucopyranosyl) carbamoyl-benzoic acid]-di-n-butyltin}carboxylate (2). These two compounds were then characterized by IR, NMR and MS. In vitro tests showed that both compounds have high cytotoxicity in four tumor cell lines (P388, HL-60, A549 and BEL-7402). Clonogenic assays demonstrated that both compounds 1 and 2 have hematopoietic cell toxicity at 10−6 m.
Co-reporter:Wei Li;Zhong-Wei Zhang;Shi-Xi Wang;Su-Mei Ren
Chemical Biology & Drug Design 2009 Volume 74( Issue 1) pp:80-86
Publication Date(Web):
DOI:10.1111/j.1747-0285.2009.00831.x
A new series of potential DNA bisintercalators have been designed by linking 8-hydroxy-quinoline to the 6-CH2OH group of glucose and connecting the 1-OH group of glucose with various linkers, such as quinol, glycol, and triethylene glycol. These new compounds were well-synthesized and fully characterized. Preliminary binding assays of these compounds to calf thymus DNA (CT-DNA) were conducted using UV-absorption and fluorescence spectroscopy analysis. The primary antitumor activity of these compounds was also performed.
Co-reporter:Zhongwei Zhang, Shixi Wang, Shengbiao Wan, Sumei Ren, Wei Li, Tao Jiang
Carbohydrate Research 2009 Volume 344(Issue 3) pp:291-297
Publication Date(Web):17 February 2009
DOI:10.1016/j.carres.2008.11.015
Jusbetonin, an indolo[3,2-b]quinoline alkaloid glycoside originally isolated from Justicia betonica, and its derivatives were synthesized. The key steps in the synthetic strategy were the construction of indolo[3,2-b]quinoline skeleton and efficient coupling with the saccharides, in which the α-d-glycopyranosyl bromides were shown to be effective donors. Primary screening showed that all synthesized compounds possessed moderate proliferation inhibitory activity.
Co-reporter:Sheng Biao Wan, Zhe Lin Liu, Di Chen, Qing Ping Dou, Tao Jiang
Chinese Chemical Letters 2007 Volume 18(Issue 10) pp:1179-1181
Publication Date(Web):October 2007
DOI:10.1016/j.cclet.2007.08.014
11-Oxo-10,11-dihydroxy-5H-indolo[3,2,b]quinoline7-carboxylic acid was obtained specifically by polyphosphorous acid catalyzed cyclization with optimal reaction conditions. Biological assays showed that it potentially inhibits the proteasomal chymotrypsin-like activity in vitro and suppresses breast cancer cell growth.
Co-reporter:Kong De-Xin;Jiang Tao;Guan Hua-Shi
Chinese Journal of Chemistry 2005 Volume 23(Issue 7) pp:
Publication Date(Web):16 AUG 2005
DOI:10.1002/cjoc.200590816
Antioxidants are of great interest because of their involvement in many important biological and industrial processes. It is meaningful to study their structure-antioxidant activity relationship (SAAR) and design novel, efficient and low-toxicity antioxidant. In this paper, Eigen Value Analysis (EVA), a 3-dimensional quantitative structure activity relationship (3-D QSAR) method, was employed to study antioxidant SAAR. Significant relational models were obtained with all the PLS cross-validate values being larger than 0.5, meaning that the models have sound predictive power. Compared with other QSAR methods, EVA possesses several advantages, especially that it does not need alignment. It should be believed that EVA will be an efficient approach to SAAR.
Co-reporter:Zhong-Wei Zhang;Su-Mei Ren;Yan-Xia Zhang;Jing-Sheng Yu
Chinese Journal of Chemistry 2005 Volume 23(Issue 12) pp:1655-1658
Publication Date(Web):22 DEC 2005
DOI:10.1002/cjoc.200591655
Di-n-butyltin oxide reacted with p-[N,N-bis(2-chloroethyl)amino]benzoic acid to yield the compounds {{4-[(ClCH2CH2)2N]C6H4COOSnBu2}2O}2 (1) and {4-[(ClCH2CH2)2N]C6H4COO}2SnBu2 (2), which have been characterized by IR and 1H NMR spectra. The X-ray diffractional studies of 1 reveal the structure of the molecule to be a dimer, in which the two Bu2Sn groups were linked via two bridging oxygen atoms to form a central Bu4Sn2O2 unit. And the tin atom adopts two carbons from two n-butyl groups and three oxygen atoms from the acid and the bridging oxygen. In vitro test showed compound 1 to exhibit high cytotoxicity against P388 and HL-60 cell lines.
