Co-reporter:Candy S. Hwang and Kim D. Janda
Biochemistry October 24, 2017 Volume 56(Issue 42) pp:5625-5625
Publication Date(Web):October 10, 2017
DOI:10.1021/acs.biochem.7b00948
Co-reporter:Paul T. Bremer, Sabine Pellett, James P. Carolan, William H. Tepp, Lisa M. Eubanks, Karen N. Allen, Eric A. Johnson, and Kim D. Janda
Journal of the American Chemical Society May 31, 2017 Volume 139(Issue 21) pp:7264-7264
Publication Date(Web):May 5, 2017
DOI:10.1021/jacs.7b01084
Botulinum neurotoxin serotype A (BoNT/A) causes a debilitating and potentially fatal illness known as botulism. The toxin is also a bioterrorism threat, yet no pharmacological antagonist to counteract its effects has reached clinical approval. Existing strategies to negate BoNT/A intoxication have looked to antibodies, peptides, or organic small molecules as potential therapeutics. In this work, a departure from the traditional drug discovery mindset was pursued, in which the enzyme’s susceptibility to metal ions was exploited. A screen of a series of metal salts showed marked inhibitory activity of group 11 and 12 metals against the BoNT/A light chain (LC) protease. Enzyme kinetics revealed that copper (I) and (II) cations displayed noncompetitive inhibition of the LC (Ki ≈ 1 μM), while mercury (II) cations were 10-fold more potent. Crystallographic and mutagenesis studies elucidated a key binding interaction between Cys165 on BoNT/A LC and the inhibitory metals. As potential copper prodrugs, ligand-copper complexes were examined in a cell-based model and were found to prevent BoNT/A cleavage of the endogenous protein substrate, SNAP-25, even at low μM concentrations of complexes. Further investigation of the complexes suggested a bioreductive mechanism causing intracellular release of copper, which directly inhibited the BoNT/A protease. In vivo experiments demonstrated that copper (II) dithiocarbamate and bis(thiosemicarbazone) complexes could delay BoNT/A-mediated lethality in a rodent model, indicating their potential for treating the harmful effects of BoNT/A intoxication. Our studies illustrate that metals can be therapeutically viable enzyme inhibitors; moreover, enzymes that share homology with BoNT LCs may be similarly targeted with metals.
Co-reporter:Paul T. Bremer, Joel E. Schlosburg, Matthew L. Banks, Floyd. F. Steele, Bin Zhou, Justin L. Poklis, and Kim D. Janda
Journal of the American Chemical Society June 28, 2017 Volume 139(Issue 25) pp:8601-8601
Publication Date(Web):June 2, 2017
DOI:10.1021/jacs.7b03334
Heroin is a highly abused opioid and incurs a significant detriment to society worldwide. In an effort to expand the limited pharmacotherapy options for opioid use disorders, a heroin conjugate vaccine was developed through comprehensive evaluation of hapten structure, carrier protein, adjuvant and dosing. Immunization of mice with an optimized heroin-tetanus toxoid (TT) conjugate formulated with adjuvants alum and CpG oligodeoxynucleotide (ODN) generated heroin “immunoantagonism”, reducing heroin potency by >15-fold. Moreover, the vaccine effects proved to be durable, persisting for over eight months. The lead vaccine was effective in rhesus monkeys, generating significant and sustained antidrug IgG titers in each subject. Characterization of both mouse and monkey antiheroin antibodies by surface plasmon resonance (SPR) revealed low nanomolar antiserum affinity for the key heroin metabolite, 6-acetylmorphine (6AM), with minimal cross reactivity to clinically used opioids. Following a series of heroin challenges over six months in vaccinated monkeys, drug-sequestering antibodies caused marked attenuation of heroin potency (>4-fold) in a schedule-controlled responding (SCR) behavioral assay. Overall, these preclinical results provide an empirical foundation supporting the further evaluation and potential clinical utility of an effective heroin vaccine in treating opioid use disorders.
Co-reporter:Atsushi Kimishima;Cody J. Wenthur;Bin Zhou;Kim D. Janda
ACS Chemical Biology January 20, 2017 Volume 12(Issue 1) pp:36-40
Publication Date(Web):November 17, 2016
DOI:10.1021/acschembio.6b00977
Prescription opioids (POs) such as oxycodone and hydrocodone are highly effective medications for pain management, yet they also present a substantial risk for abuse and addiction. The consumption of POs has been escalating worldwide, resulting in tens of thousands of deaths due to overdose each year. Pharmacokinetic strategies based upon vaccination present an attractive avenue to suppress PO abuse. Herein, the preparation of two active PO vaccines is described that were found to elicit high-affinity antiopioid antibodies through a structurally congruent drug-hapten design. Administration of these vaccines resulted in a significant blockade of opioid analgesic activity, along with an unprecedented increase in drug serum half-life and protection against lethal overdose.
Co-reporter:Paul T. Bremer;Michael Adler;Cecilia H. Phung;Ajay K. Singh;Kim D. Janda
Journal of Medicinal Chemistry January 12, 2017 Volume 60(Issue 1) pp:338-348
Publication Date(Web):December 14, 2016
DOI:10.1021/acs.jmedchem.6b01393
Botulinum neurotoxin A (BoNT/A) is one of the most deadly toxins and is the etiological agent of the potentially fatal condition, botulism. Herein, we investigated 8-hydroxyquinoline (quinolin-8-ol) as a potential inhibitor scaffold for preventing the deadly neurochemical effects of the toxin. Quinolinols are known chelators that can disrupt the BoNT/A metalloprotease zinc-containing active site, thus impeding its proteolysis of the endogenous protein substrate, synaptosomal-associated protein 25 (SNAP-25). By use of this information, the structure–activity relationship (SAR) of the quinolinol-5-sulfonamide scaffold was explored through preparation of a crude sulfonamide library and evaluation of the library in a BoNT/A LC enzymatic assay. Potency optimization of the sulfonamide hit compounds was undertaken as informed by docking studies, granting a lead compound with a submicromolar Ki. These quinolinol analogues demonstrated inhibitory activity in a cell-based model for SNAP-25 cleavage and an ex vivo assay for BoNT/A-mediated muscle paralysis.
Co-reporter:NIcholas T. Jacob;Kensaku Anraku;Atsushi Kimishima;Bin Zhou;Karen C. Collins;Jonathan W. Lockner;Beverley A. Ellis;Kim D. Janda
Chemical Communications 2017 vol. 53(Issue 58) pp:8156-8159
Publication Date(Web):2017/07/18
DOI:10.1039/C7CC04055E
A method for potentiating the response to an anti-cocaine vaccine by leveraging xenoreactive antibodies against the carbohydrate epitope Galα1,3-Gal (GAL) was found to result in a highly specific anti-cocaine response that was able to significantly attenuate cocaine-induced locomotion at 20 mg kg−1 with superior efficacy compared to a standard conjugate.
Co-reporter:Kensaku Anraku;Shun Sato;Nicholas T. Jacob;Lisa M. Eubanks;Beverly A. Ellis;Kim D. Janda
Organic & Biomolecular Chemistry 2017 vol. 15(Issue 14) pp:2979-2992
Publication Date(Web):2017/04/05
DOI:10.1039/C7OB00448F
Carbohydrate antigens displaying Galα(1,3)Gal epitopes are recognized by naturally occurring antibodies in humans. These anti-Gal antibodies comprise up to 1% of serum IgG and have been viewed as detrimental as they are responsible for hyperacute organ rejections. In order to model this condition, α(1,3)galactosyltransferase-knockout mice are inoculated against the Galα(1,3)Gal epitope. In our study, two α-Gal trisaccharide epitopes composed of either Galα(1,3)Galβ(1,4)GlcNAc or Galα(1,3)Galβ(1,4)Glc linked to a squaric acid ester moiety were examined for their ability to elicit immune responses in KO mice. Both target epitopes were synthesized using a two-component enzymatic system using modified disaccharide substrates containing a linker moiety for coupling. While both glycoconjugate vaccines induced the required high anti-Gal IgG antibody titers, it was found that this response had exquisite specificity for the Galα(1,3)Galβ(1,4)GlcNAc hapten used, with little cross reactivity with the Galα(1,3)Galβ(1,4)Glc hapten. Our findings indicate that while homogenous glycoconjugate vaccines provide high IgG titers, the carrier and adjuvanting factors can deviate the specificity to an antigenic determinant outside the purview of interest.
Co-reporter:Song Xue, Hajime Seki, Marek Remes, Peter Šilhár, Kim Janda
Bioorganic & Medicinal Chemistry Letters 2017 Volume 27, Issue 22(Issue 22) pp:
Publication Date(Web):15 November 2017
DOI:10.1016/j.bmcl.2017.10.021
Botulinum neurotoxins (BoNT) are among the most toxic known substances and currently there are no effective treatments for intraneuronal BoNT intoxication. Chicoric acid (ChA) was previously reported as a BoNT/A inhibitor that binds to the enzyme’s α-exosite. Herein, we report the synthesis and structure-activity relationships (SARs) of a series of ChA derivatives, which revealed essential binding interactions between ChA and BoNT/A. Moreover, several ChA-based inhibitors with improved potency against the BoNT/A were discovered.Download high-res image (103KB)Download full-size image
Co-reporter:Cody J. Wenthur, Xiaoqing Cai, Beverly A. Ellis, Kim D. Janda
Bioorganic & Medicinal Chemistry Letters 2017 Volume 27, Issue 16(Issue 16) pp:
Publication Date(Web):15 August 2017
DOI:10.1016/j.bmcl.2017.07.014
Given the need for further improvements in anti-cocaine vaccination strategies, a chimeric hapten (GNET) was developed that combines chemically-stable structural features from steady-state haptens with the hydrolytic functionality present in transition-state mimetic haptens. Additionally, as a further investigation into the generation of an improved bifunctional antibody pool, sequential vaccination with steady-state and transition-state mimetic haptens was undertaken. While GNET induced the formation of catalytically-active antibodies, it did not improve overall behavioral efficacy. In contrast, the resulting pool of antibodies from GNE/GNT co-administration demonstrated intermediate efficacy as compared to antibodies developed from either hapten alone. Overall, improved antibody catalytic efficiency appears necessary to achieve the synergistic benefits of combining cocaine hydrolysis with peripheral sequestration.Download high-res image (168KB)Download full-size image
Co-reporter:Hajime Seki; Song Xue; Sabine Pellett; Peter Šilhár; Eric A. Johnson;Kim D. Janda
Journal of the American Chemical Society 2016 Volume 138(Issue 17) pp:5568-5575
Publication Date(Web):April 12, 2016
DOI:10.1021/jacs.5b12929
Botulium neurotoxins (BoNTs) are among the most lethal toxins known to man. They are comprised of seven serotypes with BoNT/A being the most deadly; yet, there is no approved therapeutic for their intoxication or one that has even advanced to clinical trials. Botulinum neurotoxicity is ultimately governed through light chain (LC) protease SNARE protein cleavage leading to a loss of neurotransmitter release. Pharmacological attempts to ablate BoNT/A intoxication have sought to either nullify cellular toxin entry or critical biochemical junctions found within its intricate mechanism of action. In these regards, reports have surfaced of nonpeptidic small molecule inhibitors, but few have demonstrated efficacy in neutralizing cellular toxicity, a key prerequisite before rodent lethality studies can be initiated. On the basis of a lead discovered in our BoNT/A cellular assay campaign, we investigated a family of N-hydroxysuccinimide inhibitors grounded upon structure activity relationship (SAR) fundamentals. Molecules stemming from this SAR exercise were theorized to be protease inhibitors. However, this proposition was overturned on the basis of extensive kinetic analysis. Unexpectedly, inhibitor data pointed to thioredoxin reductase (TrxR), an essential component required for BoNT protease translocation. Also unforeseen was the inhibitors’ mechanism of action against TrxR, which was found to be brokered through a suicide-mechanism utilizing quinone methide as the inactivating element. This new series of TrxR inhibitors provides an alternative means to negate the etiological agent responsible for BoNT intoxication, the LC protease.
Co-reporter:T. L. Harris, C. J. Wenthur, A. Diego-Taboada, G. Mackenzie, T. S. Corbitt and K. D. Janda
Chemical Communications 2016 vol. 52(Issue 22) pp:4187-4190
Publication Date(Web):18 Feb 2016
DOI:10.1039/C6CC00615A
3,4-Diaminopyridine has shown promise in reversing botulinum intoxication, but poor pharmacokinetics and a narrow therapeutic window limit its clinical utility. Thus, we developed a pH-dependent oral delivery platform using club moss spore exines. These exine microcapsules slowed 3,4-diaminopyridine absorption, limited its seizure activity, and enabled delivery of doses which prolonged mouse survival after botulism neurotoxin A intoxication.
Co-reporter:Nicholas T. Jacob; Jonathan W. Lockner; Joel E. Schlosburg; Beverly A. Ellis; Lisa M. Eubanks;Kim D. Janda
Journal of Medicinal Chemistry 2016 Volume 59(Issue 6) pp:2523-2529
Publication Date(Web):February 26, 2016
DOI:10.1021/acs.jmedchem.5b01676
Despite efforts to produce suitable smoking cessation aids, addiction to nicotine continues to carry a substantive risk of recidivism. An attractive alternative to current therapies is the pharmacokinetic strategy of antinicotine vaccination. A major hurdle in the development of the strategy has been to elicit a sufficiently high antibody concentration to curb nicotine distribution to the brain. Herein, we detail investigations into a new hapten design, which was able to elicit an antibody response of significantly higher specificity for nicotine. We also explore the use of a mutant flagellin carrier protein with adjuvanting properties. These studies underlie the feasibility of improvement in antinicotine vaccine formulations to move toward clinical efficacy.
Co-reporter:Karen C. Collins; Joel E. Schlosburg; Paul T. Bremer;Kim D. Janda
Journal of Medicinal Chemistry 2016 Volume 59(Issue 8) pp:3878-3885
Publication Date(Web):April 7, 2016
DOI:10.1021/acs.jmedchem.6b00084
Methamphetamine (MA) addiction is a serious public health problem, and current methods to abate addiction and relapse are currently ineffective for mitigating this growing global epidemic. Development of a vaccine targeting MA would provide a complementary strategy to existing behavioral therapies, but this has proven challenging. Herein, we describe optimization of both hapten design and formulation, identifying a vaccine that elicited a robust anti-MA immune response in mice, decreasing methamphetamine-induced locomotor activity.
Co-reporter:Atsushi Kimishima, Cody J. Wenthur, Lisa M. Eubanks, Shun Sato, and Kim D. Janda
Molecular Pharmaceutics 2016 Volume 13(Issue 11) pp:3884-3890
Publication Date(Web):October 7, 2016
DOI:10.1021/acs.molpharmaceut.6b00682
Although cocaine abuse and addiction continue to cause serious health and societal problems, an FDA-approved medication to treat cocaine addiction has yet to be developed. Employing a pharmacokinetic strategy, an anticocaine vaccine provides an attractive avenue to address these issues; however, current vaccines have shown varying degrees of efficacy, indicating that further formulation is necessary. As a means to improve vaccine efficacy, we examined the effects of varying anticocaine vaccine formulations by combining a Toll-like receptor 9 (TLR9) agonist with a TLR5 agonist in the presence of alum. The TLR9 agonist used was cytosine-guanine oligodeoxynucleotide 1826 (CpG 1826), while the TLR5 agonist was flagellin (FliC). Formulations with the TLR9 agonist elicited superior anticocaine antibody titers and blockade of hyperlocomotor effects compared to vaccines without CpG 1826. This improvement was seen regardless of whether the TLR5 agonist, FliC, or the nonadjuvanting Tetanus Toxoid (TT) was used as the carrier protein. Additional insights into the value of FliC as a carrier versus adjuvant was also investigated by generating two unique formats of the protein, wild-type and mutated flagellin (mFliC). While the mFliC conjugate retained its ability to stimulate mTLR5, it yielded reduced cocaine sequestration and functional blockade relative to FliC and TT. Overall, this work indicates that activation of TLR9 can improve the function of cocaine vaccines in the presence of TLR5 activation by FliC, with any potential additive effects limited by the inefficiency of FliC as a carrier protein as compared to TT.Keywords: addiction; adjuvant; carrier protein; cocaine; CpG; flagellin; TLR5; TLR9; vaccine;
Co-reporter:Bin Zhou, Lisa M. Eubanks, Nicholas T. Jacob, Beverly Ellis, Amanda J. Roberts, Kim D. Janda
Bioorganic & Medicinal Chemistry Letters 2016 Volume 26(Issue 20) pp:5078-5081
Publication Date(Web):15 October 2016
DOI:10.1016/j.bmcl.2016.08.081
There is currently no clinically-approved antidote for cocaine overdose. Efforts to develop a therapy via passive immunization have resulted in a human monoclonal antibody, GNCgzk, with a high affinity for cocaine (Kd = 0.18 nM). Efforts to improve the production of antibody manifolds based on this antibody are disclosed. The engineering of an HRV 3C protease cleavage site into the GNCgzk IgG has allowed for increased production of a F(ab′)2 with a 20% superior capacity to reduce mortality for cocaine overdose in mice.
Co-reporter:Karen C. Collins, Kyoji Tsuchikama, Colin A. Lowery, Jie Zhu, Kim D. Janda
Tetrahedron 2016 Volume 72(Issue 25) pp:3593-3598
Publication Date(Web):23 June 2016
DOI:10.1016/j.tet.2015.08.063
Autoinducer-2 (AI-2)-mediated quorum sensing (QS) is utilised for both intra- and inter-species communication by a wide variety of bacteria. An understanding of the mechanism of this communication has the potential to elucidate new targets for antibacterial therapeutics. Herein, we report the synthesis of DPD analogues with modified dynamic equilibria and the evaluation of their behaviour in Gram-negative bacteria. None of the compounds showed modulation of QS in Salmonella Typhimurium, and although no antagonism of Vibrio harveyi was observed, chloro-analogue C5-Cl-DPD showed modest agonism in this marine bacterium. This raises the possibility that access to a cyclic form of DPD may not be required for AI-2-mediated QS in V. harveyi.
Co-reporter:Major D. Gooyit and Kim D. Janda
ACS Infectious Diseases 2016 Volume 2(Issue 7) pp:465
Publication Date(Web):May 25, 2016
DOI:10.1021/acsinfecdis.6b00061
Cysteine protease Cwp84 is responsible for surface-layer processing in Clostridium difficile and was also shown to cleave several human extracellular matrix components in vitro. To enable the facile identification and characterization of Cwp84 inhibitors, we developed a fluorogenic 10-mer peptide based on the enzyme’s natural substrate SlpA that is amenable for use in FRET-based high-throughput screening. The design of substrate-mimetic inhibitors led to epoxysuccinate 8c, which displayed an inactivation efficiency (kinact/KI) of (4.7 ± 0.3) × 104 M–1 min–1. Further evaluation of 8c demonstrated its ability to inhibit fibronectin cleavage and, more importantly, subvert surface-layer biogenesis in C. difficile.Keywords: Clostridium difficile; Cwp84; FRET assay; S-layer; substrate mimetics
Co-reporter:Dr. Kyoji Tsuchikama;Dr. Major Gooyit;Dr. Tyler L. Harris;Dr. Jie Zhu;Dr. Daniel Globisch;Dr. Gunnar F. Kaufmann;Dr. Kim D. Ja
Angewandte Chemie 2016 Volume 128( Issue 12) pp:4070-4074
Publication Date(Web):
DOI:10.1002/ange.201511911
Abstract
Reported herein is that (4S)-4,5-dihydroxy-2,3-pentanedione (DPD) can undergo a previously undocumented non-enzymatic glycation reaction. Incubation of DPD with viral DNA or the antibiotic gramicidin S resulted in significant biochemical alterations. A protein-labeling method was consequently developed that facilitated the identification of unrecognized glycation targets of DPD in a prokaryotic system. These results open new avenues toward tracking and understanding the fate and function of the elusive quorum-sensing signaling molecule.
Co-reporter:Dr. Kyoji Tsuchikama;Dr. Major Gooyit;Dr. Tyler L. Harris;Dr. Jie Zhu;Dr. Daniel Globisch;Dr. Gunnar F. Kaufmann;Dr. Kim D. Ja
Angewandte Chemie International Edition 2016 Volume 55( Issue 12) pp:4002-4006
Publication Date(Web):
DOI:10.1002/anie.201511911
Abstract
Reported herein is that (4S)-4,5-dihydroxy-2,3-pentanedione (DPD) can undergo a previously undocumented non-enzymatic glycation reaction. Incubation of DPD with viral DNA or the antibiotic gramicidin S resulted in significant biochemical alterations. A protein-labeling method was consequently developed that facilitated the identification of unrecognized glycation targets of DPD in a prokaryotic system. These results open new avenues toward tracking and understanding the fate and function of the elusive quorum-sensing signaling molecule.
Co-reporter:Paul T. Bremer;Dr. Atsushi Kimishima;Dr. Joel E. Schlosburg;Dr. Bin Zhou;Dr. Karen C. Collins ;Dr. Kim D. Ja
Angewandte Chemie International Edition 2016 Volume 55( Issue 11) pp:3772-3775
Publication Date(Web):
DOI:10.1002/anie.201511654
Abstract
Fentanyl is an addictive prescription opioid that is over 80 times more potent than morphine. The synthetic nature of fentanyl has enabled the creation of dangerous “designer drug” analogues that escape toxicology screening, yet display comparable potency to the parent drug. Alarmingly, a large number of fatalities have been linked to overdose of fentanyl derivatives. Herein, we report an effective immunotherapy for reducing the psychoactive effects of fentanyl class drugs. A single conjugate vaccine was created that elicited high levels of antibodies with cross-reactivity for a wide panel of fentanyl analogues. Moreover, vaccinated mice gained significant protection from lethal fentanyl doses. Lastly, a surface plasmon resonance (SPR)-based technique was established enabling drug-specificity profiling of antibodies derived directly from serum. Our newly developed fentanyl vaccine and analytical methods may assist in the battle against synthetic opioid abuse.
Co-reporter:Paul T. Bremer;Dr. Atsushi Kimishima;Dr. Joel E. Schlosburg;Dr. Bin Zhou;Dr. Karen C. Collins ;Dr. Kim D. Ja
Angewandte Chemie 2016 Volume 128( Issue 11) pp:3836-3839
Publication Date(Web):
DOI:10.1002/ange.201511654
Abstract
Fentanyl is an addictive prescription opioid that is over 80 times more potent than morphine. The synthetic nature of fentanyl has enabled the creation of dangerous “designer drug” analogues that escape toxicology screening, yet display comparable potency to the parent drug. Alarmingly, a large number of fatalities have been linked to overdose of fentanyl derivatives. Herein, we report an effective immunotherapy for reducing the psychoactive effects of fentanyl class drugs. A single conjugate vaccine was created that elicited high levels of antibodies with cross-reactivity for a wide panel of fentanyl analogues. Moreover, vaccinated mice gained significant protection from lethal fentanyl doses. Lastly, a surface plasmon resonance (SPR)-based technique was established enabling drug-specificity profiling of antibodies derived directly from serum. Our newly developed fentanyl vaccine and analytical methods may assist in the battle against synthetic opioid abuse.
Co-reporter:Song Xue; Joel E. Schlosburg;Kim D. Janda
Journal of the American Chemical Society 2015 Volume 137(Issue 32) pp:10136-10139
Publication Date(Web):August 3, 2015
DOI:10.1021/jacs.5b06605
Smoking is the leading cause of preventable diseases; thus, effective smoking cessation aids are crucial for reducing the prevalence of cigarette smoking and smoking-related illnesses. In our current campaign we offer a nicotine-degrading enzyme from Pseudomonas putida, NicA2, a flavin-containing protein. To explore its potential, a kinetic evaluation of the enzyme was conducted, which included determination of Km, kcat, buffer/serum half-life, and thermostability. Additionally, the catabolism profile of NicA2 was elucidated to assess the potential toxicity of the nicotine-derived products. In characterizing the enzyme, a favorable biochemical profile of the enzyme was discovered, making NicA2 a prospective therapeutic candidate. This approach provides a new avenue for the field of nicotine addiction therapy.
Co-reporter:Hajime Seki, Song Xue, Mark S. Hixon, Sabine Pellett, Marek Reme, Eric A. Johnson and Kim D. Janda
Chemical Communications 2015 vol. 51(Issue 28) pp:6226-6229
Publication Date(Web):11 Mar 2015
DOI:10.1039/C5CC00677E
Dyngo-4a™ has been found to be an endocytic inhibitor of BoNT/A neurotoxicity through dynamin inhibition. Herein, we demonstrate this molecule to have a previously unrecognized dual activity against BoNT/A, dynamin–protease inhibition. To establish the importance of this dual activity, detailed kinetic analysis of Dyngo-4a's inhibition of BoNT/A metalloprotease as well as cellular and animal toxicity studies have been described. The research presented is the first polypharmacological approach to counteract BoNT/A intoxication.
Co-reporter:Jonathan W. Lockner; Jenny M. Lively; Karen C. Collins; Janaína C. M. Vendruscolo; Marc R. Azar;Kim D. Janda
Journal of Medicinal Chemistry 2015 Volume 58(Issue 2) pp:1005-1011
Publication Date(Web):December 10, 2014
DOI:10.1021/jm501625j
A leading nicotine conjugate vaccine was only efficacious for one-third of clinical trial participants, likely due in part to its use of racemic nicotine hapten, (±)-3′-AmNic. Immunization of male Wistar rats with (+)-, (−)-, or (±)-3′-AmNicSucTT and subsequent antibody immunoassays suggest that a vaccine using enantiopure (−)-3′-AmNic hapten imparts superior capacity to bind (−)-nicotine. Future nicotine vaccine clinical candidates must incorporate this design consideration (i.e., hapten enantiopurity) in order to maximize efficacy.
Co-reporter:Jie Zhu, Xiaoqing Cai, Tyler L. Harris, Major Gooyit, Malcolm Wood, Matthew Lardy, Kim D. Janda
Chemistry & Biology 2015 Volume 22(Issue 4) pp:483-491
Publication Date(Web):23 April 2015
DOI:10.1016/j.chembiol.2015.03.012
