Co-reporter:Roel Van Beeumen, David B. Williams-Young, Joseph M. Kasper, Chao Yang, Esmond G. Ng, and Xiaosong Li
Journal of Chemical Theory and Computation October 10, 2017 Volume 13(Issue 10) pp:4950-4950
Publication Date(Web):September 1, 2017
DOI:10.1021/acs.jctc.7b00402
The ab initio description of the spectral interior of the absorption spectrum poses both a theoretical and computational challenge for modern electronic structure theory. Due to the often spectrally dense character of this domain in the quantum propagator’s eigenspectrum for medium-to-large sized systems, traditional approaches based on the partial diagonalization of the propagator often encounter oscillatory and stagnating convergence. Electronic structure methods which solve the molecular response problem through the solution of spectrally shifted linear systems, such as the complex polarization propagator, offer an alternative approach which is agnostic to the underlying spectral density or domain location. This generality comes at a seemingly high computational cost associated with solving a large linear system for each spectral shift in some discretization of the spectral domain of interest. In this work, we present a novel, adaptive solution to this high computational overhead based on model order reduction techniques via interpolation. Model order reduction reduces the computational complexity of mathematical models and is ubiquitous in the simulation of dynamical systems and control theory. The efficiency and effectiveness of the proposed algorithm in the ab initio prediction of X-ray absorption spectra is demonstrated using a test set of challenging water clusters which are spectrally dense in the neighborhood of the oxygen K-edge. On the basis of a single, user defined tolerance we automatically determine the order of the reduced models and approximate the absorption spectrum up to the given tolerance. We also illustrate that, for the systems studied, the automatically determined model order increases logarithmically with the problem dimension, compared to a linear increase of the number of eigenvalues within the energy window. Furthermore, we observed that the computational cost of the proposed algorithm only scales quadratically with respect to the problem dimension.
Co-reporter:Hongbin Liu, Carl K. Brozek, Shichao Sun, David B. Lingerfelt, Daniel R. Gamelin, and Xiaosong Li
The Journal of Physical Chemistry C November 22, 2017 Volume 121(Issue 46) pp:26086-26086
Publication Date(Web):October 23, 2017
DOI:10.1021/acs.jpcc.7b08224
We present a general model for describing the properties of excess electrons in multiply charged quantum dots (QDs). Key factors governing Fermi-level energies and electron density distributions are investigated by treating carrier densities, charge compensation, and various material and dielectric medium properties as independently tunable parameters. Electronic interactions are described using a mean-field electrostatic potential calculable through Gauss’s Law by treating the quantum dot as a sphere of uniform charge density. This classical approximation modifies the “Particle in a Sphere” Schrödinger equation for a square well potential and reproduces the broken degeneracy and Fermi-level energies expected from experiment and first-principles methods. Several important implications emerge from this model: (i) excess electron density drifts substantially toward the QD surfaces with high electron densities and large radii and when solvated by a high dielectric medium. (ii) The maximum density of the conduction-band electrons depends strongly on the dielectric strength of the solvent and the electron affinity and dielectric strength of the QD material. (iii) Fermi-level energies stabilize with charge-balancing cations in close proximity to the QD surface.
Co-reporter:Greta Donati, Andrew Wildman, Stefano Caprasecca, David B. Lingerfelt, Filippo Lipparini, Benedetta Mennucci, and Xiaosong Li
The Journal of Physical Chemistry Letters November 2, 2017 Volume 8(Issue 21) pp:5283-5283
Publication Date(Web):October 10, 2017
DOI:10.1021/acs.jpclett.7b02320
Real-time time-dependent density functional theory (RT-TDDFT) is a powerful tool for obtaining spectroscopic observables and understanding complex, time-dependent properties. Currently, performing RT-TDDFT calculations on large, fully quantum mechanical systems is not computationally feasible. Previously, polarizable mixed quantum mechanical and molecular mechanical (QM/MMPol) models have been successful in providing accurate, yet efficient, approximations to a fully quantum mechanical system. Here we develop a coupling scheme between induced dipole based QM/MMPol and RT-TDDFT. Our approach is validated by comparing calculated spectra with both real-time and linear-response TDDFT calculations. The model developed within provides an accurate method for performing RT-TDDFT calculations on extended systems while accounting for mutual polarization between the quantum mechanical and molecular mechanical regions.
