Co-reporter:Kelli A. Ogawa; Adam E. Goetz
Journal of the American Chemical Society 2015 Volume 137(Issue 4) pp:1400-1403
Publication Date(Web):January 8, 2015
DOI:10.1021/ja512073m
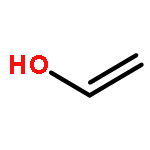
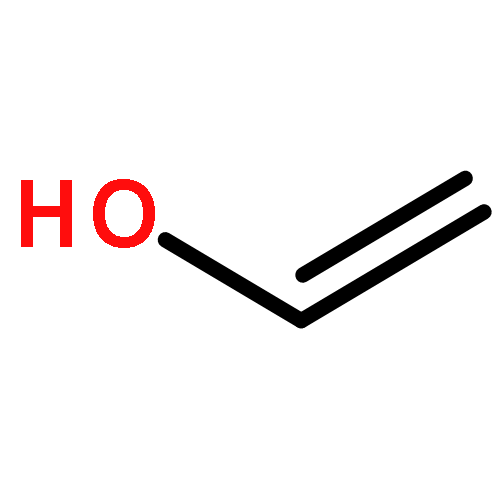
We have developed a method to achieve ring-opening metathesis polymerization (ROMP) mediated by oxidation of organic initiators in the absence of any transition metals. Radical cations, generated via one-electron oxidation of vinyl ethers, were found to react with norbornene to give polymeric species with microstructures essentially identical to those traditionally obtained via metal-mediated ROMP. We found that vinyl ether oxidation could be accomplished under mild conditions using an organic photoredox mediator. This led to high yields of polymer and generally good correlation between Mn values and initial monomer to catalyst loadings. Moreover, temporal control over reinitiation of polymer growth was achieved during on/off cycles of light exposure. This method demonstrates the first metal-free method for controlled ROMP.
Co-reporter:Adam E. Goetz
Journal of the American Chemical Society 2015 Volume 137(Issue 24) pp:7572-7575
Publication Date(Web):June 8, 2015
DOI:10.1021/jacs.5b03665
Metal-free ring-opening metathesis polymerization (ROMP) utilizes organic photoredox mediators as alternatives to traditional metal-based ROMP initiators to allow the preparation of polymers without residual metal contamination. Herein we report studies exploring the use of endo-dicyclopentadiene (DCPD), a common ROMP monomer, to form linear polyDCPD and copolymers with norbornene. Subsequent cross-linking of the linear polyDCPD using thiol–ene chemistry allows for a completely metal-free preparation of cross-linked polyDCPD. Furthermore, the examination of a number of structurally related monomers offers insights into mechanistic details of this polymerization and demonstrates new monomers that can be utilized for metal-free ROMP.
Co-reporter:Gregory I. Peterson, Michael B. Larsen, Mark A. Ganter, Duane W. Storti, and Andrew J. Boydston
ACS Applied Materials & Interfaces 2015 Volume 7(Issue 1) pp:577
Publication Date(Web):December 5, 2014
DOI:10.1021/am506745m
We describe the preparation and characterization of photo- and mechanochromic 3D-printed structures using a commercial fused filament fabrication printer. Three spiropyran-containing poly(ε-caprolactone) (PCL) polymers were each filamentized and used to print single- and multicomponent tensile testing specimens that would be difficult, if not impossible, to prepare using traditional manufacturing techniques. It was determined that the filament production and printing process did not degrade the spiropyran units or polymer chains and that the mechanical properties of the specimens prepared with the custom filament were in good agreement with those from commercial PCL filament. In addition to printing photochromic and dual photo- and mechanochromic PCL materials, we also prepare PCL containing a spiropyran unit that is selectively activated by mechanical impetus. Multicomponent specimens containing two different responsive spiropyrans enabled selective activation of different regions within the specimen depending on the stimulus applied to the material. By taking advantage of the unique capabilities of 3D printing, we also demonstrate rapid modification of a prototype force sensor that enables the assessment of peak load by simple visual assessment of mechanochromism.Keywords: additive manufacturing; mechanochemistry; polymers; responsive materials
Co-reporter:Michael B. Larsen
Journal of the American Chemical Society 2014 Volume 136(Issue 4) pp:1276-1279
Publication Date(Web):January 13, 2014
DOI:10.1021/ja411891x
We have developed a mechanochemically responsive material capable of successively releasing small organic molecules from a cross-linked network upon repeated compressions. The use of a flex activated mechanophore that does not lead to main chain scission and an elastomeric polyurethane enabled consecutive compressions with incremental increases in the % mechanophore activation. Additionally, we examined the effect of multiple applications of compressive stress on both mechanophore activity and the mechanical behavior of the elastomeric matrix in which the mechanophore is embedded.
Co-reporter:Kelli A. Ogawa and Andrew J. Boydston
Organic Letters 2014 Volume 16(Issue 7) pp:1928-1931
Publication Date(Web):March 14, 2014
DOI:10.1021/ol500459x
A method has been developed for the direct conversion of aldehydes to thioesters via integration of organocatalysis and electrosynthesis. The thiazolium precatalyst was found to facilitate oxidation of thiolate anions, leading to deleterious formation of disulfide byproducts. By circumventing this competing reaction, thioesters were obtained in good-to-excellent yields for a broad range of aldehyde and thiol substrates. This approach provides an atom-efficient thioesterification that circumvents the need for stoichiometric exogenous oxidants, high cell potentials, or redox mediators.
Co-reporter:Derek C. Church, Gregory I. Peterson, and Andrew J. Boydston
ACS Macro Letters 2014 Volume 3(Issue 7) pp:648
Publication Date(Web):June 17, 2014
DOI:10.1021/mz5003068
The effect of star versus linear polymer architecture on the rates of mechanochemically induced bond scission has been explored. We determined rate constants for chain scission of parent linear and star polymers, from which daughter fragments were cleanly resolved. These studies confirm a mechanistic interpretation of star polymer chain scission that is governed by the spanning rather than total molecular weight. We further demonstrate the preserved rate of site-selective mechanophore activation across two different polymer structures. Specifically, we observed consistent activation rate constants from three-arm star and linear polymer analogues, despite the Mn of the star polymer being 1.5 times greater than that of the linear system.