•A LasB inhibitor motif was identified using computational modeling•C. elegans virulence factor infection model•LasB knockout strain attenuated virulence in C. elegans•Visualization of LasB virulence in C. elegans through TEMThe emergence of antibiotic resistance places a sense of urgency on the development of alternative antibacterial strategies, of which targeting virulence factors has been regarded as a “second generation” antibiotic approach. In the case of Pseudomonas aeruginosa infections, a proteolytic virulence factor, LasB, is one such target. Unfortunately, we and others have not been successful in translating in vitro potency of LasB inhibitors to in vivo efficacy in an animal model. To overcome this obstacle, we now integrate in silico and in vitro identification of the mercaptoacetamide motif as an effective class of LasB inhibitors with full in vivo characterization of mercaptoacetamide prodrugs using Caenorhabditis elegans. We show that one of our mercaptoacetamide prodrugs has a good selectivity profile and high in vivo efficacy, and confirm that LasB is a promising target for the treatment of bacterial infections. In addition, our work highlights that the C. elegans infection model is a user-friendly and cost-effective translational tool for the development of anti-virulence compounds.Figure optionsDownload full-size imageDownload high-quality image (152 K)Download as PowerPoint slide
Co-reporter:Jonathan W. Lockner, Lisa M. Eubanks, Jennifer L. Choi, Jenny M. Lively, Joel E. Schlosburg, Karen C. Collins, Daniel Globisch, Robin J. Rosenfeld-Gunn, Ian A. Wilson, and Kim D. Janda
Molecular Pharmaceutics 2015 Volume 12(Issue 2) pp:653-662
Publication Date(Web):December 22, 2014
DOI:10.1021/mp500520r
Cocaine abuse is problematic, directly and indirectly impacting the lives of millions, and yet existing therapies are inadequate and usually ineffective. A cocaine vaccine would be a promising alternative therapeutic option, but efficacy is hampered by variable production of anticocaine antibodies. Thus, new tactics and strategies for boosting cocaine vaccine immunogenicity must be explored. Flagellin is a bacterial protein that stimulates the innate immune response via binding to extracellular Toll-like receptor 5 (TLR5) and also via interaction with intracellular NOD-like receptor C4 (NLRC4), leading to production of pro-inflammatory cytokines. Reasoning that flagellin could serve as both carrier and adjuvant, we modified recombinant flagellin protein to display a cocaine hapten termed GNE. The resulting conjugates exhibited dose-dependent stimulation of anti-GNE antibody production. Moreover, when adjuvanted with alum, but not with liposomal MPLA, GNE-FliC was found to be better than our benchmark GNE-KLH. This work represents a new avenue for exploration in the use of hapten-flagellin conjugates to elicit antihapten immune responses.
Co-reporter:Major Gooyit, Nancy Tricoche, Sacha Javor, Sara Lustigman, and Kim D. Janda
ACS Medicinal Chemistry Letters 2015 Volume 6(Issue 3) pp:339
Publication Date(Web):January 20, 2015
DOI:10.1021/ml500516r
Onchocerciasis is an infection caused by the filarial worm Onchocerca volvulus, which can eventually result in blindness. The lack of an effective macrofilaricide and the possible development of ivermectin-resistant strains of O. volvulus necessitate the need for alternative treatment strategies. We have shown that targeting the L3-stage-specific chitinase OvCHT1 impairs the shedding of the filarial cuticle. In our continued efforts to discover OvCHT1 inhibitors, we identified the β-carboline alkaloid scaffolding as a chitinase inhibitor that is capable of penetrating the worm cuticle. Herein, we disclose the rich polypharmacology of the β-carboline class of compounds as an approach to abrogate the molting of the parasite and thus the initiation of infection in the human host.Keywords: chitinase inhibitor; Onchocerciasis; polypharmacology; β-carbolines
Co-reporter:Daniel Globisch, Sabine Specht, Kenneth M. Pfarr, Lisa M. Eubanks, Achim Hoerauf, Kim D. Janda
Bioorganic & Medicinal Chemistry Letters 2015 Volume 25(Issue 24) pp:5804-5807
Publication Date(Web):15 December 2015
DOI:10.1016/j.bmcl.2015.10.037
The neglected tropical disease onchocerciasis affects more than 35 million people worldwide with over 95% in Africa. Disease infection initiates from the filarial parasitic nematode Onchocerca volvulus, which is transmitted by the blackfly vector Simulium sp. carrying infectious L3 larvae. New treatments and diagnostics are required to eradicate this parasitic disease. Herein, we describe that a previously discovered biomarker for onchocerciasis, N-acetyltyramine-O-glucuronide (NATOG) is also present in urine samples of jirds infected with the onchocerciasis model nematode Litomosoides sigmodontis. Increased NATOG values paralleled a progressing infection and demonstrated that quantification of NATOG in this rodent model can be utilized to track its infectivity. Moreover, our findings suggest how NATOG monitoring may be used for evaluating potential drug candidates.
Co-reporter:Xiaoqing Cai, Daniel Globisch, Pamela Thompson, Jia Lin, Dorin Toader, Nazzareno Dimasi, Changshou Gao, Kim D. Janda
Tetrahedron Letters 2015 Volume 56(Issue 34) pp:4960
Publication Date(Web):19 August 2015
DOI:10.1016/j.tetlet.2015.06.088
Co-reporter:Xiaoqing Cai, Kim D. Janda
Tetrahedron Letters 2015 Volume 56(Issue 23) pp:3172-3175
Publication Date(Web):3 June 2015
DOI:10.1016/j.tetlet.2014.12.025
An efficient chemical synthesis of UDP-N-azidoacetylgalactosamine (UDP-GalNAz) is presented, while the value of this molecule was demonstrated through its attachment to an antibody Fc domain. Thus, the antibody was first degalactosylated, which was followed by loading of the UDP-GalNAz with a recombinant galactosyltransferase. This engineered Azide-Fc-N-glycan antibody was subsequently ‘clicked’ by a strain-promoted alkyne-azide cycloaddition reaction for site-specific attachment of a fluorescent probe. The principles detailed will allow for the facile preparation of chemically defined homogeneous antibody–drug conjugates (ADCs).
Co-reporter:Major Gooyit, Tyler L. Harris, Nancy Tricoche, Sacha Javor, Sara Lustigman, and Kim D. Janda
ACS Infectious Diseases 2015 Volume 1(Issue 5) pp:198
Publication Date(Web):March 17, 2015
DOI:10.1021/acsinfecdis.5b00017
The anthelmintic closantel has shown promise in abrogating the L3 molting of Onchocerca volvulus, the causative agent of the infectious disease onchocerciasis. In our search for alternative scaffolds, we utilized a fragment replacement/modification approach to generate novel chemotypes with improved chitinase inhibitory properties. Further evaluation of the compounds unveiled the potential of urea-tropolones as potent inhibitors of O. volvulus L3 molting.Keywords: molting; onchocerciasis; tropolones
Co-reporter:K. C. Collins, J. E. Schlosburg, J. W. Lockner, P. T. Bremer, B. A. Ellis and K. D. Janda
Chemical Communications 2014 vol. 50(Issue 31) pp:4079-4081
Publication Date(Web):03 Mar 2014
DOI:10.1039/C4CC00682H
The immunopotentiator tucaresol was modified for incorporation into liposomes, where it was found to be a superior adjuvant to MPLA for vaccination against methamphetamine.
Co-reporter:Major Gooyit ; Nancy Tricoche ; Sara Lustigman ;Kim D. Janda
Journal of Medicinal Chemistry 2014 Volume 57(Issue 13) pp:5792-5799
Publication Date(Web):June 11, 2014
DOI:10.1021/jm5006435
The L3-stage-specific chitinase OvCHT1 has been implicated in the development of Onchocerca volvulus, the causative agent of onchocerciasis. Closantel, a known anthelmintic drug, was previously discovered as a potent and specific OvCHT1 inhibitor. As closantel is also a known protonophore, we performed a simple scaffold modulation to map out the structural features that are relevant for its individual or dual biochemical roles. Furthermore, we present that either OvCHT1 inhibition or protonophoric activity was capable of affecting O. volvulus L3 molting and that the presence of both activities in a single molecule yielded more potent inhibition of the nematode’s developmental process.
Co-reporter:Karen C. Collins and Kim D. Janda
Bioconjugate Chemistry 2014 Volume 25(Issue 3) pp:593
Publication Date(Web):February 12, 2014
DOI:10.1021/bc500016k
Vaccines for drugs of abuse have yet to achieve full clinical relevance, largely due to poor/inconsistent immune responses in patients. The use of multivalent scaffolding as a means to tailor drug–hapten density and clustering was examined in the context of drug-immune response modulation. A modular trivalent hapten containing a diglycine spacer, triAM1(Gly)2, was synthesized and shown to elicit anti-nicotine antibodies at equivalent affinity and concentration to the monovalent AM1 analog, despite in this instance having a lower effective hapten density. Augmenting this data, the corresponding monovalent hapten AM1(Gly)2 resulted in enhanced antibody affinity and concentration. Drug-hapten clustering represents a new vaccine paradigm, and, while examined only in the context of nicotine, it should be readily translatable to other drugs of abuse.
Co-reporter:Tyler L. Harris, Colin A. Lowery, Mark S. Hixon, and Kim D. Janda
ACS Chemical Neuroscience 2014 Volume 5(Issue 8) pp:632
Publication Date(Web):July 7, 2014
DOI:10.1021/cn500135h
Botulinum neurotoxicity is characterized by peripheral neuromuscular blockade/flaccid paralysis that can lead to respiratory failure and ultimately death. Current therapeutic options provide relief in a pre-exposure scenario, but there are no clinically approved postexposure medical countermeasures. Here, we introduce a platform that utilizes a combination of a toxin sequestering agent and a pharmacological antagonist to ablate botulinum neurotoxicity in a well-defined mouse lethality assay. The platform was constructed to allow for ready exchange of sequestering agent and/or pharmacological antagonist for therapeutic optimization. As such, we attempted to improve upon the pharmacological antagonist, a potassium channel blocker, 3,4-diaminopyridine, through a prodrug approach; thus, a complete kinetic decomposition pathway is described. These experiments provide the first proof-of-principle that a synergistic combination strategy can be used to reduce toxin burden in the peripheral using a sequestering antibody, while restoring muscle action via a pharmacological small molecule antagonist.Keywords: antibody; Botulinum neurotoxin; drug combination; K+ channel blocker; prodrug
Co-reporter:Paul T. Bremer, Joel E. Schlosburg, Jenny M. Lively, and Kim D. Janda
Molecular Pharmaceutics 2014 Volume 11(Issue 3) pp:1075-1080
Publication Date(Web):February 11, 2014
DOI:10.1021/mp400631w
Active immunization is an effective means of blocking the pharmacodynamic effects of drugs and holds promise as a treatment for heroin addiction. Previously, we demonstrated the efficacy of our first-generation vaccine in blocking heroin self-administration in rats, however, many vaccine components can be modified to further improve performance. Herein we examine the effects of varying heroin vaccine injection route and adjuvant formulation. Mice immunized via subcutaneous (sc) injection exhibited inferior anti-heroin titers compared to intraperitoneal (ip) and sc/ip coadministration injection routes. Addition of TLR9 agonist cytosine-guanine oligodeoxynucleotide 1826 (CpG ODN 1826) to the original alum adjuvant elicited superior antibody titers and opioid affinities compared to alum alone. To thoroughly assess vaccine efficacy, full dose–response curves were generated for heroin-induced analgesia in both hot plate and tail immersion tests. Mice treated with CpG ODN 1826 exhibited greatly shifted dose–response curves (10–13-fold vs unvaccinated controls) while non-CpG ODN vaccine groups did not exhibit the same robust effect (2–7-fold shift for ip and combo, 2–3-fold shift for sc). Our results suggest that CpG ODN 1826 is a highly potent adjuvant, and injection routes should be considered for development of small molecule–protein conjugate vaccines. Lastly, this study has established a new standard for assessing drugs of abuse vaccines, wherein a full dose–response curve should be performed in an appropriate behavioral task.Keywords: adjuvant; analgesia; antibody; CpG ODN; dose−response; heroin; route of administration; vaccine;
Co-reporter:Song Xue, Sacha Javor, Mark S. Hixon, and Kim D. Janda
Biochemistry 2014 Volume 53(Issue 43) pp:
Publication Date(Web):October 8, 2014
DOI:10.1021/bi500950x
Botulinum neurotoxin serotype A (BoNT/A) is one of the most lethal toxins known. Its extreme toxicity is due to its light chain (LC), a zinc protease that cleaves SNAP-25, a synaptosome-associated protein, leading to the inhibition of neuronal activity. Studies on BoNT/A LC have revealed that two regions, termed exosites, can play an important role in BoNT catalytic activity. A clear understanding of how these exosites influence neurotoxin catalytic activity would provide a critical framework for deciphering the mechanism of SNAP-25 cleavage and the design of inhibitors. Herein, based on the crystallographic structure of BoNT/A LC complexed with its substrate, we designed an α-exosite binding probe. Experiments with this unique probe demonstrated that α-exosite binding enhanced both catalytic activity and stability of the LC. These data help delineate why α-exosite binding is needed for SNAP-25 cleavage and also provide new insights into the extended lifetime observed for BoNT/A LC in vivo.
Co-reporter:Hajime Seki, Sabine Pellett, Peter Šilhár, G. Neil Stowe, Beatriz Blanco, Matthew A. Lardy, Eric A. Johnson, Kim D. Janda
Bioorganic & Medicinal Chemistry 2014 Volume 22(Issue 3) pp:1208-1217
Publication Date(Web):1 February 2014
DOI:10.1016/j.bmc.2013.11.053
Botulinum neurotoxin A (BoNT/A) is the most potent toxin known. Unfortunately, it is also a potential bioweapon in terrorism, which is without an approved therapeutic treatment once cellular intoxication takes place. Previously, we reported how hydroxamic acid prodrug carbamates increased cellular uptake, which translated to successful inhibition of this neurotoxin. Building upon this research, we detail BoNT/A protease molecular modeling studies accompanied by the construction of small library of hydroxamic acids based on 2,4-dichlorocinnamic hydroxamic acid scaffold and their carbamate prodrug derivatization along with the evaluation of these molecules in both enzymatic and cellular models.
Co-reporter:Lisa M. Eubanks, Beverly A. Ellis, Xiaoqing Cai, Joel E. Schlosburg, Kim D. Janda
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 19) pp:4664-4666
Publication Date(Web):1 October 2014
DOI:10.1016/j.bmcl.2014.08.035
Cocaine abuse remains prevalent worldwide and continues to be a major health concern; nonetheless, there is no effective therapy. Immunopharmacotherapy has emerged as a promising treatment strategy by which anti-cocaine antibodies bind to the drug blunting its effects. Previous passive immunization studies using our human monoclonal antibody, GNCgzk, resulted in protection against cocaine overdose and acute toxicity. To further realize the clinical potential of this antibody, a recombinant IgG form of the antibody has been produced in mammalian cells. This antibody displayed a high binding affinity for cocaine (low nanomolar) in line with the superior attributes of the GNCgzk antibody and reduced cocaine-induced ataxia in a cocaine overdose model.
Co-reporter:Jonathan R. Hart;Amanda L. Garner;Jing Yu;Yoshihiro Ito;Minghao Sun;Lynn Ueno;Jin-Kyu Rhee;Michael M. Baksh;Eduard Stefan;Markus Hartl;Klaus Bister;Peter K. Vogt;Kim D. Janda
PNAS 2014 Volume 111 (Issue 34 ) pp:12556-12561
Publication Date(Web):2014-08-26
DOI:10.1073/pnas.1319488111
In a fluorescence polarization screen for the MYC–MAX interaction, we have identified a novel small-molecule inhibitor of
MYC, KJ-Pyr-9, from a Kröhnke pyridine library. The Kd of KJ-Pyr-9 for MYC in vitro is 6.5 ± 1.0 nM, as determined by backscattering interferometry; KJ-Pyr-9 also interferes with
MYC–MAX complex formation in the cell, as shown in a protein fragment complementation assay. KJ-Pyr-9 specifically inhibits
MYC-induced oncogenic transformation in cell culture; it has no or only weak effects on the oncogenic activity of several
unrelated oncoproteins. KJ-Pyr-9 preferentially interferes with the proliferation of MYC-overexpressing human and avian cells
and specifically reduces the MYC-driven transcriptional signature. In vivo, KJ-Pyr-9 effectively blocks the growth of a xenotransplant
of MYC-amplified human cancer cells.
Co-reporter:Paul T. Bremer, Mark S. Hixon, Kim D. Janda
Bioorganic & Medicinal Chemistry 2014 22(15) pp: 3971-3981
Publication Date(Web):
DOI:10.1016/j.bmc.2014.06.004
Co-reporter:Nicholas T. Jacob;Dr. Jonathan W. Lockner;Dr. Vladimir V. Kravchenko ;Dr. Kim D. Ja
Angewandte Chemie International Edition 2014 Volume 53( Issue 26) pp:6628-6631
Publication Date(Web):
DOI:10.1002/anie.201402133
Abstract
Tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) is an immunosurveillance cytokine that kills cancer cells but demonstrates little toxicity against normal cells. While investigating the TRAIL-inducing imidazolinopyrimidinone TIC10, a misassignment of its active structure was uncovered. Syntheses of the two isomers, corresponding to the published and reassigned structures, are reported. The ability of each to induce TRAIL expression in macrophages was investigated and it was found that only the compound corresponding to the reassigned structure shows the originally reported activity; the compound corresponding to the published structure is inactive. Importantly, this structural reassignment has furnished a previously unknown antitumor pharmacophore.
Co-reporter:Nicholas T. Jacob;Dr. Jonathan W. Lockner;Dr. Vladimir V. Kravchenko ;Dr. Kim D. Ja
Angewandte Chemie 2014 Volume 126( Issue 26) pp:6746-6749
Publication Date(Web):
DOI:10.1002/ange.201402133
Abstract
Tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) is an immunosurveillance cytokine that kills cancer cells but demonstrates little toxicity against normal cells. While investigating the TRAIL-inducing imidazolinopyrimidinone TIC10, a misassignment of its active structure was uncovered. Syntheses of the two isomers, corresponding to the published and reassigned structures, are reported. The ability of each to induce TRAIL expression in macrophages was investigated and it was found that only the compound corresponding to the reassigned structure shows the originally reported activity; the compound corresponding to the published structure is inactive. Importantly, this structural reassignment has furnished a previously unknown antitumor pharmacophore.
Co-reporter:Xiaoqing Cai ; Kyoji Tsuchikama ;Kim D. Janda
Journal of the American Chemical Society 2013 Volume 135(Issue 8) pp:2971-2974
Publication Date(Web):February 11, 2013
DOI:10.1021/ja400356g
Cocaine addiction is a long-lasting relapsing illness characterized by cycles of abuse, abstinence, and reinstatement, and antibody-based therapies could be a powerful therapeutic approach. Herein, we explored the possibility of using halogenated cocaine haptens to enhance the immunological properties of anti-cocaine vaccines. Three fluorine-containing cocaine haptens (GNF, GNCF and GN5F) and one chlorine-containing cocaine hapten (GNCl) were designed and synthesized, based upon the chemical scaffold of the only hapten that has reached clinical trials, succinyl norcocaine (SNC). Hapten GNF was found to retain potent cocaine affinity, and also elicit antibodies in a higher concentration than the parent structure SNC. Our data suggests that not only could strategic hapten fluorination be useful for improving upon the current cocaine vaccine undergoing clinical trials, but it may also be a valuable new approach, with application to any of the vaccines being developed for the treatment of drugs of abuse.
Co-reporter:Amanda L. Garner ; Jessica L. Fullagar ; Joshua A. Day ; Seth M. Cohen ;Kim D. Janda
Journal of the American Chemical Society 2013 Volume 135(Issue 27) pp:10014-10017
Publication Date(Web):June 28, 2013
DOI:10.1021/ja404180x
Streptococcus pneumoniae relies on a number of virulence factors, including immunoglobulin A1 protease (IgA1P), a Zn2+ metalloprotease produced on the extracellular surface of the bacteria, to promote pathogenic colonization. IgA1P exhibits a unique function, in that it catalyzes the proteolysis of human IgA1 at its hinge region to leave the bacterial cell surface masked by IgA1 Fab, enabling the bacteria to evade the host’s immune system and adhere to host epithelial cells to promote colonization. Thus, S. pneumoniae IgA1P has emerged as a promising antibacterial target; however, the lack of an appropriate screening assay has limited the investigation of this metalloprotease virulence factor. Relying on electrostatics-mediated AuNP aggregation, we have designed a promising high-throughput colorimetric assay for IgA1P. By using this assay, we have uncovered inhibitors of the enzyme that should be useful in deciphering its role in pneumococcal colonization and virulence.
Co-reporter:Jie Zhu ; Mark S. Hixon ; Daniel Globisch ; Gunnar F. Kaufmann ;Kim D. Janda
Journal of the American Chemical Society 2013 Volume 135(Issue 21) pp:7827-7830
Publication Date(Web):May 14, 2013
DOI:10.1021/ja4024989
In enteric bacteria, the kinase LsrK catalyzes the phosphorylation of the C5-hydroxyl group in the linear form of 4,5-dihydroxy-2,3-pentanedione (DPD), the precursor of the type II bacterial quorum sensing molecule (AI-2). This phosphorylation is required for AI-2 sequestration in the cytoplasm and subsequent derepression of AI-2-related genes necessary for quorum development. While LsrK is a critical enzyme within the DPD quorum sensing relay system, kinetic details of this kinase have yet to be reported. A continuous UV–vis spectrophotometric assay was developed that allowed steady-state kinetic analysis of LsrK to be undertaken with the substrates ATP and DPD. The data was most consistent with a rapid equilibrium ordered mechanism with ATP binding first: kcat (7.4 ± 0.6 s–1), Km,ATP (150 ± 30 μM) and Km(app),DPD (1.0 ± 0.2 mM). The assay also allowed a DPD substrate profile to be conducted, which provided an unexpected biochemical disconnect between the previous agonist/antagonist cell-based reporter assay and the LsrK assay presented herein. Together these findings raise the importance of LsrK and lay the foundation not only for further understanding of this enzyme and its critical biological role but also for the rational design of regulatory molecules targeting AI-2 quorum sensing in pathogenic bacteria.
Co-reporter:Jessica L. Fullagar, Amanda L. Garner, Anjali K. Struss, Joshua A. Day, David P. Martin, Jing Yu, Xiaoqing Cai, Kim D. Janda and Seth M. Cohen
Chemical Communications 2013 vol. 49(Issue 31) pp:3197-3199
Publication Date(Web):06 Mar 2013
DOI:10.1039/C3CC41191E
Tropolone emerged from the screening of a chelator fragment library (CFL) as an inhibitor of the Zn2+-dependent virulence factor, Pseudomonas aeruginosa elastase (LasB). Based on this initial hit, a series of substituted tropolone-based LasB inhibitors was prepared, and a compound displaying potent activity in vitro and in a bacterial swarming assay was identified. Importantly, this inhibitor was found to be specific for LasB over other metalloenzymes, validating the usage of tropolone as a viable scaffold for identifying first-in-class LasB inhibitors.
Co-reporter:Amanda L. Garner, Jing Yu, Anjali K. Struss, Gunnar F. Kaufmann, Vladimir V. Kravchenko and Kim D. Janda
Chemical Communications 2013 vol. 49(Issue 15) pp:1515-1517
Publication Date(Web):10 Jan 2013
DOI:10.1039/C3CC38851D
As a guide for chemical probe design, focused analogue synthetic studies were undertaken upon the lactone ring of 3-oxo-C12-homoserine lactone. We have concluded that hydrolytic instability of the heterocyclic ring is pivotal for its ability to modulate immune signaling and probe preparation was aligned with these findings.
Co-reporter:Xiaoqing Cai ; Timothy Whitfield ; Mark S. Hixon ; Yanabel Grant ; George F. Koob ;Kim D. Janda
Journal of Medicinal Chemistry 2013 Volume 56(Issue 9) pp:3701-3709
Publication Date(Web):April 29, 2013
DOI:10.1021/jm400228w
Presently, there are no FDA-approved medications to treat cocaine addiction. Active vaccination has emerged as one approach to intervene through the rapid sequestering of the circulating drug, thus terminating both psychoactive effects and drug toxicity. Herein, we report our efforts examining two complementary, but mechanistically distinct active vaccines, i.e., noncatalytic and catalytic, for cocaine treatment. A cocaine-like hapten GNE and a cocaine transition-state analogue GNT were used to generate the active vaccines, respectively. GNE–KLH (keyhole limpet hemocyannin) was found to elicit persistent high-titer, cocaine-specific antibodies and blunt cocaine-induced locomotor behaviors. Catalytic antibodies induced by GNT–KLH were also shown to produce potent titers and suppress locomotor response in mice; however, upon repeated cocaine challenges, the vaccine’s protecting effects waned. In depth kinetic analysis suggested that loss of catalytic activity was due to antibody modification by cocaine. The work provides new insights for the development of active vaccines for the treatment of cocaine abuse.
Co-reporter:Peter Šilhár ; Lisa M. Eubanks ; Hajime Seki ; Sabine Pellett ; Sacha Javor ; William H. Tepp ; Eric A. Johnson ;Kim D. Janda
Journal of Medicinal Chemistry 2013 Volume 56(Issue 20) pp:7870-7879
Publication Date(Web):October 15, 2013
DOI:10.1021/jm400873n
The botulinum neurotoxin light chain (LC) protease has become an important therapeutic target for postexposure treatment of botulism. Hydroxamic acid based small molecules have proven to be potent inhibitors of LC/A with nanomolar Ki values, yet they lack cellular activity conceivably due to low membrane permeability. To overcome this potential liability, we investigated two prodrug strategies, 1,4,2-dioxazole and carbamate, based on our 1-adamantylacetohydroxamic acid scaffold. The 1,4,2-dioxazole prodrug did not demonstrate cellular activity, however, carbamates exhibited cellular potency with the most active compound displaying an EC50 value of 20 μM. Cellular trafficking studies were conducted using a “fluorescently silent” prodrug that remained in this state until cellular uptake was complete, which allowed for visualization of the drug’s release inside neuronal cells. In sum, this research sets the stage for future studies leveraging the specific targeting and delivery of these prodrugs, as well as other antibotulinum agents, into neuronal cells.