Co-reporter:Alessio Petrone, David B. Williams-Young, David B. Lingerfelt, and Xiaosong Li
The Journal of Physical Chemistry A May 25, 2017 Volume 121(Issue 20) pp:3958-3958
Publication Date(Web):May 3, 2017
DOI:10.1021/acs.jpca.7b02905
Time-resolved Raman spectroscopy has proven useful for studying the formation of polarons in conjugated polymers, verifying the presence of reactive intermediates in photochemical reactions, investigating nonradiative transitions in the short lifetime of the photoexcited species, and resolving electron–phonon coupling strengths and exciton dissociation in crystalline materials. In this paper, we present an excited state transient Raman analysis protocol combining ab initio direct molecular dynamics, transient excited state Hessian, and excited state nonresonant Raman activities evaluations. Prototypical molecules are used as test cases, showing the evolution of the transient Raman signatures that follow electronic excitation. This protocol provides a direct route to assigning the vibrations implicated in the (photo)dynamics of several (photoactive) systems, complementary to the transient infrared analysis.
Co-reporter:Franco Egidi, David B. Williams-Young, Alberto Baiardi, Julien Bloino, Giovanni Scalmani, Michael J. Frisch, Xiaosong Li, and Vincenzo Barone
Journal of Chemical Theory and Computation June 13, 2017 Volume 13(Issue 6) pp:2789-2789
Publication Date(Web):April 28, 2017
DOI:10.1021/acs.jctc.7b00218
We present a reliable and cost-effective procedure for the inclusion of anharmonic effects in excited-state energies and spectroscopic intensities by means of second-order vibrational perturbation theory. This development is made possible thanks to a recent efficient implementation of excited-state analytic Hessians and properties within the time-dependent density functional theory framework. As illustrated in this work, by taking advantage of such algorithmic developments, it is possible to perform calculations of excited-state infrared spectra of medium-large isolated molecular systems, with anharmonicity effects included in both the energy and property surfaces. We also explore the use of this procedure for the inclusion of anharmonic effects in the simulation of vibronic bandshapes of electronic spectra and compare the results with previous, more approximate models.
Co-reporter:David B. Lingerfelt, Patrick J. Lestrange, Joseph J. Radler, Samantha E. Brown-XuPyosang Kim, Felix N. Castellano, Lin X. ChenXiaosong Li
The Journal of Physical Chemistry A 2017 Volume 121(Issue 9) pp:
Publication Date(Web):February 16, 2017
DOI:10.1021/acs.jpca.6b12099
Materials and molecular systems exhibiting long-lived electronic coherence can facilitate coherent transport, opening the door to efficient charge and energy transport beyond traditional methods. Recently, signatures of a possible coherent, recurrent electronic motion were identified in femtosecond pump–probe spectroscopy experiments on a binuclear platinum complex, where a persistent periodic beating in the transient absorption signal’s anisotropy was observed. In this study, we investigate the excitonic dynamics that underlie the suspected electronic coherence for a series of binuclear platinum complexes exhibiting a range of interplatinum distances. Results suggest that the long-lived coherence can only result when competitive electronic couplings are in balance. At longer Pt–Pt distances, the electronic couplings between the two halves of the binuclear system weaken, and exciton localization and recombination is favored on short time scales. For short Pt–Pt distances, electronic couplings between the states in the coherent superposition are stronger than the coupling with other excitonic states, leading to long-lived coherence.
Co-reporter:David Williams-Young, Joshua J. Goings, and Xiaosong Li
Journal of Chemical Theory and Computation 2016 Volume 12(Issue 11) pp:5333-5338
Publication Date(Web):October 17, 2016
DOI:10.1021/acs.jctc.6b00693
Solutions of the real-time time-dependent density functional theory (RT-TDDFT) equations provide an affordable route to understanding the electronic dynamics that underpins many spectroscopic techniques. From the solutions of the RT-TDDFT equations, it is possible to extract optical absorption and circular dichroism spectra, as well as descriptions of charge transfer and charge transport dynamics. In order to apply RT-TDDFT to increasingly large systems, it is necessary to develop methods to overcome computational bottlenecks. One current bottleneck is the cost required to form the time propagator for the RT-TDDFT equations, because of the full matrix diagonalization that is required at each time step. Here, we present a (semi)diagonalization-free formation of the propagator based on a nonrecursive Chebyshev polynomial expansion. The Chebyshev expansion relies only on matrix multiply operations which have lower computational cost and are furthermore extremely parallelizable. We demonstrate the accuracy and stability of the Chebyshev approach, and then discuss the favorable scaling of the method, compared to traditional approaches based on matrix diagonalization. The Chebyshev expansion method should enable the application of RT-TDDFT methods to large systems such as nanocrystals and biomolecules.