Co-reporter:Gregory I. Peterson
Macromolecular Rapid Communications 2014 Volume 35( Issue 18) pp:1611-1614
Publication Date(Web):
DOI:10.1002/marc.201400271
Co-reporter:Gregory I. Peterson
Macromolecular Theory and Simulations 2014 Volume 23( Issue 9) pp:
Publication Date(Web):
DOI:10.1002/mats.201470026
Co-reporter:Gregory I. Peterson
Macromolecular Theory and Simulations 2014 Volume 23( Issue 9) pp:555-563
Publication Date(Web):
DOI:10.1002/mats.201400045
A model for predicting the molecular weight distributions of mechanochemically degraded star polymers has been developed. The model was shown to be in good agreement with experimental distributions and average total molecular weights obtained from ultrasonically degraded three-arm star poly(methyl acrylate)s. Generalization of the model to four- and n-arm star polymers was also achieved. The models are straightforward to use, and thus, all calculations were completed in Microsoft Excel.
Co-reporter:Gregory I. Peterson, Derek C. Church, Neal A. Yakelis, Andrew J. Boydston
Polymer 2014 Volume 55(Issue 23) pp:5980-5985
Publication Date(Web):5 November 2014
DOI:10.1016/j.polymer.2014.09.048
•A 1,2-oxazine thermal trigger for self-immolative polymers has been demonstrated.•The oxazine is a Diels Alder adduct between carbamoylnitroso and diene moieties.•The temperature dependence of activation and depolymerization were explored.•Thermal cycloreversion appeared to dominate the triggering pathway up to 60 °C.•The triggering mechanism was supported by diene transfer experiments.We have demonstrated the site-specific thermal activation of self-immolative polymers (SIPs) using a bicyclic oxazine as a temperature-sensitive triggering moiety. The oxazine-based trigger was installed at the junction of a SIP-poly(N,N-dimethylacrylamide) (PDMA) diblock copolymer via oxidation of hydroxyurea end groups on the SIP and in situ [4 + 2] cycloaddition with cyclopentadiene-functionalized PDMA. The trigger undergoes a thermally-driven cycloreversion which ultimately leads to initiation of the depolymerization process. The temperature dependence of activation and depolymerization were investigated, along with the mechanism of activation. The relative rates of depolymerization at different temperatures suggested to us that the thermal trigger design may be a good candidate for on-demand activation of SIPs with minimal background triggering.
Co-reporter:Michael B. Larsen
Journal of the American Chemical Society 2013 Volume 135(Issue 22) pp:8189-8192
Publication Date(Web):May 20, 2013
DOI:10.1021/ja403757p
We describe studies in mechanochemical transduction that probe the activation of bonds orthogonal to an elongated polymer main chain. Compression of mechanophore-cross-linked materials resulted in the release of small molecules via cleavage of covalent bonds that were not integral components of the elongated polymer segments. The reactivity is proposed to arise from the distribution of force through the cross-linking units of the polymer network and subsequent bond bending motions that are consistent with the geometric changes in the overall reaction. This departure from contemporary polymer mechanochemistry, in which activation is achieved primarily by force-induced bond elongation, is a first step toward mechanophores capable of releasing side-chain functionalities without inherently compromising the overall macromolecular architecture.
Co-reporter:Eric E. Finney ; Kelli A. Ogawa
Journal of the American Chemical Society 2012 Volume 134(Issue 30) pp:12374-12377
Publication Date(Web):July 6, 2012
DOI:10.1021/ja304716r
A method for the catalytic formation of electroauxiliaries and subsequent anodic oxidation has been developed. The process interfaces N-heterocyclic carbene-based organocatalysis with electro-organic synthesis to achieve direct oxidation of catalytically generated electroactive intermediates. We demonstrate the applicability of this method as a one-pot conversion of aldehydes to esters for a broad range of aldehyde and alcohol substrates. Furthermore, the anodic oxidation reactions are very clean, producing only H2 gas as a result of cathodic reduction.
Co-reporter:Gregory I. Peterson, Michael B. Larsen, and Andrew J. Boydston
Macromolecules 2012 Volume 45(Issue 18) pp:7317-7328
Publication Date(Web):August 10, 2012
DOI:10.1021/ma300817v
Self-immolative polymers (SIPs) are unique macromolecules that are able to react to multiple types of environmental influences by giving amplified response outputs. When triggering moieties installed at SIP chain ends are activated by their corresponding stimuli, a spontaneous head-to-tail depolymerization ensues, often involving multitopic release of small molecules. SIP designs have evolved a high degree of modularity in each of their functional components, enabling a broad range of utility and applications-driven tuning. In this Perspective, we summarize and discuss recent progress in this nascent area of research, including (i) synthesis of different types of SIPs, (ii) design and evaluation of triggering moieties, (iii) depolymerization mechanisms and kinetics, (iv) applications of SIPs, and (v) outlook and challenges facing the field.
Co-reporter:Nebojša Momčilović ; Paul G. Clark ; Andrew J. Boydston ;Robert H. Grubbs
Journal of the American Chemical Society 2011 Volume 133(Issue 47) pp:19087-19089
Publication Date(Web):October 24, 2011
DOI:10.1021/ja208515r
A one-pot synthesis of polyrotaxanes has been developed. The method employs a supramolecular monomer comprising a polymerizable ammonium salt and crown ether, in combination with dynamic ADMet polymerization. Ultimately, highly efficient complexation, polymerization, and end-capping were accomplished in a single operation to yield polyrotaxanes with Mw up to 19.3 kDa and >80% of the repeat units being complexed.
-Washington Seattle(3).jpg)