Co-reporter:Colin A. Lowery, Susana Matamouros, Sherry Niessen, Jie Zhu, Jonathan Scolnick, Jenny M. Lively, Benjamin F. Cravatt, Samuel I. Miller, Gunnar F. Kaufmann, Kim D. Janda
Chemistry & Biology 2013 Volume 20(Issue 7) pp:903-911
Publication Date(Web):25 July 2013
DOI:10.1016/j.chembiol.2013.05.009
•Known quorum-sensing proteins were downregulated specifically in response to a QS antagonist•Developed an approach to map QS networks without reliance on genetic manipulation•No observation of clear link between QS signaling and virulence in S. typhimurium•Demonstrates need to examine QS networks at genomic, proteomic, and phenotypic levelsSmall molecule probes have been used extensively to explore biologic systems and elucidate cellular signaling pathways. In this study, we use an inhibitor of bacterial communication to monitor changes in the proteome of Salmonella enterica serovar Typhimurium with the aim of discovering unrecognized processes regulated by AI-2-based quorum-sensing (QS), a mechanism of bacterial intercellular communication that allows for the coordination of gene expression in a cell density-dependent manner. In S. typhimurium, this system regulates the uptake and catabolism of intercellular signals and has been implicated in pathogenesis, including the invasion of host epithelial cells. We demonstrate that our QS antagonist is capable of selectively inhibiting the expression of known QS-regulated proteins in S. typhimurium, thus attesting that QS inhibitors may be used to confirm proposed and elucidate previously unidentified QS pathways without relying on genetic manipulation.
Co-reporter:Anjali K. Struss, Ashlee Nunes, Jill Waalen, Colin A. Lowery, Prasanna Pullanikat, Judith R. Denery, Douglas J. Conrad, Gunnar F. Kaufmann, and Kim D. Janda
Analytical Chemistry 2013 Volume 85(Issue 6) pp:3355
Publication Date(Web):February 7, 2013
DOI:10.1021/ac400032a
The opportunistic bacterial pathogen Pseudomonas aeruginosa causes chronic lung infections in cystic fibrosis (CF) patients. Importantly, virulence factor expression and biofilm formation in P. aeruginosa is coordinated by quorum sensing (QS) and one of the key QS signaling molecules is 3-oxo-C12-HSL. Remarkably, a tetramic acid, (C12-TA), with antibacterial properties is formed spontaneously from 3-oxo-C12-HSL under physiological conditions. Seeking to better understand this relationship, we sought to investigate whether 3-oxo-C12-HSL and C12-TA may be contributing factors to the overall pathogenicity of P. aeruginosa in CF individuals and if their detection and quantitation in sputum samples might be used as an indicator to assess disease states and monitor therapy success in CF patients. To this end, 3-oxo-C12-HSL and C12-TA concentrations were initially analyzed in P. aeruginosa flow cell biofilms using liquid chromatography coupled with mass spectrometry (LC–MS). A liquid chromatography tandem mass spectrometry (LC–MS/MS)-based method was then developed and validated for their detection and quantification in the sputa of CF patients. To the best of our knowledge, this is the first report to show the presence of both the quorum sensing molecule (3-oxo-C12-HSL) and its rearranged product (C12-TA) in human clinical samples such as sputum. A total of 47 sputum samples from 20 CF and 2 non-CF individuals were analyzed. 3-Oxo-C12-HSL was detected and quantified in 45 samples with concentrations ranging from 20 to >1000 nM; C12-TA was found in 14 samples (13–900 nM). On the basis of our findings, quorum sensing autoinducers merit further investigation as biomarkers for infectious disease states.
Co-reporter:Vladimir Kravchenko, Amanda L. Garner, John Mathison, Alim Seit-Nebi, Jing Yu, Irina P. Gileva, Richard Ulevitch, and Kim D. Janda
ACS Chemical Biology 2013 Volume 8(Issue 6) pp:1117
Publication Date(Web):March 21, 2013
DOI:10.1021/cb4000184
Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) preferentially induces apoptosis in cancer cells over normal cells; however, tumor cells may develop TRAIL resistance. Here, we demonstrate that this resistance can be overcome in the presence of bacterial acylhomoserine lactones (AHLs) or AHL-producing bacteria through the combined effect of TRAIL-induced apoptosis and AHL-mediated inhibition of inflammation regulated by NF-κB signaling. This discovery unveils a previously unrecognized symbiotic link between bacteria and host immunosurveillance.
Co-reporter:Xiaoqing Cai, Timothy Whitfield, Amira Y. Moreno, Yanabel Grant, Mark S. Hixon, George F. Koob, and Kim D. Janda
Molecular Pharmaceutics 2013 Volume 10(Issue 11) pp:4176-4184
Publication Date(Web):August 8, 2013
DOI:10.1021/mp400214w
Judicious hapten design has been shown to be of importance when trying to generate a viable vaccine against a drug of abuse. Hapten design has typically been predicated upon faithfully emulating the unique chemical architecture that each drug presents. However, the need for drug–hapten congruency may also compromise vaccine immunogenicity if the drug–hapten conjugate possesses chemical epitope instability. There has been no systematic study on the impact of hapten stability as it relates to vaccine immunogenicity. As a starting point, we have probed the stability of a series of cocaine haptens through varying several of its structural elements, including functionality at the C2-position, the nature of the linker, and its site of attachment. Accordingly, a hydrolytic stability profile of four cocaine haptens (GNNA, GNNS, GNE, and GNC) was produced, and these results were compared through each hapten’s immunological properties, which were generated via active vaccination. From this group of four, three of the haptens, GNE, GNNA, and GNC, were further examined in an animal behavioral model, and findings here were again measured in relationship to hapten stability. We demonstrate a corresponding relationship between the half-life of the hapten and its immunogenicity, wherein haptens presenting a fully representative cocaine framework elicited higher concentrations of cocaine-specific IgG in sera and also conferred better protection against cocaine-induced locomotor activity. Our results indicate that hapten half-life plays an important role in vaccine immunogenicity and this in turn can impact animal behavioral effects when challenged with a drug of abuse.Keywords: anticocaine vaccine; hapten; immunogenicity; kinetics; psychomotor stimulant effects;
Co-reporter:Peter Šilhár, Matthew A. Lardy, Mark S. Hixon, Charles B. Shoemaker, Joseph T. Barbieri, Anjali K. Struss, Jenny M. Lively, Sacha Javor, and Kim D. Janda
ACS Medicinal Chemistry Letters 2013 Volume 4(Issue 2) pp:283
Publication Date(Web):December 23, 2012
DOI:10.1021/ml300428s
Botulinum neurotoxins (BoNTs) are among the most deadly poisons known, though ironically, they also are of great therapeutic utility. A number of research programs have been initiated to discover small molecule inhibitors of BoNTs metalloprotease activity. Many, though not all, of these programs have screened against a truncated and more stable form of the enzyme, that possesses comparable catalytic properties to the full length enzyme. Interestingly, several classes of inhibitors, notably the hydroxamates, display a large shift in potency between the two enzyme forms. In this report we compare the kinetics of active-site, α-exosite and β-exosite inhibitors versus truncated and full length enzyme. Molecular dynamics simulations conducted with the truncated and homology models of the full length BoNT LC/A indicate the flexibility of the C-terminus of the full length enzyme is responsible for the potency shifts of active-site proximally binding inhibitors while distal binding (α-exosite) inhibitors remain equipotent.Keywords: Botulinum neurotoxin; natural product; protease inhibitor; small molecule inhibitor; zinc-dependent metalloprotease
Co-reporter:Peter Šilhár, Nicholas R. Silvaggi, Sabine Pellett, Kateřina Čapková, Eric A. Johnson, Karen N. Allen, Kim D. Janda
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 5) pp:1344-1348
Publication Date(Web):1 March 2013
DOI:10.1016/j.bmc.2012.12.001
Botulinum neurotoxins (BoNTs) are the most lethal biotoxins known to mankind and are responsible for the neuroparalytic disease botulism. Current treatments for botulinum poisoning are all protein based and thus have a limited window of treatment opportunity. Inhibition of the BoNT light chain protease (LC) has emerged as a therapeutic strategy for the treatment of botulism as it may provide an effective post exposure remedy. Using a combination of crystallographic and modeling studies a series of hydroxamates derived from 1-adamantylacetohydroxamic acid (3a) were prepared. From this group of compounds, an improved potency of about 17-fold was observed for two derivatives. Detailed mechanistic studies on these structures revealed a competitive inhibition model, with a Ki = 27 nM, which makes these compounds some of the most potent small molecule, non-peptidic BoNT/A LC inhibitors reported to date.
Co-reporter:Jonathan W. Lockner, Sam On Ho, Karen C. McCague, Su Ming Chiang, Thai Q. Do, Gary Fujii, Kim D. Janda
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 4) pp:975-978
Publication Date(Web):15 February 2013
DOI:10.1016/j.bmcl.2012.12.048
A major liability of existing nicotine vaccine candidates is the wide variation in anti-nicotine immune responses among clinical trial participants. In order to address this liability, significant emphasis has been directed at evaluating adjuvants and delivery systems that confer more robust potentiation of the anti-nicotine immune response. Toward that end, we have initiated work that seeks to exploit the adjuvant effect of liposomes, with or without Toll-like receptor agonist(s). The results of the murine immunization study described herein support the hypothesis that a liposomal nicotine vaccine formulation may provide a means for addressing the immunogenicity challenge.
Co-reporter:Colin A. Lowery, Michael Adler, Andrew Borrell, Kim D. Janda
Bioorganic & Medicinal Chemistry Letters 2013 23(24) pp: 6743-6746
Publication Date(Web):
DOI:10.1016/j.bmcl.2013.10.029
Co-reporter:Daniel Globisch;Amira Y. Moreno;Ashlee A. K. Nunes;Mark S. Hixon;Judith R. Denery;Sabine Specht;Achim Hoerauf;Kim D. Janda
PNAS 2013 Volume 110 (Issue 11 ) pp:4218-4223
Publication Date(Web):2013-03-12
DOI:10.1073/pnas.1221969110
Onchocerciasis, also known as “river blindness”, is a neglected tropical disease infecting millions of people mainly in Africa
and the Middle East but also in South America and Central America. Disease infectivity initiates from the filarial parasitic
nematode Onchocerca volvulus, which is transmitted by the blackfly vector Simulium sp. carrying infectious third-stage larvae. Ivermectin has controlled transmission of microfilariae, with an African Program
elimination target date of 2025. However, there is currently no point-of-care diagnostic that can distinguish the burden of
infection—including active and/or past infection—and enable the elimination program to be effectively monitored. Here, we
describe how liquid chromatography-MS–based urine metabolome analysis can be exploited for the identification of a unique
biomarker, N-acetyltyramine-O,β-glucuronide (NATOG), a neurotransmitter-derived secretion metabolite from O. volvulus. The regulation of this tyramine neurotransmitter was found to be linked to patient disease infection, including the controversial
antibiotic doxycycline treatment that has been shown to both sterilize and kill adult female worms. Further clues to its regulation
have been elucidated through biosynthetic pathway determination within the nematode and its human host. Our results demonstrate
that NATOG tracks O. volvulus metabolism in both worms and humans, and thus can be considered a host-specific biomarker for onchocerciasis progression.
Liquid chromatography-MS–based urine metabolome analysis discovery of NATOG not only has broad implications for a noninvasive
host-specific onchocerciasis diagnostic but provides a basis for the metabolome mining of other neglected tropical diseases
for the discovery of distinct biomarkers and monitoring of disease progression.
Co-reporter:Kim D. Janda
&
Jennifer B. Treweek
Nature Reviews Immunology 2012 12(1) pp:67
Publication Date(Web):2011-12-16
DOI:10.1038/nri3130
The advent of vaccines targeting drugs of abuse heralded a fundamentally different approach to treating substance-related disorders. In contrast to traditional pharmacotherapies for drug abuse, vaccines act by sequestering circulating drugs and terminating the drug-induced 'high' without inducing unwanted neuromodulatory effects. Drug-targeting vaccines have entered clinical evaluation, and although these vaccines show promise from a biomedical viewpoint, the ethical and socioeconomic implications of vaccinating patients against drugs of abuse merit discussion within the scientific community.
Co-reporter:Kyoji Tsuchikama ; Jie Zhu ; Colin A. Lowery ; Gunnar F. Kaufmann ;Kim D. Janda
Journal of the American Chemical Society 2012 Volume 134(Issue 33) pp:13562-13564
Publication Date(Web):August 6, 2012
DOI:10.1021/ja305532y
Bacteria have developed cell-to-cell communication mechanisms, termed quorum sensing (QS), that regulate bacterial gene expression in a cell population-dependent manner. Autoinducer-2 (AI-2), a class of QS signaling molecules derived from (4S)-4,5-dihydroxy-2,3-pentanedione (DPD), has been identified in both Gram-negative and Gram-positive bacteria. Despite considerable interest in the AI-2 QS system, the biomolecular communication used by distinct bacterial species still remains shrouded. Herein, we report the synthesis and evaluation of a new class of DPD analogues, C4-alkoxy-5-hydroxy-2,3-pentanediones, termed C4-alkoxy-HPDs. Remarkably, two of the analogues were more potent QS agonists than the natural ligand, DPD, in Vibrio harveyi. The findings presented extend insights into ligand–receptor recognition/signaling in the AI-2 mediated QS system.
Co-reporter:Paul T. Bremer ;Kim D. Janda
Journal of Medicinal Chemistry 2012 Volume 55(Issue 23) pp:10776-10780
Publication Date(Web):November 7, 2012
DOI:10.1021/jm301262z
We challenged the performance of our previous heroin vaccine with a similar vaccine containing a more hydrolytically stable hapten analogue and a Th1 adjuvant (CpG ODN). Our results indicate that the elements of our previous vaccine are essential for its anti-heroin potency, i.e., a chemically labile hapten and an exclusively Th2 humoral response elicited by alum. Such design elements are critical for producing next-generation heroin vaccines.
Co-reporter:Jennifer B. Treweek and Kim D. Janda
Molecular Pharmaceutics 2012 Volume 9(Issue 4) pp:969-978
Publication Date(Web):March 1, 2012
DOI:10.1021/mp200588v
Not only has immunopharmacotherapy grown into a field that addresses the abuse of numerous illicit substances, but also the treatment methodologies within immunopharmacotherapy have expanded from traditional active vaccination to passive immunization with anti-drug monoclonal antibodies, optimized mAb formats, and catalytic drug-degrading antibodies. Many laboratories have focused on transitioning distinct immunopharmacotherapeutics to clinical evaluation, but with respect to the indication of cocaine abuse, only the active vaccine TA-CD, which is modeled after our original cocaine hapten GNC,(1) has been carried through to human clinical trials.(2) The successful application of murine mAb GNC92H2 to the reversal of cocaine overdose in a mouse model prompted investigations of human immunoglobulins with the clinical potential to serve as cocaine antidotes. We now report the therapeutic utility of a superior clone, human mAb GNCgzk (Kd = 0.18 nM), which offers a 10-fold improvement in cocaine binding affinity. The GNCgzk manifold was engineered for rapid cocaine clearance, and administration of the F(ab′)2 and Fab formats even after the appearance of acute behavioral signs of cocaine toxicity granted nearly complete prevention of lethality. Thus, contrary to the immunopharmacotherapeutic treatment of drug self-administration, minimal antibody doses were shown to counteract the lethality of a molar excess of circulating cocaine. Passive vaccination with drug-specific antibodies represents a viable treatment strategy for the human condition of cocaine overdose.Keywords: antidote; cocaine; drug abuse; immunopharmacotherapy; mice; monoclonal antibody; overdose; passive immunization; vaccine;
Co-reporter:Joseph S. Zakhari, Eric P. Zorrilla, Bin Zhou, Alexander V. Mayorov, and Kim D. Janda
Molecular Pharmaceutics 2012 Volume 9(Issue 2) pp:281-289
Publication Date(Web):December 7, 2011
DOI:10.1021/mp200376c
Ghrelin, an enteric peptide hormone linked to the pathophysiology of obesity has been a therapeutic target of great interest over the past decade. Many research efforts have focused on the antagonism of ghrelin’s endogenous receptor GHSR1a, which is found along ascending vagal afferent fibers, as well as in the arcuate nucleus of the hypothalamus. Additionally, peptidic inhibitors of ghrelin O-acyltransferase, the enzyme responsible for the paracrine activation of ghrelin, have recently been studied. Our research has taken an alternative immunological approach, studying both active and passive vaccination as a means to sequester ghrelin in the periphery, with the original discovery in rat of decreased feed efficiency and adiposity, as well as increased metabolic activity. Using our previous hapten designs as a stepping-stone, three monoclonal antibodies (JG2, JG3, and JG4) were procured against ghrelin and tested in vivo. While mAb JG4 had the highest affinity for ghrelin, it failed to attenuate the orexigenic effects of food deprivation on energy metabolism or food intake in mice. However, animals that were administered a combination of JG3:JG4 (termed a doublet) or JG2:JG3:JG4 (termed a triplet) demonstrated higher heat dispersion and rate of respiration (higher CO2 emission and O2 consumption) during a 24 h fast refeed. Mice administered the triplet cocktail of JG2:JG3:JG4 also demonstrated decreased food intake upon refeeding as compared to control animals. Recently, Lu and colleagues reported that a passive approach using a single, high affinity N-terminally directed monoclonal antibody did not abrogate the effects of endogenous ghrelin. Our current report corroborates this finding, yet, refutes that a monoclonal antibody approach cannot be efficacious. Rather, we find that a multiple monoclonal antibody (oligoclonal) approach can reproduce the underlying logic to previously reported efficacies using active vaccinations.Keywords: active vaccination; food intake; ghrelin; metabolism; monoclonal antibodies; passive vaccination;
Co-reporter:Amanda L. Garner, Anjali K. Struss, Jessica L. Fullagar, Arpita Agrawal, Amira Y. Moreno, Seth M. Cohen, and Kim D. Janda
ACS Medicinal Chemistry Letters 2012 Volume 3(Issue 8) pp:668
Publication Date(Web):June 19, 2012
DOI:10.1021/ml300128f
Bacterial resistance coupled to our current arsenal of antibiotics presents us with a growing threat to public health, thus warranting the exploration of alternative antibacterial strategies. In particular, the targeting of virulence factors has been regarded as a “second generation” antibiotic approach. In Pseudomonas aeruginosa, a Zn2+ metalloprotease virulence factor, LasB or P. aeruginosa elastase, has been implicated in the development of P. aeruginosa-related keratitis, pneumonia, and burn infection. Moreover, the enzyme also plays a critical role in swarming and biofilm formation, both of which are processes that have been linked to antibiotic resistance. To further validate the importance of LasB in P. aeruginosa infection, we describe our efforts toward the discovery of nonpeptidic small molecule inhibitors of LasB. Using identified compounds, we have confirmed the role that LasB plays in P. aeruginosa swarming and demonstrate the potential for LasB-targeted small molecules in studying antimicrobial-resistant P. aeruginosa phenotypes.Keywords: antibiotics; hydroxypyridinthiones; metalloprotease inhibitors; virulence factors
Co-reporter:Vladimir V. Kravchenko, Christian Gloeckner, G. Neil Stowe, Young J. Kang, Peter S. Tobias, John C. Mathison, Richard J. Ulevitch, Gunnar F. Kaufmann, Kim D. Janda
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 5) pp:2043-2045
Publication Date(Web):1 March 2012
DOI:10.1016/j.bmcl.2012.01.024
Simultaneous activation of signaling pathways requires dynamic assembly of higher-order protein complexes at the cytoplasmic domains of membrane-associated receptors in a stimulus-specific manner. Here, using the paradigm of cellular activation through cytokine and innate immune receptors, we demonstrate the proof-of-principle application of small molecule probes for the dissection of receptor-proximal signaling processes, such as activation of the transcription factor NF-κB and the protein kinase p38.
Co-reporter:Dr. Daniel Globisch;Dr. Colin A. Lowery;Dr. Karen C. McCague ; Kim D. Ja
Angewandte Chemie 2012 Volume 124( Issue 17) pp:4280-4284
Publication Date(Web):
DOI:10.1002/ange.201109149
Co-reporter:Dr. Daniel Globisch;Dr. Colin A. Lowery;Dr. Karen C. McCague ; Kim D. Ja
Angewandte Chemie 2012 Volume 124( Issue 17) pp:
Publication Date(Web):
DOI:10.1002/ange.201201407
Co-reporter:Dr. Daniel Globisch;Dr. Colin A. Lowery;Dr. Karen C. McCague ; Kim D. Ja
Angewandte Chemie International Edition 2012 Volume 51( Issue 17) pp:4204-4208
Publication Date(Web):
DOI:10.1002/anie.201109149
Co-reporter:Dr. Daniel Globisch;Dr. Colin A. Lowery;Dr. Karen C. McCague ; Kim D. Ja
Angewandte Chemie International Edition 2012 Volume 51( Issue 17) pp:
Publication Date(Web):
DOI:10.1002/anie.201201407
Co-reporter:Amira Y. Moreno ; Alexander V. Mayorov ;Kim D. Janda
Journal of the American Chemical Society 2011 Volume 133(Issue 17) pp:6587-6595
Publication Date(Web):April 7, 2011
DOI:10.1021/ja108807j
(+)-Methamphetamine (METH) use and addiction has grown at alarming rates over the past two decades, while no approved pharmacotherapy exists for its treatment. Immunopharmacotherapy has the potential to offer relief through producing highly specific antibodies that prevent drug penetration across the blood−brain barrier thus decreasing reinforcement of the behavior. Current immunotherapy efforts against methamphetamine have focused on a single hapten structure, namely linker attachment at the aromatic ring of the METH molecule. Hapten design is largely responsible for immune recognition, as it affects presentation of the target antigen and thus the quality of the response. In the current paper we report the systematic generation of a series of haptens designed to target the most stable conformations of methamphetamine as determined by molecular modeling. On the basis of our previous studies with nicotine, we show that introduction of strategic molecular constraint is able to maximize immune recognition of the target structure as evidenced by higher antibody affinity. Vaccination of GIX+ mice with six unique METH immunoconjugates resulted in high antibody titers for three particularly promising formulations (45−108 μg/mL, after the second immunization) and high affinity (82, 130, and 169 nM for MH2, MH6, and MH7 hapten-based vaccines, respectively). These findings represent a unique approach to the design of new vaccines against methamphetamine abuse.
Co-reporter:Joseph S. Zakhari ; Isao Kinoyama ; Anjali K. Struss ; Prasanna Pullanikat ; Colin A. Lowery ; Matthew Lardy ;Kim D. Janda
Journal of the American Chemical Society 2011 Volume 133(Issue 11) pp:3840-3842
Publication Date(Web):February 24, 2011
DOI:10.1021/ja111138y
The triphenyl amide/ester 12 was originally reported to be a potent mimic of the natural 3-oxo-dodecanoyl homoserine lactone quorum sensing molecule in Pseudomonas aeruginosa. However, explicit synthesis/chemical characterization was lacking, and a later report providing protein crystallographic data inferred 12 to be incorrect, with 9 now being the surmised structure. Because of these inconsistencies and our interest in quorum sensing molecules utilized by Gram-negative bacteria, we found it necessary to synthesize 9 and 12 to test for agonistic activity in a P. aeruginosa reporter assay. Despite distinct regiochemical differences, both 9 and 12 were found to have comparable EC50 values. To reconcile these unanticipated findings, modeling studies were conducted, and both compounds were revealed to have comparable properties for binding to the LasR receptor.
Co-reporter:Amanda L. Garner ; Junguk Park ; Joseph S. Zakhari ; Colin A. Lowery ; Anjali Kumari Struss ; Daisuke Sawada ; Gunnar F. Kaufmann ;Kim D. Janda
Journal of the American Chemical Society 2011 Volume 133(Issue 40) pp:15934-15937
Publication Date(Web):September 13, 2011
DOI:10.1021/ja207556d
Multivalency is a common principle in the recognition of cellular receptors, and multivalent agonists and antagonists have played a major role in understanding mammalian cell receptor biology. The study of bacterial cell receptors using similar approaches, however, has lagged behind. Herein we describe our efforts toward the development of a dendrimer-based multivalent probe for studying AI-2 quorum-sensing receptors. From these studies, we have discovered a chemical probe specific for Lsr-type AI-2 quorum-sensing receptors with the potential for enabling the identification of new bacterial species that utilize AI-2 as a quorum-sensing signaling molecule.
Co-reporter:Amanda L. Garner and Kim D. Janda
Chemical Communications 2011 vol. 47(Issue 26) pp:7512-7514
Publication Date(Web):19 May 2011
DOI:10.1039/C1CC11817J
Using our recently disclosed fluorescence-based assay to monitor acyltransferase activity, the first non-peptidic, small molecule antagonists of ghrelin O-acyltransferase (GOAT), a potential anti-obesity and anti-diabetes target, have been discovered. Each exhibits micromolar inhibition of the enzyme, and may be useful probes for future study of the ghrelin-GOAT system.
Co-reporter:Nicholas T. Salzameda, Lisa M. Eubanks, Joseph S. Zakhari, Kyoji Tsuchikama, Nicholas J. DeNunzio, Karen N. Allen, Mark S. Hixon and Kim D. Janda