Co-reporter:David Williams-Young, Franco Egidi, and Xiaosong Li
Journal of Chemical Theory and Computation 2016 Volume 12(Issue 11) pp:5379-5384
Publication Date(Web):September 26, 2016
DOI:10.1021/acs.jctc.6b00833
With the recent introduction of the particle–particle random-phase and Tamm–Dancoff approximations to ab initio theory, routine queries of traditionally difficult systems, such as diradicals and doubly excited states, have been made possible. However, although a wealth of inquiry has been directed to investigating these methods, the current formulations have been restricted to spin-collinear systems, leaving the methods incapable of treating noncollinearity and spin–orbit relativistic effects in excited states. In this work, we extend the particle–particle Tamm–Dancoff approximation to suit two-component Hamiltonians to explicitly treat relativistic effects in excited states. After reviewing the theory and computational implementation, we demonstrate the accuracy of this extension by evaluating the fine structure splittings some of atomic and molecular systems.
Co-reporter:Franco Egidi, Joshua J. Goings, Michael J. Frisch, and Xiaosong Li
Journal of Chemical Theory and Computation 2016 Volume 12(Issue 8) pp:3711-3718
Publication Date(Web):July 7, 2016
DOI:10.1021/acs.jctc.6b00474
In this work, we present a linear-response formalism of the complex two-component Hartree–Fock Hamiltonian that includes relativistic effects within the Douglas–Kroll–Hess and the Exact-Two-Component frameworks. The method includes both scalar and spin relativistic effects in the variational description of electronic ground and excited states, although it neglects the picture-change and explicit spin–orbit contributions arising from the two-electron interaction. An efficient direct formalism of solving the complex two-component response function is also presented in this work. The presence of spin–orbit couplings in the Hamiltonian and the two-component nature of the wave function and Fock operator allows the computation of excited-state zero-field splittings of systems for which relativistic effects are dominated by the one-electron term. Calculated results are compared to experimental reference values to assess the quality of the underlying approximations. The results show that the relativistic two-component linear response methods are able to capture the excited-state zero-field splittings with good agreement with experiments for the systems considered here, with all approximations exhibiting a similar performance. However, the error increases for heavy elements and for states of high orbital angular momentum, suggesting the importance of the two-electron relativistic effect in such situations.
Co-reporter:David B. Lingerfelt, David B. Williams-Young, Alessio Petrone, and Xiaosong Li
Journal of Chemical Theory and Computation 2016 Volume 12(Issue 3) pp:935-945
Publication Date(Web):February 8, 2016
DOI:10.1021/acs.jctc.5b00697
Tractable methods for studying the molecular dynamics of chemical processes driven by electronic nonadiabaticity are highly sought after to provide insight into, for example, photochemical reaction mechanisms, molecular collisions, and thermalized electronic band structures. Starting from the time-dependent Schrödinger equation for a many-body system, a direct ab initio trajectory surface-hopping (TSH) method relying on an analytical treatment of nonadiabatic couplings between electronic states is developed in this work. An approach that combines time-dependent perturbation theory and explicit time evolution via TSH to expedite calculation of nonadiabatic transition rates, namely, meta-surface-hopping dynamics, is presented, and an extrapolatory approach using time-dependent perturbation theory for recovering unbiased transition rates is assessed. The meta-surface-hopping method is applied to the problem of estimating nonradiative relaxation rates of a photoexcited iminium ion, CH2NH2+, and evidence for internal consistency of the combined dynamics/perturbation theory approach is presented.