Co-reporter:Ruijuan Yin, Meng Zhang, Cui Hao, Wei Wang, Peiju Qiu, Shengbiao Wan, Lijuan Zhang and Tao Jiang
Chemical Communications 2013 - vol. 49(Issue 76) pp:NaN8518-8518
Publication Date(Web):2013/07/25
DOI:10.1039/C3CC45203D
The synthesis of a 4 × 4 series of novel quindoline derivatives with or without boronic acid modifications and their cytotoxicities, cellular localizations, and implications on cancer cells are presented and discussed.
Co-reporter:Jiachi Chiou, Shengbiao Wan, Kin-Fai Chan, Pui-Kin So, Dandan He, Edward Wai-chi Chan, Tak-hang Chan, Kwok-yin Wong, Jiang Tao and Sheng Chen
Chemical Communications 2015 - vol. 51(Issue 46) pp:NaN9546-9546
Publication Date(Web):2015/04/20
DOI:10.1039/C5CC02594J
We report the discovery of a promising NDM-1 inhibitor, ebselen, through a cell-based screening approach. Enzymatic kinetic study and ESI-MS analysis suggested that ebselen could bind to NDM-1 by forming a S–Se bond with the Cys221 residue at the active site, thereby exhibiting a new inhibition mechanism with broad spectrum inhibitory potential.











![4-[2-[4-[(E)-3-ethoxyprop-1-enyl]phenyl]-5-[4-(propan-2-ylamino)phenyl]-3H-imidazol-4-yl]-N-propan-2-yl-aniline](http://img.cochemist.com/ccimg/216300/216227-54-2.png)
![4-[2-[4-[(E)-3-ethoxyprop-1-enyl]phenyl]-5-[4-(propan-2-ylamino)phenyl]-3H-imidazol-4-yl]-N-propan-2-yl-aniline](http://img.cochemist.com/ccimg/216300/216227-54-2_b.png)
![4-{[4,5-bis(4-methoxybenzyl)-1-methyl-1H-imidazol-2-yl]amino}-1-methyl-1H-imidazole-2,5-dione](http://img.cochemist.com/ccimg/171800/171784-01-3.png)
![4-{[4,5-bis(4-methoxybenzyl)-1-methyl-1H-imidazol-2-yl]amino}-1-methyl-1H-imidazole-2,5-dione](http://img.cochemist.com/ccimg/171800/171784-01-3_b.png)
![(7S)-7-ethyl-7-hydroxy-10H-1,3-Dioxolo[4,5-g]pyrano[3',4':6,7]indolizino[1,2-b]quinoline-8,11(7H,13H)-dione](http://img.cochemist.com/ccimg/135500/135415-73-5.png)
![(7S)-7-ethyl-7-hydroxy-10H-1,3-Dioxolo[4,5-g]pyrano[3',4':6,7]indolizino[1,2-b]quinoline-8,11(7H,13H)-dione](http://img.cochemist.com/ccimg/135500/135415-73-5_b.png)