Chemical Communications 2011 vol. 47(Issue 6) pp:1713-1715
Publication Date(Web):04 Jan 2011
DOI:10.1039/C0CC04078A
Clostridium
botulinum produces the most lethal toxins known to man, as such they are high risk terrorist threats, and alarmingly there is no approved therapeutic. We report the first cross-over small molecule inhibitor of these neurotoxins and propose a mechanism by which it may impart its inhibitory activity.
Co-reporter:Amanda L. Garner ; Christian Gloeckner ; Nancy Tricoche ; Joseph S. Zakhari ; Moses Samje ; Fidelis Cho-Ngwa ; Sara Lustigman ;Kim D. Janda
Journal of Medicinal Chemistry 2011 Volume 54(Issue 11) pp:3963-3972
Publication Date(Web):May 2, 2011
DOI:10.1021/jm200364n
Onchocerciasis, or river blindness, is a neglected tropical disease that affects more than 37 million people worldwide, primarily in Africa and Central and South America. We have disclosed evidence that the larval-stage-specific chitinase, OvCHT1, may be a potential biological target for affecting nematode development. On the basis of screening efforts, closantel, a known anthelmintic drug, was discovered as a potent and highly specific OvCHT1 inhibitor. Originally, closantel’s anthelmintic mode of action was believed to rely solely on its role as a proton ionophore; thus, the impact of each of its biological activities on O. volvulus L3 molting was investigated. Structure–activity relationship studies on an active closantel fragment are detailed, and remarkably, by use of a simple salicylanilide scaffold, compounds acting only as protonophores or chitinase inhibitors were identified. From these data, unexpected synergistic protonophore and chitinase inhibition activities have also been found to be critical for molting in O. volvulus L3 larvae.
Co-reporter:G. Neil Stowe ; Leandro F. Vendruscolo ; Scott Edwards ; Joel E. Schlosburg ; Kaushik K. Misra ; Gery Schulteis ; Alexander V. Mayorov ; Joseph S. Zakhari ; George F. Koob ;Kim D. Janda
Journal of Medicinal Chemistry 2011 Volume 54(Issue 14) pp:5195-5204
Publication Date(Web):June 21, 2011
DOI:10.1021/jm200461m
Heroin addiction is a wide-reaching problem with a spectrum of damaging social consequences. A vaccine capable of blocking heroin’s effects could provide a long-lasting and sustainable adjunct to heroin addiction therapy. Heroin, however, presents a particularly challenging immunotherapeutic target, as it is metabolized to multiple psychoactive molecules. To reconcile this dilemma, we examined the idea of a singular vaccine with the potential to display multiple drug-like antigens; thus two haptens were synthesized, one heroin-like and another morphine-like in chemical structure. A key feature in this approach is that immunopresentation with the heroin-like hapten is thought to be immunochemically dynamic such that multiple haptens are simultaneously presented to the immune system. We demonstrate the significance of this approach through the extremely rapid generation of robust polyclonal antibody titers with remarkable specificity. Importantly, both the antinociceptive effects of heroin and acquisition of heroin self-administration were blocked in rats vaccinated using the heroin-like hapten.
Co-reporter:Joseph S. Zakhari, Isao Kinoyama, Mark S. Hixon, Antonia Di Mola, Daniel Globisch, Kim D. Janda
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 21) pp:6203-6209
Publication Date(Web):1 November 2011
DOI:10.1016/j.bmc.2011.09.019
Botulism is a disease characterized by neuromuscular paralysis and is produced from botulinum neurotoxins (BoNTs) found within the Gram positive bacterium Clostridium botulinum. This bacteria produces the most deadliest toxin known, with lethal doses as low as 1 ng/kg. Due to the relative ease of production and transport, the use of these agents as potential bioterrorist weapons has become of utmost concern. No small molecule therapies against BoNT intoxication have been approved to date. However, 3,4-diaminopyridine (3,4-DAP), a potent reversible inhibitor of voltage-gated potassium channels, is an effective cholinergic agonist used in the treatment of neuromuscular degenerative disorders that require cholinergic enhancement. 3,4-DAP has also been shown to facilitate recovery of neuromuscular action potential post botulinum intoxication by blocking K+ channels. Unfortunately, 3,4-DAP displays toxicity largely due to blood–brain-barrier (BBB) penetration. As a dual-action prodrug approach to cholinergic enhancement we have designed carbamate and amide conjugates of 3,4-DAP. The carbamate prodrug is intended to be a slowly reversible inhibitor of acetylcholinesterase (AChE) along the lines of the stigmines thereby allowing increased persistence of released acetylcholine within the synaptic cleft. As a secondary activity, cleavage of the carbamate prodrug by AChE will afford the localized release of 3,4-DAP, which in turn, will enhance the pre-synaptic release of additional acetylcholine. Being a competitive inhibitor with respect to acetylcholine, the activity of the prodrug will be greatest at the synaptic junctions most depleted of acetylcholine. Here we report upon the synthesis and biochemical characterization of three new classes of prodrugs intended to limit previously reported stability and toxicity issues. Of the prodrugs examined, compound 32, demonstrated the most clinically relevant half-life of 2.76 h, while selectively inhibiting AChE over butyrylcholinesterase—a plasma-based high activity esterase. Future in vivo studies could provide validation of prodrug 32 as a potential treatment against BoNT intoxication as well as other neuromuscular disorders.
Co-reporter:Yuya Nakai, Sabine Pellett, William H. Tepp, Eric A. Johnson, Kim D. Janda
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 13) pp:4154
Publication Date(Web):1 July 2011
DOI:10.1016/j.bmc.2011.05.023
Co-reporter:G. Neil Stowe, Kim D. Janda
Tetrahedron Letters 2011 Volume 52(Issue 17) pp:2085-2087
Publication Date(Web):27 April 2011
DOI:10.1016/j.tetlet.2010.10.134
Conducting reactions using water as solvent is a highly prized goal for the organic chemist. Based upon recent literature and our continuing interest in the field of aqueous organocatalysis, we tested the scope of an enamine based Diels–Alder reaction using (±)-nornicotine, proline, and a proline derivative as aqueous organocatalysts. Unfortunately, none of the examined catalysts under aqueous conditions proved useful, leaving the aqueous Diels–Alder reaction as an elusive goal.
Co-reporter:Peter Šilhár, Sami Alakurtti, Kateřina Čapková, Feng Xiaochuan, Charles B. Shoemaker, Jari Yli-Kauhaluoma, Kim D. Janda
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 8) pp:2229-2231
Publication Date(Web):15 April 2011
DOI:10.1016/j.bmcl.2011.02.115
Botulinum neurotoxins (BoNTs) are the most toxic proteins currently known. Current treatments for botulinum poisoning are all protein based with a limited window of opportunity. Inhibition of the BoNT light chain protease (LC) has emerged as a new therapeutic strategy for the treatment of botulism as it may provide an effective post-exposure remedy. As such, a small library of 40 betulin derivatives was synthesized and screened against the light chain of BoNT serotype A (LC/A); five positive hits (IC50 <100 μM) were uncovered. Detailed evaluation of inhibition mechanism of three most active compounds revealed a competitive model, with sub-micromolar Ki value for the best inhibitor (7). Unfortunately, an in vitro cell-based assay did not show any protection of rat cerebellar neurons against BoNT/A intoxication by 7.
Co-reporter:Amanda L. Garner, Jing Yu, Anjali Kumari Struss, Colin A. Lowery, Jie Zhu, Sook Kyung Kim, Junguk Park, Alexander V. Mayorov, Gunnar F. Kaufmann, Vladimir V. Kravchenko, Kim D. Janda
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 9) pp:2702-2705
Publication Date(Web):1 May 2011
DOI:10.1016/j.bmcl.2010.11.122
Alkynyl- and azido-tagged 3-oxo-C12-acylhomoserine lactone probes have been synthesized to examine their potential utility as probes for discovering the mammalian protein target of the Pseudomonas aeruginosa autoinducer, 3-oxo-C12-acylhomoserine lactone. Although such substitutions are commonly believed to be quite conservative, from these studies, we have uncovered a drastic difference in activity between the alkynyl- and azido-modified compounds, and provide an example where such structural modification has proved to be much less than conservative.
Co-reporter:Dr. Ama L. Garner ; Kim D. Ja
ChemBioChem 2011 Volume 12( Issue 4) pp:523-525
Publication Date(Web):
DOI:10.1002/cbic.201000777
Co-reporter:Dr. Lisa M. Eubanks;Dr. G. Neil Stowe;Dr. Sra DeLamoMarin;Dr. Alexer V. Mayorov;Dr. Mark S. Hixon; Kim D. Ja
Angewandte Chemie 2011 Volume 123( Issue 45) pp:10887-10890
Publication Date(Web):
DOI:10.1002/ange.201104512
Co-reporter:Kyoji Tsuchikama, Colin A. Lowery, and Kim D. Janda
The Journal of Organic Chemistry 2011 Volume 76(Issue 17) pp:6981-6989
Publication Date(Web):June 16, 2011
DOI:10.1021/jo200882k
Bacteria have developed a cell-to-cell communication system, termed quorum sensing (QS), which allows for the population-dependent coordination of their behavior via the exchange of chemical signals. Autoinducer-2 (AI-2), a class of QS signals derived from 4,5-dihydroxy-2,3-pentandione (DPD), has been revealed as a universal signaling molecule in a variety of bacterial species. In spite of considerable interest, the study of putative AI-2 based QS systems remains a challenging topic in part due to the rapid interconversion between the linear and cyclic forms of DPD. Herein, we report the design and development of efficient syntheses of carbocyclic analogues of DPD, which are locked in the cyclic form. The synthetic analogues were evaluated for the modulation of AI-2-based QS in Vibrio harveyi and Salmonella typhimurium. No agonists were uncovered in either V. harveyi or S. typhimurium assay, whereas weak to moderate antagonists were found against V. harveyi. On the basis of NMR analyses and DFT calculations, the heterocyclic oxygen atom within DPD appears necessary to promote hydration at the C3 position of cyclic DPD to afford the active tetrahydroxy species. These results also shed light on the interaction between the heterocyclic oxygen atom and receptor proteins as well as the importance of the linear form and dynamic equilibrium of DPD as crucial requirements for activation of AI-2 based QS circuits.
Co-reporter:Dr. Lisa M. Eubanks;Dr. G. Neil Stowe;Dr. Sra DeLamoMarin;Dr. Alexer V. Mayorov;Dr. Mark S. Hixon; Kim D. Ja
Angewandte Chemie International Edition 2011 Volume 50( Issue 45) pp:10699-10702
Publication Date(Web):
DOI:10.1002/anie.201104512
Co-reporter:Peter Šilhár ; Kateřina Čapková ; Nicholas T. Salzameda ; Joseph T. Barbieri ; Mark S. Hixon ;Kim D. Janda
Journal of the American Chemical Society 2010 Volume 132(Issue 9) pp:2868-2869
Publication Date(Web):February 16, 2010
DOI:10.1021/ja910761y
A new mechanistic class of BoNT/A zinc metalloprotease inhibitors, from Echinacea, exemplified by the natural product d-chicoric acid (I1) is disclosed. A detailed evaluation of chicoric acid’s mechanism of inhibition reveals that the inhibitor binds to an exosite, displays noncompetitive partial inhibition, and is synergistic with a competitive active site inhibitor when used in combination. Other components found in Echinacea, I3 and I4, were also inhibitors of the protease.
Co-reporter:G. Neil Stowe, Peter Šilhár, Mark S. Hixon, Nicholas R. Silvaggi, Karen N. Allen, Scott T. Moe, Alan R. Jacobson, Joseph T. Barbieri and Kim D. Janda
Organic Letters 2010 Volume 12(Issue 4) pp:756-759
Publication Date(Web):January 21, 2010
DOI:10.1021/ol902820z
Botulinum neurotoxin serotype A (BoNT/A) is the most toxic protein known to man and also a bioterrorism agent. As defined by our previous research targeting the etiological agent responsible for BoNT/A intoxication, a protease, we now report on the asymmetric synthesis of four new BoNT/A inhibitors; the most potent of this series is roughly 2-fold more active than the best small molecule inhibitor currently known.
Co-reporter:Colin A. Lowery ; Nicholas T. Salzameda ; Daisuke Sawada ; Gunnar F. Kaufmann ;Kim D. Janda
Journal of Medicinal Chemistry 2010 Volume 53(Issue 21) pp:7467-7489
Publication Date(Web):July 29, 2010
DOI:10.1021/jm901742e
Co-reporter:Alexander V. Mayorov, Bert Willis, Antonia Di Mola, Derek Adler, Jennifer Borgia, Olin Jackson, Jie Wang, Yongyi Luo, Lei Tang, Richard J. Knapp, Chandra Natarajan, Michael C. Goodnough, Noam Zilberberg, Lance L. Simpson, and Kim D. Janda
ACS Chemical Biology 2010 Volume 5(Issue 12) pp:1183
Publication Date(Web):October 11, 2010
DOI:10.1021/cb1002366
Botulinum neurotoxins (BoNT) are the etiological agents responsible for botulism, a disease characterized by peripheral neuromuscular blockade and a characteristic flaccid paralysis of humans. BoNT/A is the most toxic protein known to man and has been classified by the Centers of Disease Control (CDC) as one of the six highest-risk threat agents for bioterrorism. Of particular concern is the apparent lack of clinical interventions that can reverse cellular intoxication. Efforts to uncover molecules that can act within an intoxicated cell so as to provide symptomatic relief to BoNT/A are paramount. Aminopyridines have shown clinical efficacy for multiple sclerosis treatment as well as BoNT/A intoxication; yet, aminopyridines for BoNT/A treatment has been abandoned because of blood brain barrier (BBB) penetration producing undesired neurotoxic side effects. Two aminopyridines (5 and 11) exhibited inhibitory activity toward Shaker-IR voltage-gated potassium (KV1.x) channels with potencies similar to that of the previous “gold-standard”, 3,4-diaminopyridine (3,4-DAP), including reversal of symptoms from BoNT-induced paralysis in phrenic nerve-hemidiaphragm preparations. Importantly, pharmacokinetic experiments revealed a lack of BBB penetration of 5, which is a significant advancement toward resolving the neurotoxicity issues associated with prolonged 3,4-DAP treatments. Finally, 5 was found to be as effective as 3,4-DAP in rescuing BoNT-poisoned mice in the mouse lethality assay, signifying an optimized balance between the undesired permeability across the BBB and the required permeability across lipid cellular membranes. The results demonstrate that 5 is the most promising small molecule K+ channel inhibitor discovered to date for the treatment of BoNT/A intoxication.
Co-reporter:Jennifer B. Treweek, Amanda J. Roberts, and Kim D. Janda
Molecular Pharmaceutics 2010 Volume 7(Issue 6) pp:2056-2068
Publication Date(Web):August 17, 2010
DOI:10.1021/mp900293a
While benzodiazepine intoxication alone may elicit sedative and antianxiety effects, alcohol coingestion greatly amplifies this central nervous system depression. As a result, this drug combination gained notoriety for its role in cases of facilitated sexual assault and fatal overdose. We previously validated the ability of the novel antiflunitrazepam monoclonal antibody (mAb) RCA3A3 to bind flunitrazepam (FLU) in vivo and block FLU-induced impairment of locomotion and memory. A therapeutically relevant application of this high affinity mAb (Kd,app = 200 nM), however, is to the more tenuous indication of flunitrazepam (FLU) and alcohol cointoxication. Employing a murine behavioral model, passive immunization with mAb RCA3A3 before injection of ethanol (EtOH: low-dose, 1 g/kg, or high-dose, 1.5 g/kg), FLU (0.06 mg/kg), or a cocktail of both drugs offered partial to full restoration of motor activity levels in co-drug treated and FLU-treated mouse groups (n = 12), respectively. Whereas all drug treatments left contextual learning intact, auditory cued learning was severely disrupted. Prophylactic administration of mAb RCA3A3 prevented this deficit in cued learning in FLU-treated mice but not in the FLU- and EtOH-treated mice, in which co-drug exposure exacerbated the impairment in cued fear conditioning. To substantiate this finding, a dose−response study was performed, and the changes in locomotor activity incurred by different FLU (low-dose, 0.06 mg/kg, or high-dose, 0.09 mg/kg), EtOH (1.0 g/kg, 1.5 g/kg), and mAb RCA3A3 (14.5 mg/kg, 21.8 mg/kg) dose combinations illustrated the potentiation in motor effects by concomitant exposure to FLU and EtOH. Thus, motor activity and fear conditioning results demonstrated that both the amount of FLU left unbound by antibody and the pharmacological additivity between FLU and EtOH, a GABA mimetic, were limiting factors in the therapeutic efficacy of mAb RCA3A3. In sum, our study highlights the complex nature of psychomotor impairment upon co-drug versus singular drug exposure, which may pose a unique challenge to therapeutic treatment.Keywords: alcohol; antibody; benzodiazepine; co-drug abuse; conditioned fear; ethanol; Flunitrazepam; GABAergic system; immunopharmacotherapy; locomotor activity; memory; passive immunization;
Co-reporter:Keisuke Fukuchi, Sebastian C. J. Steiniger, Elena Deryugina, Ying Liu, Colin A. Lowery, Christian Gloeckner, Bin Zhou, Gunnar F. Kaufmann, James P. Quigley and Kim D. Janda
Molecular Pharmaceutics 2010 Volume 7(Issue 1) pp:245-253
Publication Date(Web):November 16, 2009
DOI:10.1021/mp900236t
Despite significant progress and notable successes in tumor therapy, malignant disease remains an extremely difficult problem in today’s health care setting. There is, however, an increasing application of new therapies targeting proteins specifically upregulated on tumor cells. These innovative therapeutic approaches are aimed at molecules that contribute to malignant development and progression but spare normal tissues. The CUB domain containing protein 1 (CDCP1) is such a tumor-associated protein and, thus, a potential candidate for targeted cancer immunotherapy. Herein, we describe the generation of function-blocking human antibodies against CDCP1 that were obtained from human scFv phage display libraries using subtractive panning protocols on CDCP1 expressing cancer cells and immunopurified CDCP1 protein. One of the isolated anti-CDCP1 antibodies, namely, C20Fc, efficiently blocked experimental metastasis of human carcinoma cells, including HeLa cells stably transfected with CDCP1 and prostate carcinoma cells PC-hi/diss naturally expressing CDCP1, in both chick embryo and mouse model systems. The C20Fc antibody also reduced colony formation of CDCP1 expressing cells in a soft agar assay for anchorage-independent cell growth. Specific targeting of CDCP1 by C20Fc mediated the delivery of a toxin-conjugated antibody complex, thus, providing evidence for antibody internalization and specific killing of CDCP1-positive tumor cells. Our findings indicate a functional role for CDCP1 in human cancer and underscore the therapeutic potential of function-blocking anti-CDCP1 antibodies targeting both primary and metastatic carcinoma cells.Keywords: cancer; CDCP-1; human antibodies; Immunotherapy; metastasis; phage display;
Co-reporter:Amira Y. Moreno, Marc R. Azar, Noelle A. Warren, Tobin J. Dickerson, George F. Koob and Kim D. Janda
Molecular Pharmaceutics 2010 Volume 7(Issue 2) pp:431-441
Publication Date(Web):January 27, 2010
DOI:10.1021/mp900213u
(S)-Nicotine is a psychostimulant legal drug responsible for causing addiction to tobacco smoking. Tobacco smoking has been irrevocably linked to a number of serious diseases and at present is considered the leading cause of preventable death in the United States. Despite well-documented adverse medical consequences, nicotine addiction has historically been one of the hardest to break. Current therapies have offered limited success and show high rates of relapse, emphasizing the need to engineer alternative therapies to aid nicotine cessation. The current study presents a protein-based immunopharmacotherapy approach for the treatment of nicotine addiction. Immunopharmacotherapy aims to use highly specific antibodies to blunt passage of drug into the brain thus minimizing reinforcing effects on the reward pathways of the central nervous system. Generation of a successful vaccine heavily relies on appropriate optimization of hapten design, immunogenic carrier and adjuvant. Modification of a classical nicotine hapten in conjugation with three distinct carrier proteins allowed for priming of a nicotine vaccine able to elicit significant amounts of nicotine-specific antibodies. Increased self-administration with use of a high drug dose (0.03 mg/kg/infusion; ∼2 cigarettes in human) was observed in the vaccinated versus control animals suggesting a compensatory pattern and possibly reduced passage of nicotine to the brain. These results support the hypothesis that proper optimization of vaccine formulations could lead to successful nicotine vaccines for human use.Keywords: immunotherapy; Nicotine; self-administration; vaccination;
Co-reporter:Lisa M. Eubanks, Peter Šilhár, Nicholas T. Salzameda, Joseph S. Zakhari, Feng Xiaochuan, Joseph T. Barbieri, Charles B. Shoemaker, Mark S. Hixon and Kim D. Janda
ACS Medicinal Chemistry Letters 2010 Volume 1(Issue 6) pp:268
Publication Date(Web):June 9, 2010
DOI:10.1021/ml100074s
Botulinum neurotoxins (BoNTs) are the etiological agents responsible for botulism, a disease characterized by peripheral neuromuscular blockade and a characteristic flaccid paralysis of humans. BoNTs are the most lethal known poisons affecting humans and have been recognized as a potential bioterrorist threat. Current treatments for botulinum poisoning are predominately prophylactic in nature relying on passive immunization with antitoxins. Inhibition of the BoNT light chain metalloprotease (LC) has emerged as a new therapeutic strategy for the treatment of botulism that may provide an effective postexposure remedy. A high-throughput screening effort against the light chain of BoNT serotype A (LC/A) was conducted with the Johns Hopkins Clinical Compound Library composed of over 1,500 existing drugs. Lomofungin, a natural product first isolated in the late 1960s, was identified as an inhibitor of LC/A, displaying classical noncompetitive inhibition kinetics with a Ki of 6.7 ± 0.7 μM. The inhibition profile of lomofungin has been delineated by the use of both an active site inhibitor, 2,4-dichlorocinnamic hydroxamate, and a noncompetitive inhibitor d-chicoric acid. The inhibitor combination studies reveal that lomofungin binding is nonmutually exclusive (synergistic) with both inhibitors; the mechanistic implications of these observations are discussed. Lastly, cellular efficacy was investigated using a rat primary cell model which demonstrated that lomofungin can protect against SNAP-25 cleavage, the intracellular protein target of LC/A.Keywords (keywords): Botulinum neurotoxin; high-throughput screening; natural product; small molecule inhibitor; zinc-dependent metalloprotease
Co-reporter:Yuya Nakai, Sabine Pellett, William H. Tepp, Eric A. Johnson, Kim D. Janda
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 3) pp:1280-1287
Publication Date(Web):1 February 2010
DOI:10.1016/j.bmc.2009.12.030
Botulinum neurotoxins (BoNTs) are the etiological agents responsible for botulism, a disease characterized by peripheral neuromuscular blockade and a characteristic flaccid paralysis of humans. The natural product toosendanin, a limonoid, is a traditional Chinese medicine that has reported anti-botulinum properties in animal models. Toosendanin effectively inhibits the biological activity of BoNT/A in neuronal cells at concentrations of 200 nM, and partial inhibition can be observed with concentrations as low as 8 nM. Mechanistically, toosendanin’s inhibition is due to prevention of transduction of the BoNT LC through the HC channel. Intriguing questions as to the molecular architecture of toosendanin as related to its anti-botulinum properties have focused our attention on a synthesis of toosendanin’s unusual AB-ring, containing a unique bridged hemi-acetal. Within the current work, a synthetic strategy allowing access to the AB-fragment of toosendanin was achieved from a trans-decalin system. In addition, this fragment was examined for its modulation of BoNT/A intoxication in a rat spinal cord cellular assay.Toosendanin, a limonoid, inhibits the biological activity of BoNT/A. With the concept of function-oriented synthesis as a goal, the AB-ring of toosendanin was prepared sand examined for anti-botulinum properties.
Co-reporter:Kateřina Čapková, Mark S. Hixon, Sabine Pellett, Joseph T. Barbieri, Eric A. Johnson, Kim D. Janda
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 1) pp:206-208
Publication Date(Web):1 January 2010
DOI:10.1016/j.bmcl.2009.10.129
A series of benzylidene cyclopentenedione-based inhibitors, acting through covalent modification of the active site of botulinum neurotoxin A light chain metalloprotease, are reported.A series of benzylidene cyclopentenedione-based inhibitors, acting through covalent modification of the active site of botulinum neurotoxin A light chain metalloprotease, are reported.
Co-reporter:Dr. Ama L. Garner ; Kim D. Ja
Angewandte Chemie International Edition 2010 Volume 49( Issue 50) pp:9630-9634
Publication Date(Web):
DOI:10.1002/anie.201003387
Co-reporter:Dr. Ama L. Garner ; Kim D. Ja
Angewandte Chemie 2010 Volume 122( Issue 50) pp:9824-9828
Publication Date(Web):
DOI:10.1002/ange.201003387
Co-reporter:Christian Gloeckner;Amanda L. Garner;Fana Mersha;Nancy Tricoche;Sara Lustigman;Yelena Oksov;Kim D. Janda;Lisa M. Eubanks;Gunnar F. Kaufmann
PNAS 2010 Volume 107 (Issue 8 ) pp:3424-3429
Publication Date(Web):2010-02-23
DOI:10.1073/pnas.0915125107
Onchocerciasis, or river blindness, is a neglected tropical disease caused by the filarial nematode Onchocerca volvulus that affects more than 37 million people, mainly in third world countries. Currently, the only approved drug available for
mass treatment is ivermectin, however, drug resistance is beginning to emerge, thus, new therapeutic targets and agents are
desperately needed to treat and cure this devastating disease. Chitin metabolism plays a central role in invertebrate biology
due to the critical structural function of chitin for the organism. Taken together with its absence in mammals, targeting