Co-reporter:Joshua J. Goings, David B. Lingerfelt, and Xiaosong Li
The Journal of Physical Chemistry Letters 2016 Volume 7(Issue 24) pp:5193-5197
Publication Date(Web):December 1, 2016
DOI:10.1021/acs.jpclett.6b02424
We explore the question of whether mean-field or “Ehrenfest” mixed quantum-classical dynamics is capable of capturing the quantized vibrational features in photoabsorption spectra that result from infrared and Raman-active vibrational transitions. We show that vibrational and electronic absorption spectra can indeed be obtained together within a single Ehrenfest simulation. Furthermore, the electronic transitions show new sidebands that are absent in electronic dynamics simulations with fixed nuclei. Inspection of the electronic sidebands reveals that the spacing corresponds to vibrational frequencies of totally symmetric vibrational modes of the ground electronic state. A simple derivation of the time-evolving dipole in the presence of external fields and vibrational motion shows the origin of these features, demonstrating that mixed quantum-classical Ehrenfest dynamics is capable of producing infrared, Raman, and electronic absorption spectra from a single simulation.
Co-reporter:Alessio Petrone, David B. Lingerfelt, David B. Williams-Young, and Xiaosong Li
The Journal of Physical Chemistry Letters 2016 Volume 7(Issue 22) pp:4501-4508
Publication Date(Web):October 27, 2016
DOI:10.1021/acs.jpclett.6b02292
Pump probe spectroscopy techniques have enabled the direct observation of a variety of transient molecular species in both ground and excited electronic states. Time-resolved vibrational spectroscopy is becoming an indispensable tool for investigating photoinduced nuclear dynamics of chemical systems of all kinds. On the other hand, a complete picture of the chemical dynamics encoded in these spectra cannot be achieved without a full temporal description of the structural relaxation, including the explicit time-dependence of vibrational coordinates that are substantially displaced from equilibrium by electronic excitation. Here we present a transient vibrational analysis protocol combining ab initio direct molecular dynamics and time-integrated normal modes introduced in this work, relying on the recent development of analytic time-dependent density functional theory (TDDFT) second derivatives for excited states. Prototypical molecules will be used as test cases, showing the evolution of the vibrational signatures that follow electronic excitation. This protocol provides a direct route to assigning the vibrations implicated in the (photo)dynamics of several (photoactive) systems.
Co-reporter:Erica Q. Chong, David B. Lingerfelt, Alessio Petrone, and Xiaosong Li
The Journal of Physical Chemistry C 2016 Volume 120(Issue 34) pp:19434-19441
Publication Date(Web):August 16, 2016
DOI:10.1021/acs.jpcc.6b05883
Ion diffusion in semiconductor nanocrystals, or quantum dots (QDs), has gained recognition in recent years as a crucial process for advancing both energy storage and, more generally, the postsynthetic p-type doping chemistry of these materials. In this report, we present first an energetic analysis of group I cations (H+, Li+, and Na+) diffusion in (MX)84– QDs, with M = Zn, Cd and X = S, Se. The bound solutions to the corresponding one-dimensional nuclear Schrödinger equation were solved for these systems, relying on the discrete variable representation method. From this vantage, the quantum nature of the intercalating ion can be revealed. Evidence for the importance of including quantum effects in the treatment of these diffusion processes is presented, both with the density of energy eigenstates of the intercalating ion and from a comparison of the standard deviation in the population distribution of the intercalating ion to the lattice spacings of its host material. Results suggest that the use of classical mechanics for simulations of the ion diffusion processes in these and other related materials can be a questionable practice for the smallest group I cations. Trends devised herein can be useful to help guide the development of new experimental approaches to postsynthetic doping of semiconductor nanocrystals, and in designing electrode materials for next generation electrochemical energy storage devices.
Co-reporter:Greta Donati, David B. Lingerfelt, Alessio Petrone, Nadia Rega, and Xiaosong Li
The Journal of Physical Chemistry A 2016 Volume 120(Issue 37) pp:7255-7261
Publication Date(Web):August 29, 2016
DOI:10.1021/acs.jpca.6b06419
The formation of polaron pairs is one of the important photophysical processes that take place after the excitation in semiconducting organic polymers. First-principles Ehrenfest excited-state dynamics is a unique tool to investigate ultrafast photoinduced charge carrier dynamics and related nonequilibrium processes involving correlated electron–nuclear dynamics. In this work the formation of polaron pairs and their dynamical evolution in an oligomer of seven thiophene units is investigated with a combined approach of first-principles exciton-nuclear dynamics and wavelet analysis. The real-time formation of a polaron pair can be observed in the dipole evolution during the excited-state dynamics. The possible driving force of the polaron pair formation is investigated through qualitative correlation between the structural dynamics and the dipole evolution. The time-dependent characteristics and spectroscopic consequences of the polaron pair formation are probed using the wavelet analysis.