![Silane, (bicyclo[2.2.1]hept-5-en-2-ylmethoxy)triethyl-](http://img.cochemist.com/ccimg/303100/303070-43-1.png)
![Silane, (bicyclo[2.2.1]hept-5-en-2-ylmethoxy)triethyl-](http://img.cochemist.com/ccimg/303100/303070-43-1_b.png)
![Cyclohexane, [(1Z)-2-methoxyethenyl]-](http://img.cochemist.com/ccimg/80500/80434-61-3.png)
![Cyclohexane, [(1Z)-2-methoxyethenyl]-](http://img.cochemist.com/ccimg/80500/80434-61-3_b.png)




![Oxazolo[3,2-a]indole, 2,3,9,9a-tetrahydro-9,9,9a-trimethyl-](/data/chemimg/560400/36508-00-6.png)
![Oxazolo[3,2-a]indole, 2,3,9,9a-tetrahydro-9,9,9a-trimethyl-](/data/chemimg/560400/36508-00-6_b.png)
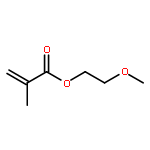
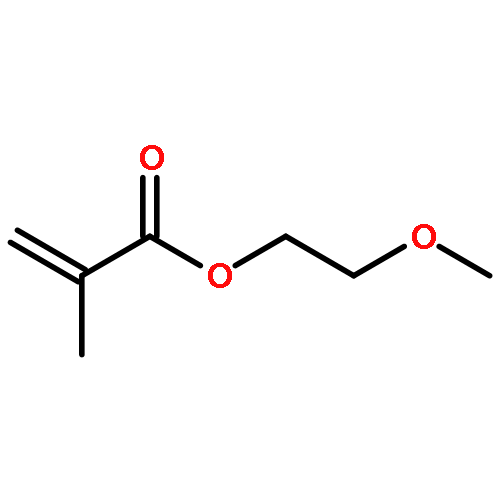