chitin is an appealing therapeutic avenue. Importantly, the chitinase OvCHT1 from O. volvulus was recently discovered, however, its exact role in the worm’s metabolism remains unknown. A screening effort against OvCHT1
was conducted using the Johns Hopkins Clinical Compound Library that contains over 1,500 existing drugs. Closantel, a veterinary
anthelmintic with known proton ionophore activities, was identified as a potent and specific inhibitor of filarial chitinases,
an activity not previously reported for this compound. Notably, closantel was found also to completely inhibit molting of
O. volvulus infective L3 stage larvae. Closantel appears to target two important biochemical processes essential to filarial parasites.
To begin to unravel closantel’s effects, a retro-fragment-based study was used to define structural elements critical for
closantel’s chitinase inhibitor function. As resources towards the development of new agents that target neglected tropical
diseases are scant, the finding of an existing drug with impact against O. volvulus provides promise in the hunt for new therapies against river blindness.
Co-reporter:Jennifer B. Treweek, Tobin J. Dickerson and Kim D. Janda
Accounts of Chemical Research 2009 Volume 42(Issue 5) pp:659
Publication Date(Web):March 10, 2009
DOI:10.1021/ar800247d
Nicotine and methamphetamine are frequently abused in modern society, despite the increasing evidence of their addictive, neuropharmacological, and toxic effects. Tobacco, the most widely abused substance, is the leading cause of preventable death in the United States, killing nearly half a million Americans annually. A methamphetamine epidemic has also spread during the past decade; severe neurotoxicity and addictiveness contribute to the drug’s notoriety. Although the majority of research on these two drugs is of pharmacological and neurobiological motivation, further study of these molecules from a chemical perspective may provide novel mechanistic insight into either their addictive potential or their pathological effects. For example, nicotine and methamphetamine share a common structural feature, a secondary amine, suggesting that these molecules could possess similar (or analogous) in vivo reactivity. Discoveries concerning the synthetic requirements for aqueous aldol catalysis and the feasibility of the enamine mechanism under physiological conditions have given rise to the hypothesis that ingested molecules, such as abused drugs, could participate in reactions utilizing an enamine intermediate in vivo. The chemical reactivity of exogenous drugs with amine functionalities was initially examined in the context of the Maillard reaction, or nonenzymatic browning. The heating of reducing sugars with amino acids yields a brown solution; studies of this reaction were originally applied to food chemistry for the production of distinct flavors and aromas. Further research has since revealed numerous instances in which the in vivo production of advanced glycation end products (AGEs) through the Maillard reaction contribute to the pathology of disease states. Specifically, the modification of long-lived proteins by glycation and glycoxidation and the accumulation of these AGEs compromise the original function of such proteins and change the mechanical properties of affected tissue. In this Account, we summarize our investigations into the capacity for exogenous compounds to initiate the Maillard reaction and the corresponding physiological and immunological impact of the drug-conjugated AGEs that form. Many of the pathological components of diabetes, atherosclerosis, cancer, macular degeneration, Alzheimer’s disease, and even the normal aging process are attributable to AGEs and their potential for aggregate formation in the vasculature. A deeper understanding of AGEs, and particularly glycated proteins, will provide fundamental mechanistic insight into disease origins.
Co-reporter:Colin A. Lowery ; Takumi Abe ; Junguk Park ; Lisa M. Eubanks ; Daisuke Sawada ; Gunnar F. Kaufmann ;Kim D. Janda
Journal of the American Chemical Society 2009 Volume 131(Issue 43) pp:15584-15585
Publication Date(Web):October 13, 2009
DOI:10.1021/ja9066783
Quorum sensing (QS) systems have been discovered in a wide variety of bacteria, and mediate both intra- and interspecies communication. The AI-2-based QS system represents the most studied of these proposed interspecies systems and has been shown to regulate diverse functions such as bioluminescence, expression of virulence factors, and biofilm formation. As such, the development of modulatory compounds, both agonists and antagonists, is of great interest for the study of unknown AI-2-based QS systems and the potential treatment of bacterial infections. The fimbrolide class of natural products has exhibited excellent inhibitory activity against AI-2-based QS and as such may be considered the “gold standard” of AI-2 inhibitors. Thus, we sought to include a fimbrolide as a control compound for our recently developed alkyl-DPD panel of AI-2 modulators. Herein, we present a revised synthesis of a commonly studied fimbrolide as well as a direct comparison between the fimbrolide and alkyl-DPD analogues. We demonstrate that our alkyl-DPD analogues are more potent inhibitors of QS in both Vibrio harveyi and Salmonella typhimurium, the two organisms with defined AI-2 QS systems, and in doing so call into question the widely accepted use of fimbrolide-derived compounds as the “gold standard” of AI-2 inhibition.
Co-reporter:Yuya Nakai, William H. Tepp, Tobin J. Dickerson, Eric A. Johnson, Kim D. Janda
Bioorganic & Medicinal Chemistry 2009 Volume 17(Issue 3) pp:1152-1157
Publication Date(Web):1 February 2009
DOI:10.1016/j.bmc.2008.12.042
Botulinum neurotoxins (BoNTs) are the etiological agents responsible for botulism, a disease characterized by peripheral neuromuscular blockade and a characteristic flaccid paralysis of humans. The natural product toosendanin is a traditional Chinese medicine which has been reported to have anti-botulinum properties in animal models. To establish what chemical functionalities are necessary for the anti-botulinum properties found within toosendanin, a study was initiated with the goal of using function-oriented synthesis (FOS) as a strategy to begin to unravel toosendanin’s powerful anti-botulinum properties. From these studies a new synthetic strategy is put forth allowing access to a 4-acetoxy CD fragment analogue (14) of toosendanin, which was achieved from mesityl oxide and acetylacetone in 14 steps. Animal studies on this fragment are also reported.Botulinum neurotoxins (BoNTs) are the most lethal poisons known, while the natural product toosendanin has demonstrated efficacy against the BoNTs. To unravel toosendanin’s efficacy, Function-Oriented Synthesis (FOS) was examined.
Co-reporter:Nicholas T. Salzameda, Joseph T. Barbieri, Kim D. Janda
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 20) pp:5848-5850
Publication Date(Web):15 October 2009
DOI:10.1016/j.bmcl.2009.08.079
A FRET peptide substrate was synthesized and evaluated for enzymatic cleavage by the BoNT/B light chain protease. The FRET substrate was found to be useful in both a high throughput assay to uncover initial ‘hits’ and a low throughput HPLC assay to determine kinetic parameters and modes of inhibition.A FRET based synthetic substrate for the botulinum neurotoxin light chain B serotype utilized in both a high and low throughput assay for identification and characterization of inhibitors.
Co-reporter:Heyue Zhou, Bin Zhou, Sabine Pellett, Eric A. Johnson, Kim D. Janda
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 3) pp:662-664
Publication Date(Web):1 February 2009
DOI:10.1016/j.bmcl.2008.12.055
Botulinum neurotoxins (BoNTs) are causative agents for botulism and are identified as a category A bioterror agents by the Centers for Disease Control and Prevention (CDC). Current antitoxins against BoNTs intoxication have some limitations including side effects or limited supply. As an alternative, neutralizing monoclonal antibodies will play an increasing role as BoNTs therapeutics. To date, no human anti-BoNT/B neutralizing monoclonal antibodies have yet to be reported. Herein, we describe an improved selection approach and characterization of a human monoclonal antibody, F2, which is capable of binding BoNT/B with high specificity and displays neutralizing activity in an in vitro cell-based assay. Through surface plasmon resonance studies, we have determined its association and dissociation rate constants. In sum, our data demonstrate that monoclonal antibody F2 is a promising BoNT/B therapeutic lead for further development.A human neutralizing antibody was selected from a naïve phage display library that specifically binds Botulinum neurotoxin serotype B. Biochemical studies provide evidence for its therapeutic potential and further development.
Co-reporter:JenniferB. Treweek;AmiraY. Moreno;KimD. Ja Dr.
Angewandte Chemie International Edition 2009 Volume 48( Issue 3) pp:438-440
Publication Date(Web):
DOI:10.1002/anie.200804552
Co-reporter:JenniferB. Treweek;AmiraY. Moreno;KimD. Ja Dr.
Angewandte Chemie 2009 Volume 121( Issue 3) pp:446-448
Publication Date(Web):
DOI:10.1002/ange.200804552
Co-reporter:Colin A. Lowery, Tobin J. Dickerson and Kim D. Janda
Chemical Society Reviews 2008 vol. 37(Issue 7) pp:1337-1346
Publication Date(Web):01 May 2008
DOI:10.1039/B702781H
Quorum sensing (QS) has traditionally referred to a mechanism of communication within a species of bacteria. However, emerging research implicates QS in interspecies communication and competition, and such systems have been proposed in a wide variety of bacteria. This activity of bacterial QS also extends to relationships between bacteria and eukaryotes and host–pathogen interactions in both clinical and agricultural settings are of particular interest. These relationships are particularly pertinent in light of the rising prevalence of antibiotic resistant bacteria. In this tutorial review we describe bacterial QS and its capacity in interspecies and interkingdom interactions, as well as the corresponding eukaryotic responses.
Co-reporter:Kateřina Čapková, Mark S. Hixon, Laura A. McAllister and Kim D. Janda
Chemical Communications 2008 (Issue 30) pp:3525-3527
Publication Date(Web):03 Jul 2008
DOI:10.1039/B808305C
The development of a sensitive, yet reliable assay for the analysis of botulinum neurotoxin A (BoNT/A) inhibitors is described; using this assay a new protease inhibitor was characterized and found to be one of the most potent inhibitors reported to date.
Co-reporter:Jennifer B. Treweek ; Chengzao Sun ; Alexander V. Mayorov ; Longwu Qi ; Coree L. Levy ; Amanda J. Roberts ; Tobin J. Dickerson ;Kim D. Janda
Journal of Medicinal Chemistry 2008 Volume 51(Issue 21) pp:6866-6875
Publication Date(Web):October 15, 2008
DOI:10.1021/jm800506v
One approach to treating drug abuse uses antidrug antibodies to immunize subjects against the illicit substance rather than administering therapeutics that target the specific CNS site of action. At present, passive vaccination has recognized efficacy in treating certain gross symptoms of drug misuse, namely, motor activation, self-administration, and overdose. However, the potential for antibodies to prevent drug-induced changes involving finer cognitive processes, such as benzodiazepine-associated amnesia, remains unexplored. To address this concept, a flunitrazepam hapten was synthesized and employed in the generation of a panel of high affinity monoclonal antibodies. Anti-flunitrazepam mAb RCA3A3 (Kd,app = 200 nM) was tested in a mouse model of passive immunization and subsequent mole-equivalent challenge with flunitrazepam. Not only was flunitrazepam-induced sedation prevented but immunization also conferred protection to memory consolidation as assessed through contextual and cued fear conditioning paradigms. These results provide evidence that immunopharmacotherapeutic blockade of drug intoxication also preserves complex cognitive function.
Co-reporter:Bin Zhou, Charlotte Carney, Kim D. Janda
Bioorganic & Medicinal Chemistry 2008 Volume 16(Issue 4) pp:1903-1913
Publication Date(Web):15 February 2008
DOI:10.1016/j.bmc.2007.11.001
A less than adequate therapeutic plan for the treatment of anthrax in the 2001 bioterrorism attacks has highlighted the importance of developing alternative or complementary therapeutic approaches for biothreat agents. In these regards passive immunization possesses several important advantages over active vaccination and the use of antibiotics, as it can provide immediate protection against Bacillus anthracis. Herein, we report the selection and characterization of several human monoclonal neutralizing antibodies against the toxin of B. anthracis from a phage displayed human scFv library. In total 15 clones were selected with distinct sequences and high specificity to protective antigen and thus were the subject of a series of both biophysical and cell-based cytotoxicity assays. From this panel of antibodies a set of neutralizing antibodies were identified, of which clone A8 recognizes the lethal (and/or edema) factor binding domain, and clones F1, G11, and G12 recognize the cellular receptor binding domain found within the protective antigen. It was noted that all clones distinguish a conformational epitope existing on the protective antigen; this steric relationship was uncovered using a sequential epitope mapping approach. For each neutralizing antibody, the kinetic constants were determined by surface plasmon resonance, while the potency of protection was established using a two-tier macrophage cytotoxicity assay. Among the neutralizing antibodies identified, clone F1 possessed the highest affinity to protective antigen, and provided superior protection from lethal toxin in the cell cytotoxicity assay. The data presented provide the ever-growing arsenal of immunological and functional analysis of monoclonal antibodies to the exotoxins of anthrax. In addition it grants new candidates for the prophylaxis and therapeutic treatment against this toxin.The selection and characterization of human monoclonal antibodies that can neutralize the toxicity of B. anthracis is described. The studies reported highlight each antibody’s protection properties and potential neutralizing epitope(s).
Co-reporter:Yoshiyuki Yoneda, Sebastian C.J. Steiniger, Kateřina Čapková, Jenny M. Mee, Ying Liu, Gunnar F. Kaufmann, Kim D. Janda
Bioorganic & Medicinal Chemistry Letters 2008 Volume 18(Issue 5) pp:1632-1636
Publication Date(Web):1 March 2008
DOI:10.1016/j.bmcl.2008.01.060
Tumor targeting peptides are promising vehicles for site-directed cancer therapy. Pep42, a cyclic 13-mer oligopeptide that specifically binds to glucose-regulated protein 78 (GRP78) and internalized into cancer cells, represents an excellent vehicle for tumor cell-specific chemotherapy. Here, we report the synthesis and evaluation of Pep42-prodrug conjugates that contain a cathepsin B-cleavable linker, resulting in the traceless release of drug inside the cancer cells.
Co-reporter:Neri Amara;Alexander V. Mayorov;Diana I. Ruiz;Mark S. Hixon;Jason A. Moss;Jason Y. Chang;Michael M. Meijler;Eric P. Zorrilla;Kim D. Janda
PNAS 2008 Volume 105 (Issue 45 ) pp:17487-17492
Publication Date(Web):2008-11-11
DOI:10.1073/pnas.0711808105
Obesity is a chronic, costly, and globally prevalent condition, with excess caloric intake a suspected etiologic factor. Nonsurgical
treatments are modestly efficacious, and weight loss maintenance is hampered by anti-famine homeostatic mechanisms. Ghrelin,
a gastric hormone linked to meal initiation, energy expenditure, and fuel partitioning, is hypothesized to facilitate weight
gain and impede weight loss. Unique among known animal peptides, the serine-3 residue of ghrelin is posttranslationally acylated
with an n-octanoic acid, a modification important for the peptide's active blood-brain transport and growth hormone secretagogue receptor-1
agonist activity. Pharmacological degradation of ghrelin would be hypothesized to reduce ghrelin's biological effects. To
study endogenous ghrelin's role in appetite and energy expenditure, we generated antibodies that hydrolyze the octanoyl moiety
of ghrelin to form des-acyl ghrelin. The most proficient antibody catalyst, GHR-11E11, was found to display a second-order rate constant of 18 M−1·s−1 for the hydrolysis of ghrelin to des-acyl ghrelin. I.v. administration of GHR-11E11 (50 mg/kg) maintained a greater metabolic rate in fasting C57BL/6J mice as
compared with mice receiving a control antibody and suppressed 6-h refeeding after 24 h of food deprivation. Indirect respiratory
measures of metabolism after refeeding and relative fuel substrate utilization were unaffected. The results support the hypothesis
that acylated ghrelin stimulates appetite and curbs energy expenditure during deficient energy intake, whereas des-acyl ghrelin does not potently share these functions. Catalytic anti-ghrelin antibodies might thereby adjunctively aid consolidation
of caloric restriction-induced weight loss and might also be therapeutically relevant to Prader–Willi syndrome, characterized
after infancy by hyperghrelinemia, hyperphagia, and obesity.
Co-reporter:Bert Willis Dr.;LisaM. Eubanks Dr.;TobinJ. Dickerson Dr. ;KimD. Ja Dr.
Angewandte Chemie International Edition 2008 Volume 47( Issue 44) pp:8360-8379
Publication Date(Web):
DOI:10.1002/anie.200705531
Abstract
In the classic novella “The Strange Case of Dr. Jekyll and Mr. Hyde”, Robert Louis Stevenson paints a stark picture of the duality of good and evil within a single man. Botulinum neurotoxin (BoNT), the most potent known toxin, possesses an analogous dichotomous nature: It shows a pronounced morbidity and mortality, but it is used with great effect in much lower doses in a wide range of clinical scenarios. Recently, tremendous strides have been made in the basic understanding of the structure and function of BoNT, which have translated into widespread efforts towards the discovery of biomacromolecules and small molecules that specifically modulate BoNT activity. Particular emphasis has been placed on the identification of inhibitors that can counteract BoNT exposure in the event of a bioterrorist attack. This Review summarizes the current advances in the development of therapeutics, including vaccines, peptides, and small-molecule inhibitors, for the prevention and treatment of botulism.
Co-reporter:Lisa M. Eubanks, Tobin J. Dickerson and Kim D. Janda
Chemical Society Reviews 2007 vol. 36(Issue 3) pp:458-470
Publication Date(Web):11 Jan 2007
DOI:10.1039/B615227A
There is a growing need for technological advancements to combat agents of chemical and biological warfare, particularly in the context of the deliberate use of a chemical and/or biological warfare agent by a terrorist organization. In this tutorial review, we describe methods that have been developed both for the specific detection of biological and chemical warfare agents in a field setting, as well as potential therapeutic approaches for treating exposure to these toxic species. In particular, nerve agents are described as a typical chemical warfare agent, and the two potent biothreat agents, anthrax and botulinum neurotoxin, are used as illustrative examples of potent weapons for which countermeasures are urgently needed.
Co-reporter:Andrew P. Brogan, Tobin J. Dickerson and Kim D. Janda
Chemical Communications 2007 (Issue 46) pp:4952-4954
Publication Date(Web):09 Oct 2007
DOI:10.1039/B713273E
Nornicotine, a native component of tobacco and minor nicotinemetabolite, was found to catalyze the chemoselective reduction of α,β-unsaturated aldehydes under homogeneous aqueous conditions.
Co-reporter:Junguk Park, Reshma Jagasia, Gunnar F. Kaufmann, John C. Mathison, Diana I. Ruiz, Jason A. Moss, Michael M. Meijler, Richard J. Ulevitch, Kim D. Janda
Chemistry & Biology 2007 Volume 14(Issue 10) pp:1119-1127
Publication Date(Web):26 October 2007
DOI:10.1016/j.chembiol.2007.08.013
Quorum sensing (QS) is the process through which bacteria communicate utilizing small diffusible molecules termed autoinducers. It has been demonstrated that QS controls a plethora of microbial processes including the expression of virulence factors. Here we report an immunopharmacotherapeutic approach for the attenuation of QS in the Gram-positive human pathogen Staphylococcus aureus. An anti-autoinducer monoclonal antibody, AP4-24H11, was elicited against a rationally designed hapten, and efficiently inhibited QS in vitro through the sequestration of the autoinducing peptide (AIP)-4 produced by S. aureus RN4850. Importantly, AP4-24H11 suppressed S. aureus pathogenicity in an abscess formation mouse model in vivo and provided complete protection against a lethal S. aureus challenge. These findings provide a strong foundation for further investigations of immunopharmacotherapy for the treatment of bacterial infections in which QS controls the expression of virulence factors.
Co-reporter:Heyue Zhou, Bin Zhou, Hongzheng Ma, Charlotte Carney, Kim D. Janda
Bioorganic & Medicinal Chemistry Letters 2007 Volume 17(Issue 20) pp:5690-5692
Publication Date(Web):15 October 2007
DOI:10.1016/j.bmcl.2007.07.053
Abrin is a highly potent and lethal type II ribosome inactivating toxin that may be used as a biological warfare agent. To date, no human anti-Abrin antibodies have yet to be reported. Herein, we describe the selection and characterization of two human monoclonal antibodies, termed E12 and RF12, which are capable of binding native Abrin with high affinity and specificity. Through surface plasmon resonance studies, we have determined the association and dissociation rate constants and the cross-reactivity for both antibodies. In our developed Biacore-based Abrin detection system, the limit of detection of antibodies E12 and RF12 is 35 and 75 ng/mL, respectively. These concentrations are about 5 × 104-fold lower than the extrapolated Abrin human LD50. In sum, our data demonstrated the power of human antibody phage display libraries and the promise of these antibodies as detection devices for Abrin.Human antibodies were selected from a phage display library that bind the potential biological warfare agent Abrin. The utility of these antibodies is envisaged as detection devices or therapeutics.
Co-reporter:Kateřina Čapková, Yoshiyuki Yoneda, Tobin J. Dickerson, Kim D. Janda
Bioorganic & Medicinal Chemistry Letters 2007 Volume 17(Issue 23) pp:6463-6466
Publication Date(Web):1 December 2007
DOI:10.1016/j.bmcl.2007.09.103
Botulinum neurotoxins are the most toxic proteins currently known. Based on a recently identified potent lead structure, 2,4-dichlorocinnamic acid hydroxamate, herein we report on the structure–activity relationship of a series of hydroxamate BoNT/A inhibitors. Among them, 2-bromo-4-chlorocinnamic acid hydroxamate, 2-methyl-4-chlorocinnamic acid hydroxamate, and 2-trifluoromethyl-4-chlorocinnamic acid hydroxamate displayed comparable inhibitory activity to that of the lead structure.The synthesis and SAR of a series of novel hydroxamate BoNT/A inhibitors is reported.
Co-reporter:Akira Ino, Tobin J. Dickerson, Kim D. Janda
Bioorganic & Medicinal Chemistry Letters 2007 Volume 17(Issue 15) pp:4280-4283
Publication Date(Web):1 August 2007
DOI:10.1016/j.bmcl.2007.05.032
Cocaine use remains a serious problem, despite intensive efforts to curb abuse. Given the lack of effective pharmacotherapeutics for the treatment of cocaine addiction, research groups have targeted immunopharmacotherapy in which the drug user’s immune system is trained to recognize and remove cocaine prior to entry into the central nervous system. Antibody cocaine esterases and simple binders have been procured, however, rates and/or affinities still need improvement before clinical trials are warranted. Herein, we report the synthesis and testing of two new haptens for the procurement of cocaine binding antibodies and cocaine esterase catalytic antibodies. Central in the design of these haptens was the placement of the linker functionality distal from the anticipated cocaine epitopes in an attempt to bury the hapten deep within an antibody combining site to gain possible entropic and enthalpic advantages.The synthesis of two new cocaine haptens designed to bury the drug deep within an antibody combining site are reported.
Co-reporter:George F. Koob;Jennifer Treweek;Sunmee Wee;Kim D. Janda
PNAS 2007 Volume 104 (Issue 28 ) pp:11580-11584
Publication Date(Web):2007-07-10
DOI:10.1073/pnas.0701328104
Methamphetamine abuse is spreading rapidly throughout the United States and is characterized by significant health consequences.
The powerfully rewarding effects of methamphetamine are attributed to multiple neuropharmacological actions such as its ability
to block plasma membrane transporters of all monoamines, reduce dopamine transporter expression, and inhibit monoamine oxidase
activity while increasing tyrosine hydroxylase activity. However, subsequent neuroreceptor changes including monoamine deficits
complement this striking increase in monoamine release. Chronic methamphetamine abuse, as studied via self-administration
paradigms in rodents, causes progressive dopaminergic neurotoxicity, a neuroanatomical change accompanied by increasing drug
tolerance and escalating intake, two behavioral parameters of addiction. We have recently proposed that methamphetamine covalently
glycates endogenous proteins. Such an event spurs antibody production against these immunoconjugates, possibly leading to
drug sequestration by antibody binding of drug. Here we demonstrate that this drug-dependent glycation mechanism is operative
in vivo through the dose-dependent detection of antibodies against methamphetamine-derived advanced glycation end products in rats
chronically self-administering methamphetamine. Furthermore, increased levels of proinflammatory cytokines, evidence of potent
immunoactivation, were also detected. Given the known role of advanced glycation end products in the alteration of protein
function in vivo and the participation of these molecules in various diseases, methamphetamine-derived advanced glycation end products provide
an unrecognized molecular mechanism for the development of vasculitis and other cardiovascular maladies reported with high
incidence in chronic methamphetamine users.
Co-reporter:Yang Xu;Mark S. Hixon;Noboru Yamamoto;Laura A. McAllister;Anita D. Wentworth;Paul Wentworth, Jr.;Kim D. Janda;