Co-reporter:Lea Nienhaus; Joshua J. Goings; Duc Nguyen; Sarah Wieghold; Joseph W. Lyding; Xiaosong Li;Martin Gruebele
Journal of the American Chemical Society 2015 Volume 137(Issue 46) pp:14743-14750
Publication Date(Web):October 30, 2015
DOI:10.1021/jacs.5b09272
Electronically excited orbitals play a fundamental role in chemical reactivity and spectroscopy. In nanostructures, orbital shape is diagnostic of defects that control blinking, surface carrier dynamics, and other important optoelectronic properties. We capture nanometer resolution images of electronically excited PbS quantum dots (QDs) by single molecule absorption scanning tunneling microscopy (SMA-STM). Dots with a bandgap of ∼1 eV are deposited on a transparent gold surface and optically excited with red or green light to produce hot carriers. The STM tip-enhanced laser light produces a large excited-state population, and the Stark effect allows transitions to be tuned into resonance by changing the sample voltage. Scanning the QDs under laser excitation, we were able to image electronic excitation to different angular momentum states depending on sample bias. The shapes differ from idealized S- or P-like orbitals due to imperfections of the QDs. Excitation of adjacent QD pairs reveals orbital alignment, evidence for electronic coupling between dots. Electronic structure modeling of a small PbS QD, when scaled for size, reveals Stark tuning and variation in the transition moment of different parity states, supporting the simple one-electron experimental interpretation in the hot carrier limit. The calculations highlight the sensitivity of orbital density to applied field, laser wavelength, and structural fluctuations of the QD.
Co-reporter:Patrick J. Lestrange, Phu D. Nguyen, and Xiaosong Li
Journal of Chemical Theory and Computation 2015 Volume 11(Issue 7) pp:2994-2999
Publication Date(Web):June 4, 2015
DOI:10.1021/acs.jctc.5b00169
X-ray absorption spectroscopy (XAS) has become a powerful technique in chemical physics, because of advances in synchrotron technology that have greatly improved its temporal and spectroscopic resolution. Our recent work on energy-specific time-dependent density functional theory (ES-TDDFT) allows for the direct calculation of excitation energies in any region of the absorption spectrum, from UV-vis to X-ray. However, the ability of different density functional theories to model X-ray absorption spectra (XAS) of light elements has not yet been verified for ES-TDDFT. This work is a calibration of the ability of existing DFT kernels and basis sets to reproduce experimental K-edge excitation energies. Results were compared against 30 different transitions from gas-phase experiments. We focus on six commonly used density functionals (BHandHLYP, B3LYP, PBE1PBE, BP86, HSE06, LC-ωPBE) and various triple-ζ basis sets. The effects of core and diffuse functions are also investigated.
Co-reporter:Bo Peng, Patrick J. Lestrange, Joshua J. Goings, Marco Caricato, and Xiaosong Li
Journal of Chemical Theory and Computation 2015 Volume 11(Issue 9) pp:4146-4153
Publication Date(Web):August 6, 2015
DOI:10.1021/acs.jctc.5b00459
Single-reference techniques based on coupled-cluster (CC) theory, in the forms of linear response (LR) or equation of motion (EOM), are highly accurate and widely used approaches for modeling valence absorption spectra. Unfortunately, these equations with singles and doubles (LR-CCSD and EOM-CCSD) scale as , which may be prohibitively expensive for the study of high-energy excited states using a conventional eigensolver. In this paper, we present an energy-specific non-Hermitian eigensolver that is able to obtain high-energy excited states (e.g., XAS K-edge spectrum) at low computational cost. In addition, we also introduce an improved trial vector for iteratively solving the EOM-CCSD equation with a focus on high-energy eigenstates. The energy-specific EOM-CCSD approach and its low-scaling alternatives are applied to calculations of carbon, nitrogen, oxygen, and sulfur K-edge excitations. The results are compared to other implementations of CCSD for excited states, energy-specific linear response time-dependent density functional theory (TDDFT), and experimental results with multiple statistical metrics are presented and evaluated.