![1H-Pyrano[3',4':6,7]indolizino[1,2-b]quinoline-3,14(4H,12H)-dione,10-amino-4-ethyl-4-hydroxy-](http://img.cochemist.com/ccimg/130200/130194-90-0.png)
![1H-Pyrano[3',4':6,7]indolizino[1,2-b]quinoline-3,14(4H,12H)-dione,10-amino-4-ethyl-4-hydroxy-](http://img.cochemist.com/ccimg/130200/130194-90-0_b.png)
![b-D-Glucopyranose,pentakis[3,4,5-tris(phenylmethoxy)benzoate] (9CI)](http://img.cochemist.com/ccimg/122700/122625-60-9.png)
![b-D-Glucopyranose,pentakis[3,4,5-tris(phenylmethoxy)benzoate] (9CI)](http://img.cochemist.com/ccimg/122700/122625-60-9_b.png)
![1H-Imidazole-2,5-dione,4-[[4,5-bis[(4-methoxyphenyl)methyl]-1H-imidazol-2-yl]amino]-1-methyl- (9CI)](http://img.cochemist.com/ccimg/121900/121819-71-4.png)
![1H-Imidazole-2,5-dione,4-[[4,5-bis[(4-methoxyphenyl)methyl]-1H-imidazol-2-yl]amino]-1-methyl- (9CI)](http://img.cochemist.com/ccimg/121900/121819-71-4_b.png)
![1H-Imidazole-2,5-dione,4-[[5-[(3-hydroxy-4-methoxyphenyl)methyl]-4-[(4-methoxyphenyl)methyl]-1-methyl-1H-imidazol-2-yl]amino]-1-methyl-](http://img.cochemist.com/ccimg/121900/121819-68-9.png)
![1H-Imidazole-2,5-dione,4-[[5-[(3-hydroxy-4-methoxyphenyl)methyl]-4-[(4-methoxyphenyl)methyl]-1-methyl-1H-imidazol-2-yl]amino]-1-methyl-](http://img.cochemist.com/ccimg/121900/121819-68-9_b.png)
![(S)-4-Ethyl-4-hydroxy-7,8-dihydro-1H-pyrano[3,4-f]indolizine-3,6,10(4H)-trione](http://img.cochemist.com/ccimg/110400/110351-94-5.png)
![(S)-4-Ethyl-4-hydroxy-7,8-dihydro-1H-pyrano[3,4-f]indolizine-3,6,10(4H)-trione](http://img.cochemist.com/ccimg/110400/110351-94-5_b.png)
![1H-Imidazole-2,5-dione,4-[[5-[(4-hydroxyphenyl)methyl]-4-[(4-methoxyphenyl)methyl]-1-methyl-1H-imidazol-2-yl]amino]-1-methyl-](http://img.cochemist.com/ccimg/110200/110189-06-5.png)
![1H-Imidazole-2,5-dione,4-[[5-[(4-hydroxyphenyl)methyl]-4-[(4-methoxyphenyl)methyl]-1-methyl-1H-imidazol-2-yl]amino]-1-methyl-](http://img.cochemist.com/ccimg/110200/110189-06-5_b.png)
![9-chloro-4-ethyl-4-hydroxy-1H-pyrano[3',4':6,7]indolizino[1,2-b]quinoline-3,14(4H,12H)-dione](http://img.cochemist.com/ccimg/109600/109581-98-8.png)