Proceedings of the National Academy of Sciences 2007 104(10) pp:3681-3686
Publication Date(Web):February 28, 2007
DOI:10.1073/pnas.0611094104
Methamphetamine [(+)-2] abuse has emerged as a fast-rising global epidemic, with immunopharmacotherapeutic approaches being
sought for its treatment. Herein, we report the generation and characterization of a monoclonal antibody, YX1-40H10, that
catalyzes the photooxidation of (+)-2 into the nonpsychoactive compound benzaldehyde (14) under anaerobic conditions in the
presence of riboflavin (
6). Studies have revealed that the antibody facilitates the conversion of (+)-2 into 14 by binding the triplet photoexcited
state of 6 in proximity to (+)-2. The antibody binds riboflavin (
K
d = 180 μM), although this was not programmed into hapten design, and the YX1-40H10-catalyzed reaction is inhibited by molecular
oxygen via the presumed quenching of the photoexcited triplet state of 6. Given that this reaction is another highlight in
the processing of reactive intermediates by antibodies, we speculate that this process may have future significance
in vivo with programmed immunoglobulins that use flavins as cofactors to destroy selectable molecular targets under hypoxic or even
anoxic conditions.
Co-reporter:Lisa M. Eubanks;Mark S. Hixon;Wei Jin;Sukwon Hong;Colin M. Clancy;William H. Tepp;Carl J. Malizio;Michael R. Baldwin;Michael C. Goodnough;Joseph T. Barbieri;Eric A. Johnson;Dale L. Boger;Kim D. Janda
PNAS 2007 Volume 104 (Issue 8 ) pp:2602-2607
Publication Date(Web):2007-02-20
DOI:10.1073/pnas.0611213104
Among the agents classified as “Category A” by the U.S. Centers for Disease Control and Prevention, botulinum neurotoxin (BoNT)
is the most toxic protein known, with microgram quantities of the protein causing severe morbidity and mortality by oral or
i.v. routes. Given that this toxin easily could be used in a potential bioterrorist attack, countermeasures urgently are needed
to counteract the pathophysiology of BoNT. At a molecular level, BoNT exerts its paralytic effects through intracellular cleavage
of vesicle docking proteins and subsequent organism-wide autonomic dysfunction. In an effort to identify small molecules that
would disrupt the interaction between the light-chain metalloprotease of BoNT serotype A and its cognate substrate, a multifaceted
screening effort was undertaken. Through the combination of in vitro screening against an optimized variant of the light chain involving kinetic analysis, cellular protection assays, and in vivo mouse toxicity assays, molecules that prevent BoNT/A-induced intracellular substrate cleavage and extend the time to death
of animals challenged with lethal toxin doses were identified. Significantly, the two most efficacious compounds in vivo showed less effective activity in cellular assays intended to mimic BoNT exposure; indeed, one of these compounds was cytotoxic
at concentrations three orders of magnitude below its effective dose in animals. These two lead compounds have surprisingly
simple molecular structures and are readily amenable to optimization efforts for improvements in their biological activity.
The findings validate the use of high-throughput screening protocols to define previously unrecognized chemical scaffolds
for the development of therapeutic agents to treat BoNT exposure.
Co-reporter:Grant E. Boldt, Lisa M. Eubanks and Kim D. Janda
Chemical Communications 2006 (Issue 29) pp:3063-3065
Publication Date(Web):03 May 2006
DOI:10.1039/B603099H
Herein we describe a small-molecule, non-peptidic, inhibitor for botulinum neurotoxin A protease, that displays for the first time efficacy in a cell-based assay.
Co-reporter:Ligang Zhang, Hai Long, Grant E. Boldt, Kim D. Janda, George C. Schatz and Frederick D. Lewis
Organic & Biomolecular Chemistry 2006 vol. 4(Issue 2) pp:314-322
Publication Date(Web):08 Dec 2005
DOI:10.1039/B513694F
The synthesis, structure, and optical spectroscopy of hairpin oligonucleotide conjugates possessing synthetic stilbene C-nucleosides (stilbenosides) are reported. Synthetic methods for selective preparation of both the α- and β-stilbenosides have been developed. Both anomers are effective in stabilizing hairpin structures when used as capping groups at the open end of the hairpin base-pair domain. However, only the β-anomer effectively stabilizes the hairpin structure when located in the interior of the base-pair domain opposite an abasic site. Similar results are obtained for hairpins possessing two stilbenosides, either adjacent to each other or with one intervening base-pair. Molecular dynamics simulations are employed to obtain averaged structures for these conjugates. The calculated structures for the capped hairpins formed with either anomer show effective π-stacking with the adjacent base-pair. The calculated structures for the internal stilbenosides show that the α- and β-anomers form extrahelical and intrahelical structures, respectively. The relative orientations of the two stilbenes in the bis-stilbenosides have been studied using a combination of exciton-coupled circular dichroism spectroscopy and molecular modeling.
Co-reporter:Yang Xu, Jin Shi, Noboru Yamamoto, Jason A. Moss, Peter K. Vogt, Kim D. Janda
Bioorganic & Medicinal Chemistry 2006 Volume 14(Issue 8) pp:2660-2673
Publication Date(Web):15 April 2006
DOI:10.1016/j.bmc.2005.11.052
Protein–protein interfaces are prominent in many therapeutically important targets. Using small organic molecules to disrupt protein–protein interactions is a current challenge in chemical biology. An important example of protein–protein interactions is provided by the Myc protein, which is frequently deregulated in human cancers. Myc belongs to the family of basic helix–loop–helix leucine zipper (bHLH-ZIP) transcription factors. It is biologically active only as heterodimer with the bHLH-ZIP protein Max. Herein, we report a new strategy for the disruption of protein–protein interactions that has been corroborated through the design and synthesis of a small parallel library composed of ‘credit-card’ compounds. These compounds are derived from a planar, aromatic scaffold and functionalized with four points of diversity. From a 285 membered library, several hits were obtained that disrupted the c-Myc–Max interaction and cellular functions of c-Myc. The IC50 values determined for this small focused library for the disruption of Myc–Max dimerization are quite potent, especially since small molecule antagonists of protein–protein interactions are notoriously difficult to find. Furthermore, several of the compounds were active at the cellular level as shown by their biological effects on Myc action in chicken embryo fibroblast assays. In light of our findings, this approach is considered a valuable addition to the armamentarium of new molecules being developed to interact with protein–protein interfaces. Finally, this strategy for disrupting protein–protein interactions should prove applicable to other families of proteins.Protein–protein interfaces are prominent in many therapeutically important targets. Using small organic molecules to disrupt protein–protein interactions is a current challenge in chemical biology. Herein, we report a new strategy for the disruption of protein–protein interactions that has been corroborated through the design and synthesis of a small parallel library termed ‘credit-card’ library. From this 285 membered library, several hits were obtained that disrupted the c-Myc–Max interaction and cellular functions of c-Myc. This strategy for disrupting protein–protein interactions should prove applicable to other families of proteins.
Co-reporter:Andrew P. Brogan Dr. Dr.;Kim D. Ja Dr.
Angewandte Chemie International Edition 2006 Volume 45(Issue 48) pp:
Publication Date(Web):25 SEP 2006
DOI:10.1002/anie.200601392
Reactionswith,in, oronwater? Despite claims to the contrary, few examples of truly aqueous organocatalysis have been reported. Close examination of the data from recent reports reveals that these reactions likely occur in concentrated organic phases.
Co-reporter:Kathleen M. McKenzie, Michael M. Meijler, Colin A. Lowery, Grant E. Boldt and Kim D. Janda
Chemical Communications 2005 (Issue 38) pp:4863-4865
Publication Date(Web):06 Sep 2005
DOI:10.1039/B509396A
An autoinducer arising from reaction of cyclized S-DPD and carbonate is shown to induce light in V. harveyi and thus may play a previously unknown role in quorum sensing.
Co-reporter:Hana Matsushita, Noboru Yamamoto, Michael M. Meijler, Peter Wirsching, Richard A. Lerner, Masayuki Matsushita and Kim D. Janda
Molecular BioSystems 2005 vol. 1(Issue 4) pp:303-306
Publication Date(Web):20 Sep 2005
DOI:10.1039/B511408J
The chiral sensing of small molecules using a blue fluorescent antibody sensor is described.
Co-reporter:Gunnar F. Kaufmann;Michael M. Meijler Dr.;Chengzao Sun Dr.;Da-Wei Chen Dr.;David P. Kujawa;Jenny M. Mee;Timothy Z. Hoffman;Peter Wirsching Dr.;Richard A. Lerner Dr.;Kim D. Ja Dr.
Angewandte Chemie International Edition 2005 Volume 44(Issue 14) pp:
Publication Date(Web):2 MAR 2005
DOI:10.1002/anie.200461143
Antibodies as light switches: The deoxynucleotide analogue–stilbene conjugate 1 can be incorporated into nascent DNA by DNA polymerase activity. The blue fluorescence of stilbene is detected only upon binding of an antibody that specifically recognizes the stilbene-modified nucleotide. Therefore, DNA generated through PCR with 1 in the substrate pool can be used for hybridization assays in which detection is triggered by the antibody (see scheme).
Co-reporter:Tobin J. Dickerson;Kim D. Janda;Andrew P. Brogan;Grant E. Boldt
PNAS 2005 Volume 102 (Issue 30 ) pp:10433-10438
Publication Date(Web):2005-07-26
DOI:10.1073/pnas.0504721102
Retinoids (vitamin A) serve two distinct functions in higher animals: light absorption for vision and gene regulation for
growth and development. Cigarette smoking is a contributing factor for diseases that affect vision such as age-related macular
degeneration and increases the risk of birth defects; however, altered retinoid homeostasis has received little attention
as a potential mechanism for smoking-associated toxicities. Herein, we demonstrate that nornicotine, a nicotine metabolite
and component of cigarette smoke, catalyzes the Z-to-E alkene isomerization of unsaturated aldehydes and ketones, including retinals. Despite the recent explosion in the use of
organic compounds as chemical catalysts, minimal effort has been devoted to biologically relevant organocatalysis. Our study
demonstrates a system in which a lowest unoccupied molecular orbital-lowering intermediate similar to the endogenous protein
rhodopsin effectively catalyzes isomerization under biologically relevant conditions. The product of retinal isomerization
is all-E-retinal, which in the eye is a biosynthetic precursor to N-retinylidene-N-retinylethanolamine, a hallmark of age-related macular degeneration. Furthermore, 9-Z- and all-E-retinal isomers are biosynthetic precursors to 9-Z- and all-E-retinoic acids, ligands that mediate specific cellular responses by binding to transcriptional regulatory proteins critical
in growth and development. Strict maintenance of retinal isomer composition is essential for proper transcriptional regulation.
Nornicotine-catalyzed retinal isomerization implies an underlying molecular mechanism for age-related macular degeneration,
the birth defects associated with smoking, and other smoking-associated abnormalities that stem from disruption of retinoid
metabolism.
Co-reporter:Tobin J. Dickerson;Jason A. Moss;Michael M. Meijler;Kim D. Janda;Gunnar F. Kaufmann;Rafaella Sartorio;Sang-Hyeup Lee;Bruce Clapham;Andrew P. Brogan;Claude J. Rogers
PNAS 2005 Volume 102 (Issue 2 ) pp:309-314
Publication Date(Web):2005-01-11
DOI:10.1073/pnas.0408639102
Bacteria use small diffusible molecules to exchange information in a process called quorum sensing. An important class of
autoinducers used by Gram-negative bacteria is the family of N-acylhomoserine lactones. Here, we report the discovery of a previously undescribed nonenzymatically formed product from N-(3-oxododecanoyl)-L-homoserine lactone; both the N-acylhomoserine and its novel tetramic acid degradation product, 3-(1-hydroxydecylidene)-5-(2-hydroxyethyl)pyrrolidine-2,4-dione,
are potent antibacterial agents. Bactericidal activity was observed against all tested Gram-positive bacterial strains, whereas
no toxicity was seen against Gram-negative bacteria. We propose that Pseudomonas aeruginosa utilizes this tetramic acid as an interference strategy to preclude encroachment by competing bacteria. Additionally, we
have discovered that this tetramic acid binds iron with comparable affinity to known bacterial siderophores, possibly providing
an unrecognized mechanism for iron solubilization. These findings merit new attention such that other previously identified
autoinducers be reevaluated for additional biological functions.
Co-reporter:Jung-Mo Ahn, Paul Wentworth, Jr. and Kim D. Janda
Chemical Communications 2004 (Issue 4) pp:364-365
Publication Date(Web):19 Jan 2004
DOI:10.1039/B312662E
In our ongoing efforts to develop new methods for lipopolysaccharide (LPS) detoxification, we have screened lipase/esterase libraries for the ability to deacylate the 2′- and 3′-fatty acid chains from lipid A: the most active esterases were successfully employed to inactivate LPSs in a crude concentrated cell supernatant of E. Coli containing a recombinant single chain antibody (scFv).
Co-reporter:M.Rocı́o A Carrera, Jon A Ashley, Timothy Z Hoffman, Shigeki Isomura, Peter Wirsching, George F Koob, Kim D Janda
Bioorganic & Medicinal Chemistry 2004 Volume 12(Issue 3) pp:563-570
Publication Date(Web):1 February 2004
DOI:10.1016/j.bmc.2003.11.029
Despite the enormous health risks, people continue to smoke and use tobacco primarily as a result of nicotine addiction. As part of our immunopharmacotherapy research, the effects of active and passive immunizations on acute nicotine-induced locomotor activity in rats were investigated. To this end, rats were immunized with either a NIC-KLH immunoconjugate vaccine designed to elicit an antinicotine immune response, or were administered an antinicotine monoclonal antibody, NIC9D9, prior to a series of nicotine challenges and testing sessions. Vaccinated rats showed a 45% decrease in locomotor activity compared to a 16% decrease in controls. Passive immunization with NIC9D9 resulted in a 66.9% decrease in locomotor activity versus a 3.4% decrease in controls. Consistent with the behavioral data, much less nicotine was found in the brains of immunized rats. The results support the potential clinical value of immunopharmacotherapy for nicotine addiction in the context of tobacco cessation programs.Graphic
Co-reporter:Gunnar F. Kaufmann;Michael M. Meijler;George F. Koob;M. Rocio A. Carrera;Jenny M. Mee;Kim D. Janda
PNAS 2004 Volume 101 (Issue 28 ) pp:10416-10421
Publication Date(Web):2004-07-13
DOI:10.1073/pnas.0403795101
Cocaine addiction continues to be a major health and social problem in the United States and other countries. Currently used
pharmacological agents for treating cocaine abuse have proved inadequate, leaving few treatment options. An alternative is
to use protein-based therapeutics that can eliminate the load of cocaine, thereby attenuating its effects. This approach is
especially attractive because the therapeutic agents exert no pharmacodynamic action of their own and therefore have little
potential for side effects. The effectiveness of these agents, however, is limited by their inability to act directly within
the CNS. Bacteriophage have the capacity to penetrate the CNS when administered intranasally. Here, a method is presented
for engineering filamentous bacteriophage to display cocaine-binding proteins on its surface that sequester cocaine in the
brain. These antibody-displaying constructs were examined by using a locomotor activity rodent model to assess the ability
of the phage-displayed proteins to block the psychoactive effects of cocaine. Results presented demonstrate a strategy in
the continuing efforts to find effective treatments for cocaine addiction and suggest the application of this protein-based
treatment for other drug abuse syndromes.
Co-reporter:Michael M. Meijler Dr.;Louis G. Hom Dr.;Gunnar F. Kaufmann;Kathleen M. McKenzie Dr.;Chengzao Sun Dr.;Jason A. Moss;Masayuki Matsushita Dr.;Kim D. Ja Dr.
Angewandte Chemie International Edition 2004 Volume 43(Issue 16) pp:
Publication Date(Web):6 APR 2004
DOI:10.1002/anie.200353150
Chemical communication (“quorum sensing”) amongst bacteria has been studied by the synthesis and study of enantiopure (R)-4,5-dihydroxy-2,3-pentanedione (DPD, see scheme). Bioactivity assays with DPD have shown that chelation of boron by the cyclic form of DPD appears to be essential for full induction of bioluminescence, which is an example of quorum-sensing-controlled behavior.
Co-reporter:Masayuki Matsushita Dr.;Kazuhiro Yoshida Dr.;Noboru Yamamoto Dr.;Peter Wirsching Dr.;Richard A. Lerner Dr.;Kim D. Ja Dr.
Angewandte Chemie International Edition 2003 Volume 42(Issue 48) pp:
Publication Date(Web):10 DEC 2003
DOI:10.1002/anie.200352793
Blue lights the way! The rapid evaluation of enantiomeric excess in the high-throughput screening of libraries has been a hurdle to the discovery of effective catalysts. A blue-fluorescent monoclonal antibody (mAb) addresses this problem; mAb 19G2 is used as a fluorescent sensor (see picture) to evaluate a panel of Cinchona alkaloid derivatives in the synthesis of asymmetric amino acids by phase-transfer catalysis.
Co-reporter:Tobin J. Dickerson;Kim D. Janda;
Proceedings of the National Academy of Sciences 2003 100(14) pp:8182-8187
Publication Date(Web):June 18, 2003
DOI:10.1073/pnas.1332847100
The origin of Alzheimer's disease (AD) has been subjected to an intense
amount of examination; however, a clear conclusion as to the nature of this
crippling disease has yet to be identified. What is readily accepted is that a
definitive marker of this disease is the aggregation of the amyloid
β-peptide (Aβ) into neuritic plaques. The recent observation that
nicotine exposure leads to delayed onset of AD has stimulated a flurry of
research into the nature of this neuroprotective effect. This phenomenon has
been debated, but no consensus has been reached, and although these studies
have targeted nicotine, the primary alkaloid in tobacco, few studies have
considered the physiological role of nicotine metabolites in disease states.
Nornicotine is a major nicotine metabolite in the CNS and has been shown to
participate in the aberrant glycation of proteins in vivo in a
process termed nornicotine-based glycation. Herein is detailed a potentially
fortuitous role of nornicotine-based glycation in relation to the pathology of
AD. Specifically, nornicotine was found to covalently alter Aβ, leading
to reduced peptide aggregation. Potential consequences of this reaction
cascade include reduced plaque formation and/or altered clearance of the
peptide, as well as attenuated toxicity of soluble Aβ aggregates. The
findings described provide an alternative mechanism for nicotine
neuroprotection in AD and a means for the alteration of amyloid folding based
on a covalent chemical event.
Co-reporter:Bin Zhou;Peter Wirsching;Kim D. Janda
PNAS 2002 Volume 99 (Issue 8 ) pp:5241-5246
Publication Date(Web):2002-04-16
DOI:10.1073/pnas.082121599
A naïve, human single-chain Fv (scFv) phage-display library was used in bio-panning against live, native spores of Bacillus subtilis IFO 3336 suspended in solution. A direct in vitro panning and enzyme-linked immunosorbent assay-based selection afforded a panel of nine scFv-phage clones of which two, 5B
and 7E, were chosen for further study. These two clones differed in their relative specificity and affinity for spores of
B. subtilis IFO 3336 vs. a panel of spores from 11 other Bacillus species/strains. A variety of enzyme-linked immunosorbent assay protocols indicated these scFv-phage clones recognized different
spore epitopes. Notably, some spore epitopes markedly changed between the free and microtiter-plate immobilized state as revealed
by antibody-phage binding. An additional library selection procedure also was examined by constructing a Fab chain-shuffled
sublibrary from the nine positive clones and by using a subtractive panning strategy to remove crossreactivity with B. licheniformis 5A24. The Fab-phage clone 52 was improved compared with 5B and was comparable to 7E in binding B. subtilis IFO 3336 vs. B. licheniformis 5A24, yet showed a distinctive crossreactivity pattern with other spores. We also developed a method to directly detect individual
spores by using fluorescently labeled antibody-phage. Finally, a variety of “powders” that might be used in deploying spores
of B. anthracis were examined for antibody-phage binding. The strategies described provide a foundation to discover human antibodies specific
for native spores of B. anthracis that can be developed as diagnostic and therapeutic reagents.
Co-reporter:Tobin J. Dickerson;Kim D. Janda;
Proceedings of the National Academy of Sciences 2002 99(23) pp:15084-15088
Publication Date(Web):October 28, 2002
DOI:10.1073/pnas.222561699
Over the past 20 years, protein glycation has been implicated in a variety of pathological states. Although smoking also can
contribute to many of these diseases, the precise mechanism by which this occurs is not known. Previously, we have demonstrated
that nornicotine, a constituent of tobacco and metabolite of nicotine, can catalyze aldol reactions under aqueous conditions.
This finding has caused us to question whether this reaction has physiological consequences. We now report that nornicotine
causes aberrant protein glycation and catalyzes the covalent modification of certain prescription drugs such as the commonly
used steroid, prednisone. Furthermore, we show that the plasma of smokers as compared with nonsmokers contains higher concentrations
of nornicotine-modified proteins, suggesting an unrecognized pathway for the development of the pathology of tobacco abuse.
Co-reporter:Changshou Gao;Shenlan Mao;Gunnar Kaufmann;Peter Wirsching;Richard A. Lerner;Kim D. Janda;
Proceedings of the National Academy of Sciences 2002 99(20) pp:12612-12616
Publication Date(Web):September 18, 2002
DOI:10.1073/pnas.192467999
For more than a decade, phage displayed combinatorial antibody libraries have been used to generate and select a wide variety
of antibodies. We previously reported that the phage coat proteins pVII and pIX could be used to display the heterodimeric
structure of the antibody Fv region. Herein, aspects of this technology were invoked and extended to construct a large, human
single-chain Fv (scFv) library of 4.5 × 109 members displayed on pIX of filamentous bacteriophage. Furthermore, the diversity, quality, and utility of the library were
demonstrated by the selection of scFv clones against six different protein antigens. Notably, more than 90% of the selected
clones showed positive binding for their respective antigens after as few as three rounds of panning. Analyzed scFvs were
also found to be of high affinity. For example, kinetic analysis (BIAcore) revealed that scFvs against staphylococcal enterotoxin
B and cholera toxin B subunit had a nanomolar and subnanomolar dissociation constant, respectively, affording affinities comparable
to, or exceeding that, of mAbs obtained from immunization. High specificity was also attained, not only between very distinct
proteins, but also in the case of the Ricinus communis (“ricin”) agglutinins (RCA60 and RCA120), despite >80% sequence homology between the two. The results suggested that the performance of pIX-display libraries can
potentially exceed that of the pIII-display format and make it ideally suited for panning a wide variety of target antigens.
Co-reporter:Kenneth M. Nicholas;Paul Wentworth, Jr.;Curtis W. Harwig;Anita D. Wentworth;Asher Shafton;Kim D. Janda;
Proceedings of the National Academy of Sciences 2002 99(5) pp:2648-2653
Publication Date(Web):2002-03-05
DOI:10.1073/pnas.052001099
A strategy for the preparation of semisynthetic copper(II)-based catalytic metalloproteins is described in which a metal-binding
bis-imidazole cofactor is incorporated into the combining site of the aldolase antibody 38C2. Antibody 38C2 features a large
hydrophobic-combining site pocket with a highly nucleophilic lysine residue, LysH93, that can be covalently modified. A comparison of several lactone and anhydride reagents shows that the latter are the most
effective and general derivatizing agents for the 38C2 Lys residue. A bis-imidazole anhydride (5) was efficiently prepared
from N-methyl imidazole. The 38C2–5-Cu conjugate was prepared by either (i) initial derivatization of 38C2 with 5 followed by metallation with CuCl2, or (ii) precoordination of 5 with CuCl2 followed by conjugation with 38C2. The resulting 38C2–5-Cu conjugate was an active catalyst for the hydrolysis of the coordinating
picolinate ester 11, following Michaelis–Menten kinetics [kcat(11) = 2.3 min−1 and Km(11) 2.2 mM] with a rate enhancement [kcat(11)kuncat(11)] of 2.1 × 105. Comparison of the second-order rate constants of the modified 38C2 and the Cu(II)-bis-imidazolyl complex k(6-CuCl2) gives a rate enhancement of 3.5 × 104 in favor of the antibody complex with an effective molarity of 76.7 M, revealing a significant catalytic benefit to the binding
of the bis-imidazolyl ligand into 38C2.
Co-reporter:Oliver Brümmer, Timothy Z. Hoffman, Da-Wei Chen and Kim D. Janda
Chemical Communications 2001 (Issue 1) pp:19-20
Publication Date(Web):11 Dec 2000
DOI:10.1039/B008065I
A catalytic antibody has been discovered that degrades
oligomeric ester substrates.
Co-reporter:Da-Wei Chen, Robert J. Kubiak, Jon A. Ashley and Kim D. Janda
Organic & Biomolecular Chemistry 2001 (Issue 21) pp:2796-2803
Publication Date(Web):09 Oct 2001
DOI:10.1039/B105412K
In the search for biocatalysts for degradation of nonnatural polymers, reactive immunization with haptens 7 and 11 was used to prepare catalytic antibodies capable of cleaving short oligomeric esters, as well as the insoluble polyester 25. These antibodies were found to be highly specific and efficient esterases for oligomers. Triester 24 was preferentially hydrolyzed by an endo-cleavage pathway, however, with a higher molecular weight polymer 25 no site specificity could be observed. Catalytic efficiency of the antibodies towards the insoluble polymer 25 was limited due to physical constraints.