.jpg)







![2,1,3-Benzothiadiazole, 4-bromo-5,6-difluoro-7-(5'-hexyl[2,2'-bithiophen]-5-yl)-](/data/chemimg/3782300/1774364-87-2.png)
![2,1,3-Benzothiadiazole, 4-bromo-5,6-difluoro-7-(5'-hexyl[2,2'-bithiophen]-5-yl)-](/data/chemimg/3782300/1774364-87-2_b.png)
![Stannane, tributyl(6-octylthieno[3,2-b]thien-2-yl)-](/data/chemimg/3782400/1623751-72-3.png)
![Stannane, tributyl(6-octylthieno[3,2-b]thien-2-yl)-](/data/chemimg/3782400/1623751-72-3_b.png)
![2,1,3-Benzothiadiazole, 4-bromo-5-fluoro-7-(5'-hexyl[2,2'-bithiophen]-5-yl)-](/data/chemimg/2259900/1402460-83-6.png)
![2,1,3-Benzothiadiazole, 4-bromo-5-fluoro-7-(5'-hexyl[2,2'-bithiophen]-5-yl)-](/data/chemimg/2259900/1402460-83-6_b.png)








![Poly[2,6-(4,4-bis-(2-ethylhexyl)-4H-cyclopenta[2,1-b;3,4-b']dithiophene)-alt-4,7(2,1,3-benzothiadiazole)]](/data/chemimg/102300/920515-34-0.png)
![Poly[2,6-(4,4-bis-(2-ethylhexyl)-4H-cyclopenta[2,1-b;3,4-b']dithiophene)-alt-4,7(2,1,3-benzothiadiazole)]](/data/chemimg/102300/920515-34-0_b.png)
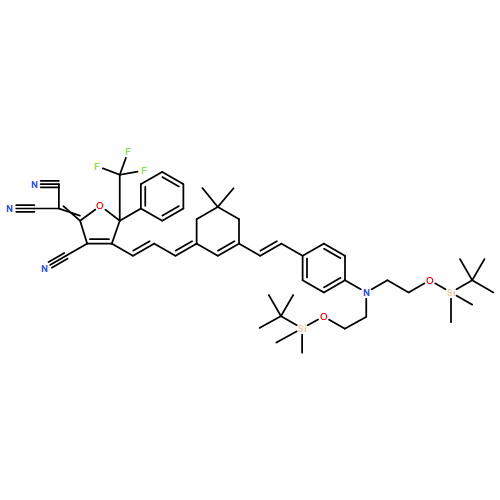



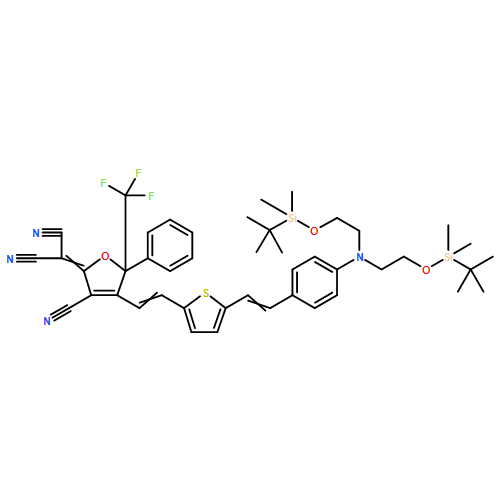