![9-chloro-4-ethyl-4-hydroxy-1H-pyrano[3',4':6,7]indolizino[1,2-b]quinoline-3,14(4H,12H)-dione](http://img.cochemist.com/ccimg/109600/109581-98-8_b.png)
![4-ethyl-4-hydroxy-7,8-dihydro-1h-pyrano[3,4-f]indolizine-3,6,10(4 H)-trione](http://img.cochemist.com/ccimg/103000/102978-40-5.png)
![4-ethyl-4-hydroxy-7,8-dihydro-1h-pyrano[3,4-f]indolizine-3,6,10(4 H)-trione](http://img.cochemist.com/ccimg/103000/102978-40-5_b.png)
![(S)-10-Amino-4-ethyl-4-hydroxy-1H-pyrano[3',4':6,7]indolizino[1,2-b]quinoline-3,14(4H,12H)-dione](http://img.cochemist.com/ccimg/91500/91421-43-1.png)
![(S)-10-Amino-4-ethyl-4-hydroxy-1H-pyrano[3',4':6,7]indolizino[1,2-b]quinoline-3,14(4H,12H)-dione](http://img.cochemist.com/ccimg/91500/91421-43-1_b.png)




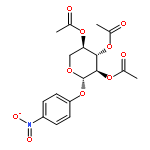
![[(2r,3r,4s,5r,6s)-4,5-diacetyloxy-6-(4-nitrophenoxy)-3-[(2s,3r,4s,5s,6r)-3,4,5-triacetyloxy-6-(acetyloxymethyl)oxan-2-yl]oxyoxan-2-yl]methyl Acetate](http://img.cochemist.com/ccimg/84100/84034-75-3.png)
![[(2r,3r,4s,5r,6s)-4,5-diacetyloxy-6-(4-nitrophenoxy)-3-[(2s,3r,4s,5s,6r)-3,4,5-triacetyloxy-6-(acetyloxymethyl)oxan-2-yl]oxyoxan-2-yl]methyl Acetate](http://img.cochemist.com/ccimg/84100/84034-75-3_b.png)
![Carbamicacid, N-[3-[[(4-methylphenyl)sulfonyl]oxy]propyl]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/81000/80909-96-2.png)
![Carbamicacid, N-[3-[[(4-methylphenyl)sulfonyl]oxy]propyl]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/81000/80909-96-2_b.png)
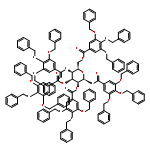
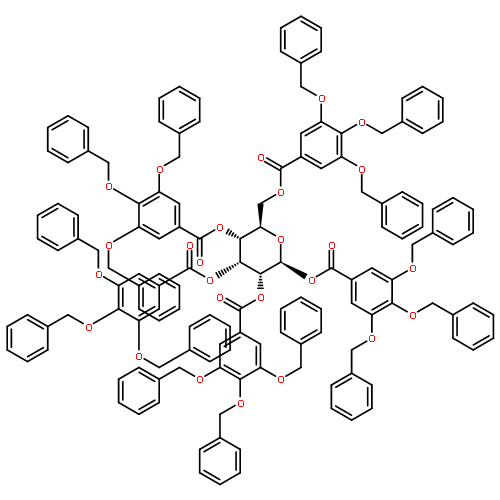
![11-chloro-4,12-diethyl-1H-pyrano[3',4':6,7]indolizino[1,2-b]quinoline-3,14(4H,12H)-dione](http://img.cochemist.com/ccimg/60000/59910-30-4.png)
![11-chloro-4,12-diethyl-1H-pyrano[3',4':6,7]indolizino[1,2-b]quinoline-3,14(4H,12H)-dione](http://img.cochemist.com/ccimg/60000/59910-30-4_b.png)
![8-methyl-9-oxo-9,11-dihydroindolizino[1,2-b]quinoline-7-carboxylic acid](http://img.cochemist.com/ccimg/57800/57764-77-9.png)
![8-methyl-9-oxo-9,11-dihydroindolizino[1,2-b]quinoline-7-carboxylic acid](http://img.cochemist.com/ccimg/57800/57764-77-9_b.png)