Co-reporter:M. Rocío A. Carrera;Jon A. Ashley;Peter Wirsching;George F. Koob;Kim D. Janda
PNAS 2001 Volume 98 (Issue 4 ) pp:1988-1992
Publication Date(Web):2001-02-13
DOI:10.1073/pnas.98.4.1988
The effects of immunization with the second-generation cocaine
immunoconjugate GND-keyhole limpet hemocyanin (KLH) or with the
anti-cocaine mAb GNC92H2 were assessed in a model of acute
cocaine-induced locomotor activity. After i.p. administration of
cocaine⋅HCl (15 mg/kg), rats were tested in photocell cages, and
stereotypy was rated to determine preimmunization drug response
(baseline). Experimental animals were subjected to an immunization
protocol with GND-KLH or treated with the mAb GNC92H2. Rats were then
challenged with systemic cocaine, and their locomotor responses were
again measured. Active immunization with GND-KLH produced a 76%
decrease in the ambulatory measure (crossovers) in the experimental
group and a 12% increase in the control group compared with baseline
values. Also, stereotypic behavior was significantly suppressed in the
vaccinated animals. Decreases in both measures were seen in the
experimental group on two subsequent challenges. The maximum effect was
observed at the time of the second challenge with a dramatic 80%
decrease in crossovers. Treatment with GNC92H2 resulted in a 69%
decrease in crossovers compared with baseline. This effect persisted
across two additional challenges over 11 days with decreases of
46–47%. In contrast, the control group showed increases of up to
28%. Significant differences between groups were observed in the
stereotypic measure in all three challenges. The results indicate that
these immunopharmacotherapeutic agents have significant
cocaine-blockade potential and therefore may offer an effective
strategy for the treatment of cocaine abuse.
Co-reporter:M. Rocío A. Carrera;Jon A. Ashley;Bin Zhou;Peter Wirsching;George F. Koob;Kim D. Janda
PNAS 2000 Volume 97 (Issue 11 ) pp:6202-6206
Publication Date(Web):2000-05-23
DOI:10.1073/pnas.97.11.6202
The efficacy of active immunization with the cocaine immunogen GNC-keyhole limpet hemocyanin (KLH) in preventing cocaine self-administration
reinstatement was assessed in rats. An animal model of relapse was used where rats were trained to self-administer cocaine,
subjected to a period of extinction by substituting the drug for saline, vaccinated, and re-exposed to cocaine. Compared with
controls, animals immunized with GNC-KLH did not reinstate cocaine self-administration behavior when given a noncontingent
cocaine infusion on two consecutive days. Upon double and triple infusions, 38–62% of vaccinated animals failed to reinstate
as compared with full reinstatement in all control animals. Exposure to ad libitum cocaine reinstated baseline values in control
animals and resulted in double to triple the baseline values of self-infusions in vaccinated animals, suggesting a partial
antibody-mediated blockade of cocaine access to the central nervous system. This compensating effect was blocked by passive
immunization pretreatment with the monoclonal IgG GNC92H2 in both vaccinated and control groups. To further assess the surmountability
potential of GNC-KLH-induced antibody titers by cocaine self-administration, and the capacity of these titers to block the
reinforcing effects of the drug, rats were tested at various doses of cocaine (0.015–0.5 mg/infusion). Active immunization
with GNC-KLH produced approximately an 8-fold rightward shift of the dose-effect function for cocaine. The results reported
suggest that immunopharmacotherapy may offer a promising means to treat cocaine abuse by aiding in the prevention of relapse.
Co-reporter:Andrew R. Vaino;Kim D. Janda
PNAS 2000 Volume 97 (Issue 14 ) pp:7692-7696
Publication Date(Web):2000-07-05
DOI:10.1073/pnas.97.14.7692
A method for the encoding of split/mix combinatorial chemical
libraries based on Euclidean shapes is described. The shapes are
fashioned from a polymeric matrix designed to swell in common organic
solvents while retaining their unique forms, and exhibit good
mechanical strength. The lightly crosslinked gel-type polymer was
processed into an array of Euclidean forms that serve as encoding
elements in the synthesis of combinatorial chemical libraries by using
the split/pool methodology. To assess the viability of this approach,
a library of compounds based on a urea scaffold was prepared. The
validity of this methodology was demonstrated through correct
deconvolution of the library mixture by shape discrimination.
Furthermore, because the shapes used have a large surface area to
volume ratio, each monolith can act as an independent chemical reactor.
This simplifies the analytical identification process because each
compound can be prepared in significant quantities and isolated as
single entities. Given the high loading capacity of the monoliths and
the conceptually simple encoding strategy, it is envisioned that these
Euclidean forms will find significant application in combinatorial and
high-throughput synthetic chemistry.
Co-reporter:Kateřina Čapková, Nicholas T. Salzameda, Kim D. Janda
Toxicon (October 2009) Volume 54(Issue 5) pp:575-582
Publication Date(Web):1 October 2009
DOI:10.1016/j.toxicon.2009.03.016
Botulinum neurotoxins (BoNTs), proteins secreted by the bacteria genus Clostridium, represent a group of extremely lethal toxins and a potential bioterrorism threat. As the current therapeutic options are of a predominantly prophylactic nature and cannot be used en masse, new strategies and ultimately potential treatments are desperately needed to combat any widespread release of these neurotoxins. In these regards, our laboratory has been working on developing new alternatives to treat botulinum intoxication through the development of inhibitors of the light chain proteases, the etiological agent which causes BoNT intoxication. Such a strategy has required the construction of two high-throughput screens and small molecule non-peptidic libraries; excitingly, inhibitors of the BoNT/A protease have been uncovered and are being optimized via structure activity relationship studies.
Co-reporter:Suzette M. Evans, Richard W. Foltin, Martin J. Hicks, Jonathan B. Rosenberg, Bishnu P. De, Kim D. Janda, Stephen M. Kaminsky, Ronald G. Crystal
Pharmacology Biochemistry and Behavior (November–December 2016) Volumes 150–151() pp:76-86
Publication Date(Web):1 November 2016
DOI:10.1016/j.pbb.2016.09.008
•Second study to evaluate immunotherapy on drug self-administration in monkeys•Vaccination attenuated ongoing cocaine self-administration in 25% of the monkeys.•Vaccination retarded reacquisition of cocaine self-administration.•The dAdGNE vaccine may have therapeutic potential for relapse prevention.Immunopharmacotherapy offers an approach for treating cocaine abuse by specifically targeting the cocaine molecule and preventing its access to the CNS. dAd5GNE is a novel cocaine vaccine that attenuates the stimulant and the reinforcing effects of cocaine in rats. The goal of this study was to extend and validate dAd5GNE vaccine efficacy in non-human primates. Six experimentally naïve adult female rhesus monkeys (Macaca mulatta) were trained to self-administer 0.1 mg/kg/injection intravenous (i.v.) cocaine or receive candy; then 4 monkeys were administered the vaccine and 2 monkeys were administered vehicle intramuscularly, with additional vaccine boosts throughout the study. The reinforcing effects of cocaine were measured during self-administration, extinction, and reacquisition (relapse) phases. Serum antibody titers in the vaccinated monkeys remained high throughout the study. There was no change in the preference for cocaine over candy over a 20-week period in 5 of the 6 monkeys; only one of the 4 (25%) vaccinated monkeys showed a decrease in cocaine choice. All 6 monkeys extinguished responding for cocaine during saline extinction testing; vaccinated monkeys tended to take longer to extinguish responding than control monkeys (17.5 vs. 7.0 sessions). Vaccination substantially retarded reacquisition of cocaine self-administration; control monkeys resumed cocaine self-administration within 6–41 sessions and 1 vaccinated monkey resumed cocaine self-administration in 19 sessions. The other 3 vaccinated monkeys required between 57 and 94 sessions to resume cocaine self-administration even in the context of employing several manipulations to encourage cocaine reacquisition. These data suggest that the dAdGNE vaccine may have therapeutic potential for humans who achieve cocaine abstinence as part of a relapse prevention strategy.
Co-reporter:Gunnar F. Kaufmann, Junguk Park, Jenny M. Mee, Richard J. Ulevitch, Kim D. Janda
Molecular Immunology (May 2008) Volume 45(Issue 9) pp:2710-2714
Publication Date(Web):1 May 2008
DOI:10.1016/j.molimm.2008.01.010
The Gram-negative bacterium Pseudomonas aeruginosa, an opportunistic human pathogen, uses acyl-homoserine lactone-based quorum sensing systems to control its pathogenicity. One of its quorum sensing factors, N-3-oxo-dodecanoyl-homoserine lactone, has been shown not only to mediate bacterial quorum sensing but also to exert cytotoxic effects on mammalian cells. The monoclonal antibody RS2-1G9 generated against a 3-oxo-dodecanoyl-homoserine lactone analogue hapten was able to protect murine bone marrow-derived macrophages from the cytotoxic effects and also prevented the activation of the mitogen-activated protein kinase p38. These data demonstrate that an immunopharmacotherapeutic approach to combat P. aeruginosa infections might be a viable therapeutic option as the monoclonal antibody RS2-1G9 can readily sequester bacterial N-3-oxo-dodecanoyl-homoserine lactone molecules, thus interfering with their biological effects in prokaryotic and eukaryotic systems.
Co-reporter:Sebastian C.J. Steiniger, Laurence J. Altobell III, Bin Zhou, Kim D. Janda
Molecular Immunology (April 2007) Volume 44(Issue 10) pp:2749-2755
Publication Date(Web):1 April 2007
DOI:10.1016/j.molimm.2006.11.011
The protective antigen (PA83) of Bacillus anthracis is the dominant antigen in natural and vaccine-induced immunity to anthrax infection. Three human single-chain variable fragments (scFvs) against cell bound PA were isolated from an antibody phage display library. Specifically, the antibodies were evaluated for their ability to bind to cell bound heptameric PA and ultimately protect against the cytotoxicity of lethal toxin. In total, all three scFvs possessed neutralizing activity against the cytotoxic effects of lethal toxin in a macrophage lysis assay. The Kd values of the Fabs were determined, interestingly their protective effects did not parallel their affinities; hence, a simple binding argument alone to PA63 cannot be used as the distinguishing feature for the prediction of their neutralization abilities. Immunofluorescent microscopy experiments were conducted and provided strong evidence for Fab binding to oligomeric PA on the cell surface and thus a plausible mechanism for the toxin neutralization activity that was observed. The results of this study presented herein suggest that our antibodies compete with LF–PA cell surface interactions, and thus may provide potential application of human antibodies as passive immunization prophylactics in cases of B. anthracis exposure and infection.
Co-reporter:Bin Zhou, Sabine Pellett, William H. Tepp, Heyue Zhou, ... Kim D. Janda
FEBS Letters (30 April 2008) Volume 582(Issue 10) pp:1526-1531
Publication Date(Web):30 April 2008
DOI:10.1016/j.febslet.2008.03.047
Botulinum neurotoxins (BoNT) are the etiological agents responsible for botulism and are acknowledged terrorist threat agents. Passive immunotherapy may provide one countermeasure. Importantly, in the virtually unlimited repertoire of antibody specificities, enzyme linked immunosorbent assays (ELISA) has become an indispensable method for antibody selection. We report that of the BoNTs, BoNT/E is highly susceptible to polystyrene induced denaturation. To further dissect this result and the potential susceptibility of other BoNTs to denaturation we selected a thermal platform, which could be readily quantified using surface plasmon resonance (SPR), a primary rat spinal cord cell-based assay and an animal lethality model.
Co-reporter:Kathleen M. McKenzie, Jenny M. Mee, Claude J. Rogers, Mark S. Hixon, ... Kim D. Janda
Journal of Molecular Biology (19 January 2007) Volume 365(Issue 3) pp:722-731
Publication Date(Web):19 January 2007
DOI:10.1016/j.jmb.2006.10.031
Cocaine is a powerful and addictive stimulant whose abuse remains a prevalent health and societal crisis. Unfortunately, no pharmacological therapies exist and therefore alternative protein-based therapies have been examined. One such approach is immunopharmacotherapy, wherein antibodies are utilized to either bind or hydrolyze cocaine thereby blocking it from exerting its euphoric effect. Towards this end, antibodies capable of binding and hydrolyzing cocaine were identified by phage display from a biased single chain antibody library generated from the spleens of mice previously immunized with a cocaine phosphonate transition state analog hapten. Two classes of antibodies emerged based on sequence homology and mode of action. Alanine scanning mutagenesis and kinetic analysis revealed that residues H97, H99, and L96 are crucial for antibodies 3F5 and 3H9 to accelerate the hydrolysis of cocaine. Antibodies 3F1 through 3F4, which are similar to our previously identified 3A6 class of antibodies, catalyze hydrolysis through transition state stabilization by tyrosine or histidine residues H50 and L94. Mutation of either one or both tyrosine residues to histidine conferred hydrolytic activity on previously inactive antibody 3F4. Mutational analysis of residue H50 of antibody 3F3 resulted in a glutamine mutant with a rate enhancement three times greater than wild-type. A double mutant, containing glutamineH50 and lysineH52, showed a tenfold rate enhancement over wild-type. These results indicate the power of initial selection of catalytic antibodies from a biased antibody library in both rapid generation and screening of mutants for improved catalysis.
Co-reporter:Erik W. Debler, Gunnar F. Kaufmann, Robert N. Kirchdoerfer, Jenny M. Mee, ... Ian A. Wilson
Journal of Molecular Biology (18 May 2007) Volume 368(Issue 5) pp:1392-1402
Publication Date(Web):18 May 2007
DOI:10.1016/j.jmb.2007.02.081
A large number of Gram-negative bacteria employ N-acyl homoserine lactones (AHLs) as signaling molecules in quorum sensing, which is a population density-dependent mechanism to coordinate gene expression. Antibody RS2-1G9 was elicited against a lactam mimetic of the N-acyl homoserine lactone and represents the only reported monoclonal antibody that recognizes the naturally-occuring N-acyl homoserine lactone with high affinity. Due to its high cross-reactivity, RS2-1G9 showed remarkable inhibition of quorum sensing signaling in Pseudomonas aeruginosa, a common opportunistic pathogen in humans. The crystal structure of Fab RS2-1G9 in complex with a lactam analog revealed complete encapsulation of the polar lactam moiety in the antibody-combining site. This mode of recognition provides an elegant immunological solution for tight binding to an aliphatic, lipid-like ligand with a small head group lacking typical haptenic features, such as aromaticity or charge, which are often incorporated into hapten design to generate high-affinity antibodies. The ability of RS2-1G9 to discriminate between closely related AHLs is conferred by six hydrogen bonds to the ligand. Conversely, cross-reactivity of RS2-1G9 towards the lactone is likely to originate from conservation of these hydrogen bonds as well as an additional hydrogen bond to the oxygen of the lactone ring. A short, narrow tunnel exiting at the protein surface harbors a portion of the acyl chain and would not allow entry of the head group. The crystal structure of the antibody without its cognate lactam or lactone ligands revealed a considerably altered antibody-combining site with a closed binding pocket. Curiously, a completely buried ethylene glycol molecule mimics the lactam ring and, thus, serves as a surrogate ligand. The detailed structural delineation of this quorum-quenching antibody will aid further development of an antibody-based therapy against bacterial pathogens by interference with quorum sensing.
Co-reporter:Paul T. Bremer, Song Xue and Kim D. Janda
Chemical Communications 2016 - vol. 52(Issue 84) pp:NaN12524-12524
Publication Date(Web):2016/09/30
DOI:10.1039/C6CC06749B
In developing small-molecule inhibitors of botulinum neurotoxin serotype A light chain (BoNT/A LC), substituted picolinic acids were identified. Extensive investigation into the SAR of the picolinic acid scaffold revealed 5-(1-butyl-4-chloro-1H-indol-2-yl)picolinic acid (CBIP), which possessed low micromolar activity against BoNT/A. Kinetic and docking studies demonstrated binding of CBIP to the β-exosite: a largely unexplored site on the LC that holds therapeutic relevance for botulism treatment.
Co-reporter:Nicholas T. Salzameda, Lisa M. Eubanks, Joseph S. Zakhari, Kyoji Tsuchikama, Nicholas J. DeNunzio, Karen N. Allen, Mark S. Hixon and Kim D. Janda
Chemical Communications 2011 - vol. 47(Issue 6) pp:NaN1715-1715
Publication Date(Web):2011/01/04
DOI:10.1039/C0CC04078A
Clostridium
botulinum produces the most lethal toxins known to man, as such they are high risk terrorist threats, and alarmingly there is no approved therapeutic. We report the first cross-over small molecule inhibitor of these neurotoxins and propose a mechanism by which it may impart its inhibitory activity.
Co-reporter:T. L. Harris, C. J. Wenthur, A. Diego-Taboada, G. Mackenzie, T. S. Corbitt and K. D. Janda
Chemical Communications 2016 - vol. 52(Issue 22) pp:NaN4190-4190
Publication Date(Web):2016/02/18
DOI:10.1039/C6CC00615A
3,4-Diaminopyridine has shown promise in reversing botulinum intoxication, but poor pharmacokinetics and a narrow therapeutic window limit its clinical utility. Thus, we developed a pH-dependent oral delivery platform using club moss spore exines. These exine microcapsules slowed 3,4-diaminopyridine absorption, limited its seizure activity, and enabled delivery of doses which prolonged mouse survival after botulism neurotoxin A intoxication.
Co-reporter:Amanda L. Garner and Kim D. Janda
Chemical Communications 2011 - vol. 47(Issue 26) pp:NaN7514-7514
Publication Date(Web):2011/05/19
DOI:10.1039/C1CC11817J
Using our recently disclosed fluorescence-based assay to monitor acyltransferase activity, the first non-peptidic, small molecule antagonists of ghrelin O-acyltransferase (GOAT), a potential anti-obesity and anti-diabetes target, have been discovered. Each exhibits micromolar inhibition of the enzyme, and may be useful probes for future study of the ghrelin-GOAT system.
Co-reporter:Andrew P. Brogan, Tobin J. Dickerson and Kim D. Janda
Chemical Communications 2007(Issue 46) pp:NaN4954-4954
Publication Date(Web):2007/10/09
DOI:10.1039/B713273E
Nornicotine, a native component of tobacco and minor nicotinemetabolite, was found to catalyze the chemoselective reduction of α,β-unsaturated aldehydes under homogeneous aqueous conditions.
Co-reporter:Jessica L. Fullagar, Amanda L. Garner, Anjali K. Struss, Joshua A. Day, David P. Martin, Jing Yu, Xiaoqing Cai, Kim D. Janda and Seth M. Cohen
Chemical Communications 2013 - vol. 49(Issue 31) pp:NaN3199-3199
Publication Date(Web):2013/03/06
DOI:10.1039/C3CC41191E
Tropolone emerged from the screening of a chelator fragment library (CFL) as an inhibitor of the Zn2+-dependent virulence factor, Pseudomonas aeruginosa elastase (LasB). Based on this initial hit, a series of substituted tropolone-based LasB inhibitors was prepared, and a compound displaying potent activity in vitro and in a bacterial swarming assay was identified. Importantly, this inhibitor was found to be specific for LasB over other metalloenzymes, validating the usage of tropolone as a viable scaffold for identifying first-in-class LasB inhibitors.
Co-reporter:Kensaku Anraku, Shun Sato, Nicholas T. Jacob, Lisa M. Eubanks, Beverly A. Ellis and Kim D. Janda
Organic & Biomolecular Chemistry 2017 - vol. 15(Issue 14) pp:NaN2992-2992
Publication Date(Web):2017/03/08
DOI:10.1039/C7OB00448F
Carbohydrate antigens displaying Galα(1,3)Gal epitopes are recognized by naturally occurring antibodies in humans. These anti-Gal antibodies comprise up to 1% of serum IgG and have been viewed as detrimental as they are responsible for hyperacute organ rejections. In order to model this condition, α(1,3)galactosyltransferase-knockout mice are inoculated against the Galα(1,3)Gal epitope. In our study, two α-Gal trisaccharide epitopes composed of either Galα(1,3)Galβ(1,4)GlcNAc or Galα(1,3)Galβ(1,4)Glc linked to a squaric acid ester moiety were examined for their ability to elicit immune responses in KO mice. Both target epitopes were synthesized using a two-component enzymatic system using modified disaccharide substrates containing a linker moiety for coupling. While both glycoconjugate vaccines induced the required high anti-Gal IgG antibody titers, it was found that this response had exquisite specificity for the Galα(1,3)Galβ(1,4)GlcNAc hapten used, with little cross reactivity with the Galα(1,3)Galβ(1,4)Glc hapten. Our findings indicate that while homogenous glycoconjugate vaccines provide high IgG titers, the carrier and adjuvanting factors can deviate the specificity to an antigenic determinant outside the purview of interest.
Co-reporter:NIcholas T. Jacob, Kensaku Anraku, Atsushi Kimishima, Bin Zhou, Karen C. Collins, Jonathan W. Lockner, Beverley A. Ellis and Kim D. Janda
Chemical Communications 2017 - vol. 53(Issue 58) pp:NaN8159-8159
Publication Date(Web):2017/06/27
DOI:10.1039/C7CC04055E
A method for potentiating the response to an anti-cocaine vaccine by leveraging xenoreactive antibodies against the carbohydrate epitope Galα1,3-Gal (GAL) was found to result in a highly specific anti-cocaine response that was able to significantly attenuate cocaine-induced locomotion at 20 mg kg−1 with superior efficacy compared to a standard conjugate.
Co-reporter:Kateřina Čapková, Mark S. Hixon, Laura A. McAllister and Kim D. Janda
Chemical Communications 2008(Issue 30) pp:NaN3527-3527
Publication Date(Web):2008/07/03
DOI:10.1039/B808305C
The development of a sensitive, yet reliable assay for the analysis of botulinum neurotoxin A (BoNT/A) inhibitors is described; using this assay a new protease inhibitor was characterized and found to be one of the most potent inhibitors reported to date.
Co-reporter:Colin A. Lowery, Tobin J. Dickerson and Kim D. Janda
Chemical Society Reviews 2008 - vol. 37(Issue 7) pp:NaN1346-1346
Publication Date(Web):2008/05/01
DOI:10.1039/B702781H
Quorum sensing (QS) has traditionally referred to a mechanism of communication within a species of bacteria. However, emerging research implicates QS in interspecies communication and competition, and such systems have been proposed in a wide variety of bacteria. This activity of bacterial QS also extends to relationships between bacteria and eukaryotes and host–pathogen interactions in both clinical and agricultural settings are of particular interest. These relationships are particularly pertinent in light of the rising prevalence of antibiotic resistant bacteria. In this tutorial review we describe bacterial QS and its capacity in interspecies and interkingdom interactions, as well as the corresponding eukaryotic responses.
Co-reporter:Hajime Seki, Song Xue, Mark S. Hixon, Sabine Pellett, Marek Reme, Eric A. Johnson and Kim D. Janda
Chemical Communications 2015 - vol. 51(Issue 28) pp:NaN6229-6229
Publication Date(Web):2015/03/11
DOI:10.1039/C5CC00677E
Dyngo-4a™ has been found to be an endocytic inhibitor of BoNT/A neurotoxicity through dynamin inhibition. Herein, we demonstrate this molecule to have a previously unrecognized dual activity against BoNT/A, dynamin–protease inhibition. To establish the importance of this dual activity, detailed kinetic analysis of Dyngo-4a's inhibition of BoNT/A metalloprotease as well as cellular and animal toxicity studies have been described. The research presented is the first polypharmacological approach to counteract BoNT/A intoxication.
Co-reporter:Amanda L. Garner, Jing Yu, Anjali K. Struss, Gunnar F. Kaufmann, Vladimir V. Kravchenko and Kim D. Janda
Chemical Communications 2013 - vol. 49(Issue 15) pp:NaN1517-1517
Publication Date(Web):2013/01/10
DOI:10.1039/C3CC38851D
As a guide for chemical probe design, focused analogue synthetic studies were undertaken upon the lactone ring of 3-oxo-C12-homoserine lactone. We have concluded that hydrolytic instability of the heterocyclic ring is pivotal for its ability to modulate immune signaling and probe preparation was aligned with these findings.
Co-reporter:K. C. Collins, J. E. Schlosburg, J. W. Lockner, P. T. Bremer, B. A. Ellis and K. D. Janda
Chemical Communications 2014 - vol. 50(Issue 31) pp:NaN4081-4081
Publication Date(Web):2014/03/03
DOI:10.1039/C4CC00682H
The immunopotentiator tucaresol was modified for incorporation into liposomes, where it was found to be a superior adjuvant to MPLA for vaccination against methamphetamine.
Co-reporter:Lisa M. Eubanks, Tobin J. Dickerson and Kim D. Janda
Chemical Society Reviews 2007 - vol. 36(Issue 3) pp:NaN470-470
Publication Date(Web):2007/01/11
DOI:10.1039/B615227A
There is a growing need for technological advancements to combat agents of chemical and biological warfare, particularly in the context of the deliberate use of a chemical and/or biological warfare agent by a terrorist organization. In this tutorial review, we describe methods that have been developed both for the specific detection of biological and chemical warfare agents in a field setting, as well as potential therapeutic approaches for treating exposure to these toxic species. In particular, nerve agents are described as a typical chemical warfare agent, and the two potent biothreat agents, anthrax and botulinum neurotoxin, are used as illustrative examples of potent weapons for which countermeasures are urgently needed.
.jpg)