![Propanedinitrile,2-[4-[(1E,3E)-3-[3-[(1E)-2-[4-[bis[2-[[(1,1-dimethylethyl)dimethylsilyl]oxy]ethyl]amino]phenyl]ethenyl]-5,5-dimethyl-2-cyclohexen-1-ylidene]-1-propen-1-yl]-3-cyano-5,5-dimethyl-2(5H)-furanylidene]-](http://img.cochemist.com/ccimg/368900/368874-13-9.png)
![Propanedinitrile,2-[4-[(1E,3E)-3-[3-[(1E)-2-[4-[bis[2-[[(1,1-dimethylethyl)dimethylsilyl]oxy]ethyl]amino]phenyl]ethenyl]-5,5-dimethyl-2-cyclohexen-1-ylidene]-1-propen-1-yl]-3-cyano-5,5-dimethyl-2(5H)-furanylidene]-](http://img.cochemist.com/ccimg/368900/368874-13-9_b.png)
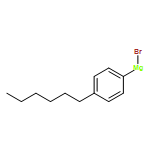



















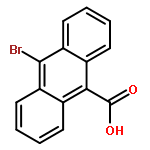
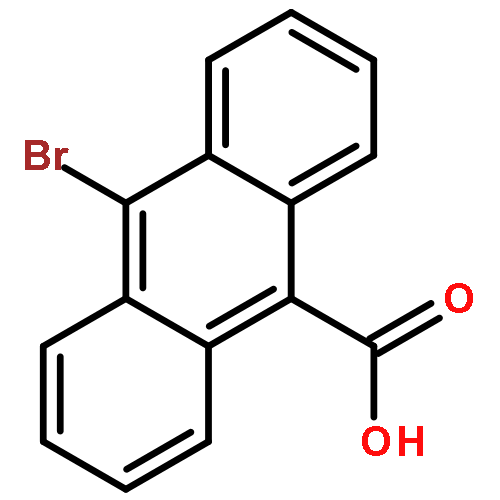







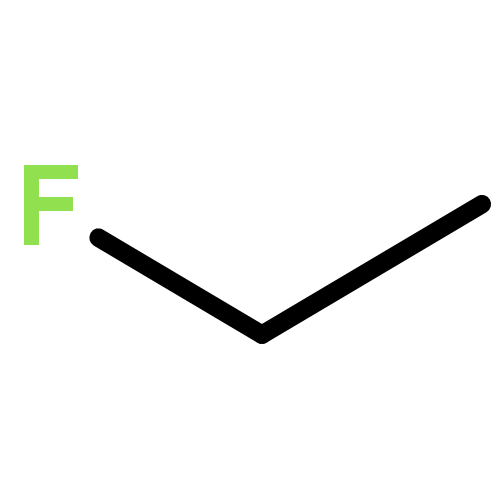


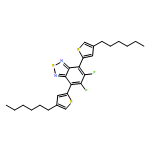
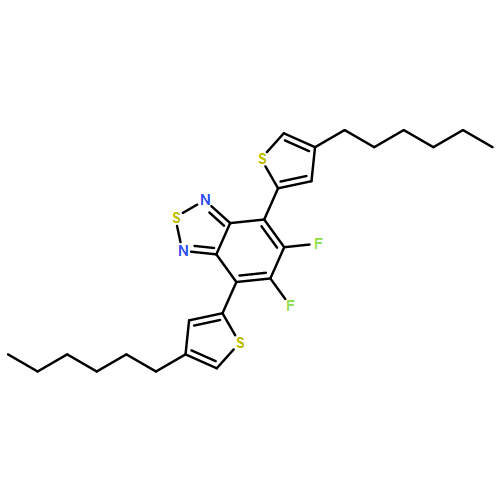
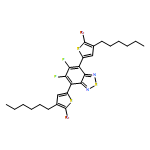
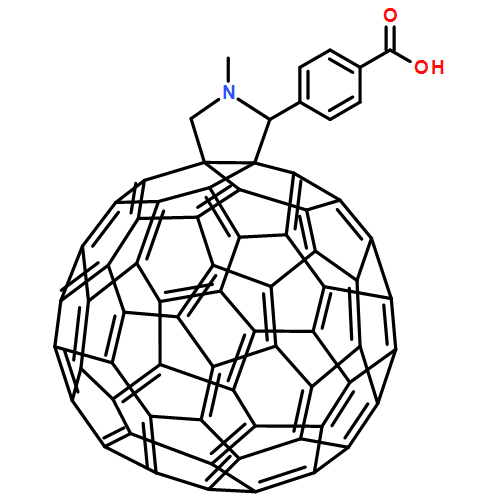
![Dithieno[2,3-d:2',3'-d']-s-indaceno[1,2-b:5,6-b']dithiophene, 6,6,12,12-tetrakis(4-hexylphenyl)-6,12-dihydro-](/data/chemimg/3782000/1420071-64-2.png)
![Dithieno[2,3-d:2',3'-d']-s-indaceno[1,2-b:5,6-b']dithiophene, 6,6,12,12-tetrakis(4-hexylphenyl)-6,12-dihydro-](/data/chemimg/3782000/1420071-64-2_b.png)