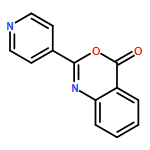
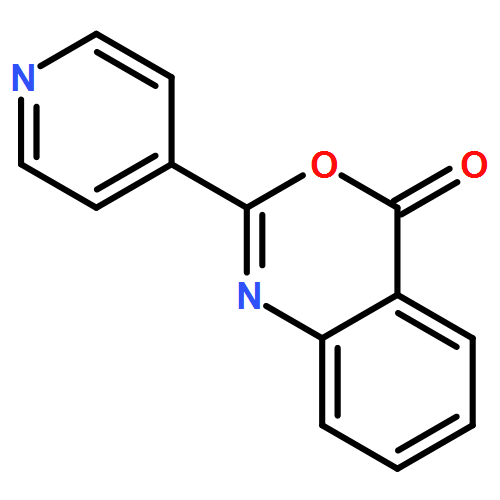
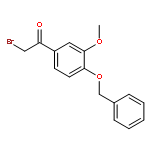
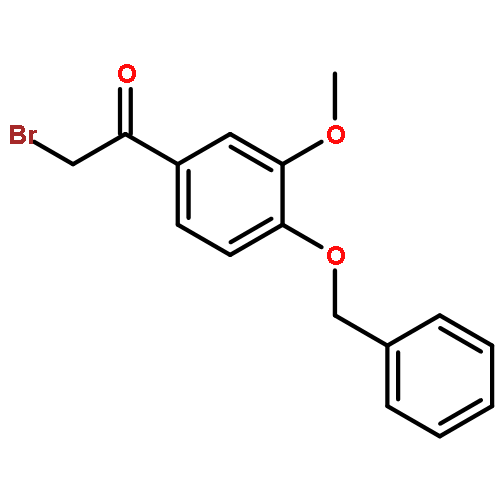
![Ethanone, 1-[2-methoxy-4-(phenylmethoxy)phenyl]-](http://img.cochemist.com/ccimg/56900/56879-12-0.png)
![Ethanone, 1-[2-methoxy-4-(phenylmethoxy)phenyl]-](http://img.cochemist.com/ccimg/56900/56879-12-0_b.png)
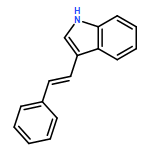
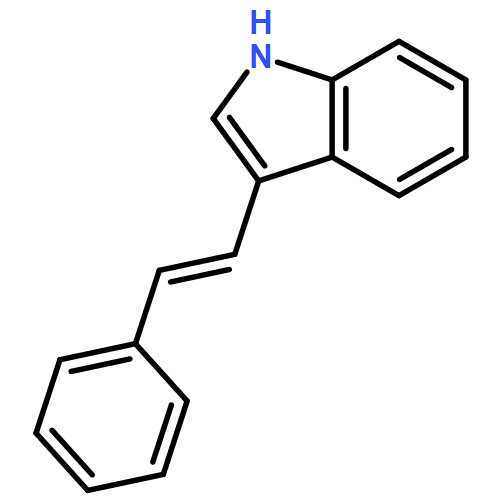
![3-[(1E)-2-nitroethenyl]-1H-Indole](http://img.cochemist.com/ccimg/51900/51870-94-1.png)
![3-[(1E)-2-nitroethenyl]-1H-Indole](http://img.cochemist.com/ccimg/51900/51870-94-1_b.png)












![[(2r,3r,4s,5r,6s)-3,4,5,6-tetrakis[(3,4,5-trihydroxybenzoyl)oxy]oxan-2-yl]methyl 3,4,5-trihydroxybenzoate](http://img.cochemist.com/ccimg/15000/14937-32-7.png)
![[(2r,3r,4s,5r,6s)-3,4,5,6-tetrakis[(3,4,5-trihydroxybenzoyl)oxy]oxan-2-yl]methyl 3,4,5-trihydroxybenzoate](http://img.cochemist.com/ccimg/15000/14937-32-7_b.png)
![Benzoic acid,2-[(2-chloroacetyl)amino]-](http://img.cochemist.com/ccimg/14500/14422-49-2.png)
![Benzoic acid,2-[(2-chloroacetyl)amino]-](http://img.cochemist.com/ccimg/14500/14422-49-2_b.png)











![[(2r,3s,4s,5r,6s)-3,4,5-triacetyloxy-6-(4-nitrophenoxy)oxan-2-yl]methyl Acetate](http://img.cochemist.com/ccimg/2900/2872-66-4.png)
![[(2r,3s,4s,5r,6s)-3,4,5-triacetyloxy-6-(4-nitrophenoxy)oxan-2-yl]methyl Acetate](http://img.cochemist.com/ccimg/2900/2872-66-4_b.png)
![1H-Isoindole-1,3(2H)-dione, 2-[2-(4-methoxyphenyl)-2-oxoethyl]-](http://img.cochemist.com/ccimg/1000/970-56-9.png)
![1H-Isoindole-1,3(2H)-dione, 2-[2-(4-methoxyphenyl)-2-oxoethyl]-](http://img.cochemist.com/ccimg/1000/970-56-9_b.png)

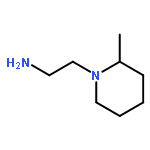
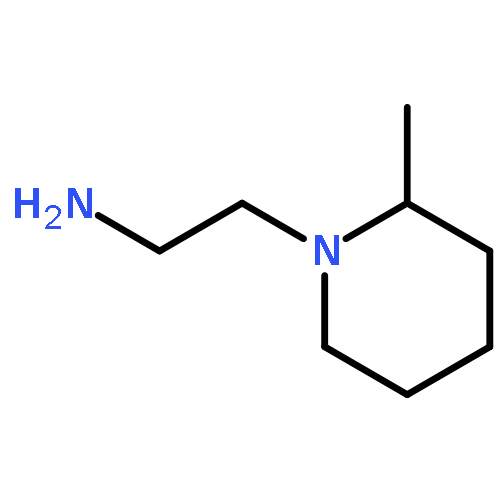
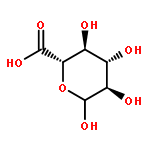
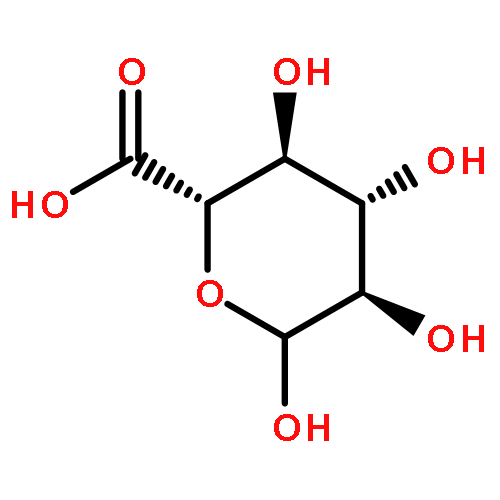




![3,6-diamino-9-[2-(methoxycarbonyl)phenyl]xanthylium chloride](/data/chemimg/622800/62669-70-9.png)
![3,6-diamino-9-[2-(methoxycarbonyl)phenyl]xanthylium chloride](/data/chemimg/622800/62669-70-9_b.png)