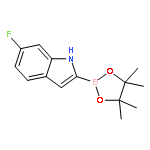






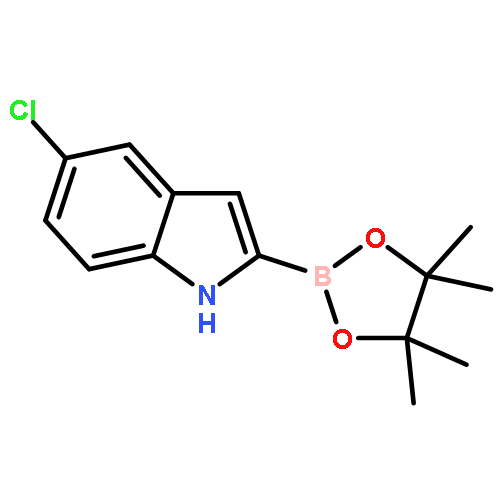
















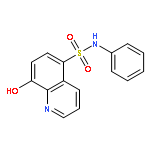


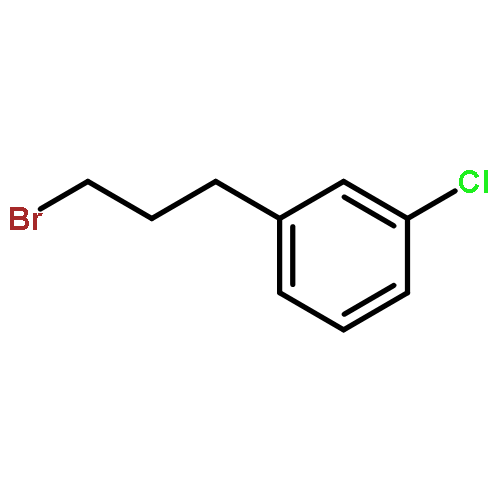


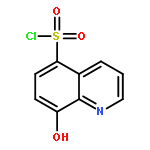
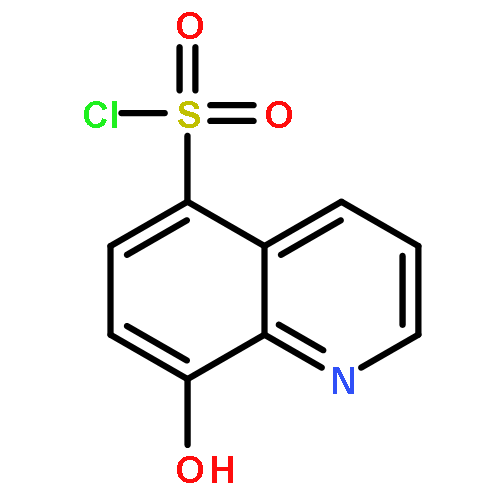















![8-Azabicyclo[3.2.1]octane-2-carboxylicacid, 3-(benzoyloxy)-8-methyl-, methyl ester, hydrochloride (1:1),(1R,2R,3S,5S)-](http://img.cochemist.com/ccimg/100/53-21-4.png)
![8-Azabicyclo[3.2.1]octane-2-carboxylicacid, 3-(benzoyloxy)-8-methyl-, methyl ester, hydrochloride (1:1),(1R,2R,3S,5S)-](http://img.cochemist.com/ccimg/100/53-21-4_b.png)














![Phosphoric acid, [4-(acetyloxy)phenyl]methyl diethyl ester](http://img.cochemist.com/ccimg/640300/640296-77-1.png)
![Phosphoric acid, [4-(acetyloxy)phenyl]methyl diethyl ester](http://img.cochemist.com/ccimg/640300/640296-77-1_b.png)
![[(2R,3S)-1-methyl-2-pyridin-3-ylpyrrolidin-3-yl]methanamine](http://img.cochemist.com/ccimg/623600/623579-03-3.png)
![[(2R,3S)-1-methyl-2-pyridin-3-ylpyrrolidin-3-yl]methanamine](http://img.cochemist.com/ccimg/623600/623579-03-3_b.png)
![[1,1'-Biphenyl]-4-carboxamide, 4'-[2-(2-furanyl)-6-phenyl-4-pyridinyl]-](http://img.cochemist.com/ccimg/581100/581074-60-4.png)
![[1,1'-Biphenyl]-4-carboxamide, 4'-[2-(2-furanyl)-6-phenyl-4-pyridinyl]-](http://img.cochemist.com/ccimg/581100/581074-60-4_b.png)
![Benzamide, 4-[2-(2-furanyl)-6-(4-nitrophenyl)-4-pyridinyl]-](http://img.cochemist.com/ccimg/581100/581073-80-5.png)
![Benzamide, 4-[2-(2-furanyl)-6-(4-nitrophenyl)-4-pyridinyl]-](http://img.cochemist.com/ccimg/581100/581073-80-5_b.png)
![2-Furancarboxamide, 5-[2-(2-furanyl)-6-(4-methoxyphenyl)-4-pyridinyl]-](http://img.cochemist.com/ccimg/581100/581073-21-4.png)
![2-Furancarboxamide, 5-[2-(2-furanyl)-6-(4-methoxyphenyl)-4-pyridinyl]-](http://img.cochemist.com/ccimg/581100/581073-21-4_b.png)
![2-Furancarboxamide, 5-[2-(2-furanyl)-6-(4-nitrophenyl)-4-pyridinyl]-](http://img.cochemist.com/ccimg/581100/581073-18-9.png)
![2-Furancarboxamide, 5-[2-(2-furanyl)-6-(4-nitrophenyl)-4-pyridinyl]-](http://img.cochemist.com/ccimg/581100/581073-18-9_b.png)


















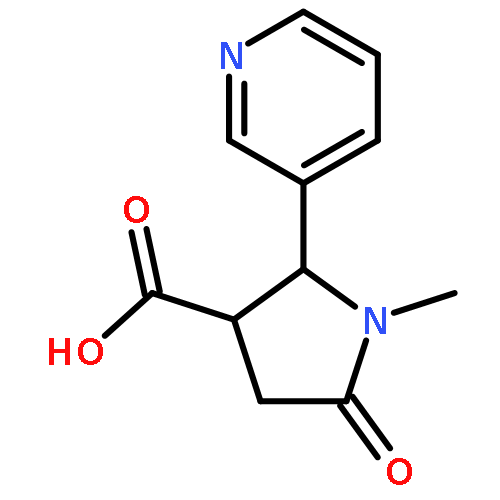






![Hexanoic acid, 6-[(methylsulfonyl)oxy]-, ethyl ester](http://img.cochemist.com/ccimg/124700/124668-93-5.png)
![Hexanoic acid, 6-[(methylsulfonyl)oxy]-, ethyl ester](http://img.cochemist.com/ccimg/124700/124668-93-5_b.png)
![PENTANAMIDE, N-PHENYL-N-[1-(2-PHENYLETHYL)-4-PIPERIDINYL]-](http://img.cochemist.com/ccimg/122900/122882-90-0.png)
![PENTANAMIDE, N-PHENYL-N-[1-(2-PHENYLETHYL)-4-PIPERIDINYL]-](http://img.cochemist.com/ccimg/122900/122882-90-0_b.png)







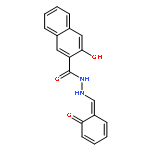





![[4-(HYDROXYMETHYL)PHENYL] 2-METHYLPROPANOATE](http://img.cochemist.com/ccimg/70400/70362-64-0.png)
![[4-(HYDROXYMETHYL)PHENYL] 2-METHYLPROPANOATE](http://img.cochemist.com/ccimg/70400/70362-64-0_b.png)


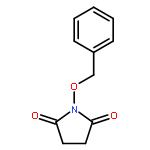







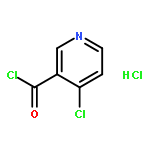
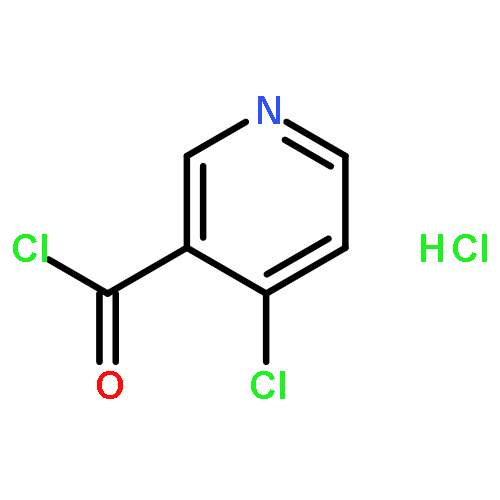
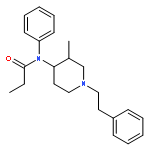


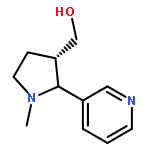







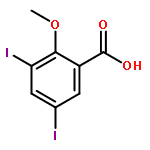

![1,9-dimethyl-9H-Pyrido[3,4-b]indole](http://img.cochemist.com/ccimg/16500/16498-64-9.png)
![1,9-dimethyl-9H-Pyrido[3,4-b]indole](http://img.cochemist.com/ccimg/16500/16498-64-9_b.png)


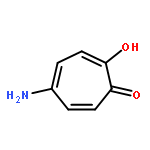


![[4-(hydroxymethyl)phenyl] Acetate](http://img.cochemist.com/ccimg/6400/6309-46-2.png)
![[4-(hydroxymethyl)phenyl] Acetate](http://img.cochemist.com/ccimg/6400/6309-46-2_b.png)
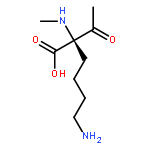











![3-Oxabicyclo[3.2.0]heptane-2,4-dione](http://img.cochemist.com/ccimg/4500/4462-96-8.png)
![3-Oxabicyclo[3.2.0]heptane-2,4-dione](http://img.cochemist.com/ccimg/4500/4462-96-8_b.png)


![Acetamide,N-phenyl-N-[1-(2-phenylethyl)-4-piperidinyl]-](http://img.cochemist.com/ccimg/3300/3258-84-2.png)
![Acetamide,N-phenyl-N-[1-(2-phenylethyl)-4-piperidinyl]-](http://img.cochemist.com/ccimg/3300/3258-84-2_b.png)


![PROPANAMIDE, N-(4-METHYLPHENYL)-N-[1-(2-PHENYLETHYL)-4-PIPERIDINYL]-](http://img.cochemist.com/ccimg/1900/1838-67-1.png)
![PROPANAMIDE, N-(4-METHYLPHENYL)-N-[1-(2-PHENYLETHYL)-4-PIPERIDINYL]-](http://img.cochemist.com/ccimg/1900/1838-67-1_b.png)
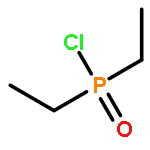



![9H-Pyrido[3,4-b]indol-7-ol,1-methyl-](http://img.cochemist.com/ccimg/500/487-03-6.png)
![9H-Pyrido[3,4-b]indol-7-ol,1-methyl-](http://img.cochemist.com/ccimg/500/487-03-6_b.png)
![9H-Pyrido[3,4-b]indole,1-methyl-](http://img.cochemist.com/ccimg/500/486-84-0.png)
![9H-Pyrido[3,4-b]indole,1-methyl-](http://img.cochemist.com/ccimg/500/486-84-0_b.png)


![N-phenyl-n-[1-(2-phenylethyl)piperidin-4-yl]propanamide](http://img.cochemist.com/ccimg/500/437-38-7.png)
![N-phenyl-n-[1-(2-phenylethyl)piperidin-4-yl]propanamide](http://img.cochemist.com/ccimg/500/437-38-7_b.png)
![4'-Chloro-[1,1'-biphenyl]-4-amine](http://img.cochemist.com/ccimg/200/135-68-2.png)
![4'-Chloro-[1,1'-biphenyl]-4-amine](http://img.cochemist.com/ccimg/200/135-68-2_b.png)




![Dextroamphetamine sulfate [USAN]](http://img.cochemist.com/ccimg/100/51-63-8.png)
![Dextroamphetamine sulfate [USAN]](http://img.cochemist.com/ccimg/100/51-63-8_b.png)
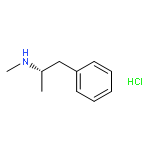


![D-Galactose, 2-[(2-azidoacetyl)amino]-2-deoxy-](/data/chemimg/115300/869186-83-4.png)
![D-Galactose, 2-[(2-azidoacetyl)amino]-2-deoxy-](/data/chemimg/115300/869186-83-4_b.png)
![D-Galactopyranose, 2-[(2-azidoacetyl)amino]-2-deoxy-, 1,3,4,6-tetraacetate](/data/chemimg/115200/653600-56-7.png)
![D-Galactopyranose, 2-[(2-azidoacetyl)amino]-2-deoxy-, 1,3,4,6-tetraacetate](/data/chemimg/115200/653600-56-7_b.png)








![Methyl (3s,4r)-3-benzoyloxy-8-methyl-8-azabicyclo[3.2.1]octane-4-carboxylate](http://img.cochemist.com/ccimg/100/50-36-2.png)
![Methyl (3s,4r)-3-benzoyloxy-8-methyl-8-azabicyclo[3.2.1]octane-4-carboxylate](http://img.cochemist.com/ccimg/100/50-36-2_b.png)