![Bicyclo[2.2.1]hept-5-ene-2-methanol,a,a-dimethyl-](http://img.cochemist.com/ccimg/22500/22497-08-1.png)
![Bicyclo[2.2.1]hept-5-ene-2-methanol,a,a-dimethyl-](http://img.cochemist.com/ccimg/22500/22497-08-1_b.png)
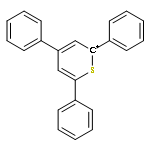
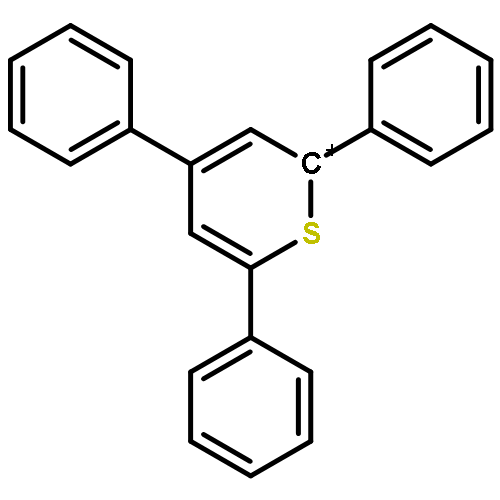


![Bicyclo[2.2.1]hept-2-ene, 5-(methoxymethyl)-](http://img.cochemist.com/ccimg/10500/10471-23-5.png)
![Bicyclo[2.2.1]hept-2-ene, 5-(methoxymethyl)-](http://img.cochemist.com/ccimg/10500/10471-23-5_b.png)




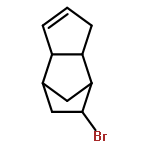
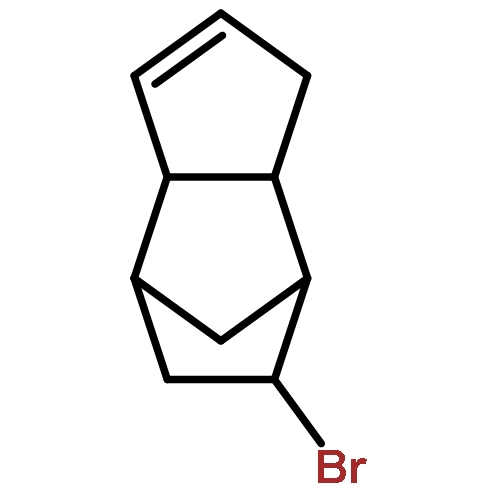






![Poly[(octahydro-1,3-pentalenediyl)-1,2-ethanediyl]](http://img.cochemist.com/ccimg/171200/171188-08-2.png)
![Poly[(octahydro-1,3-pentalenediyl)-1,2-ethanediyl]](http://img.cochemist.com/ccimg/171200/171188-08-2_b.png)
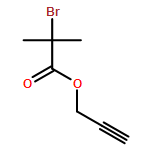









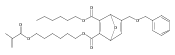
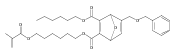

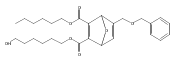
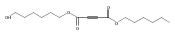
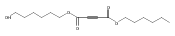



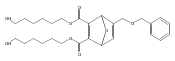
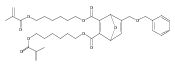
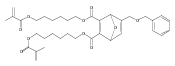
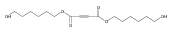
















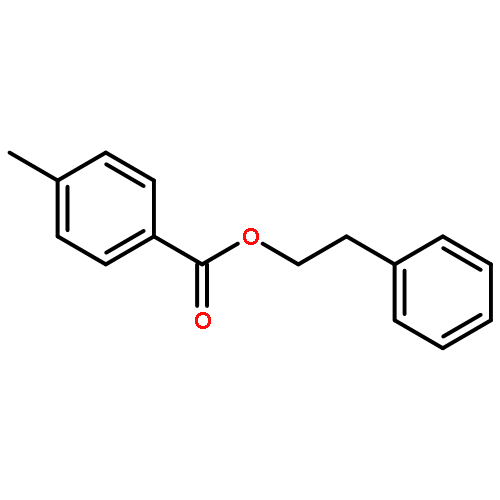


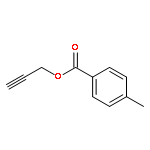
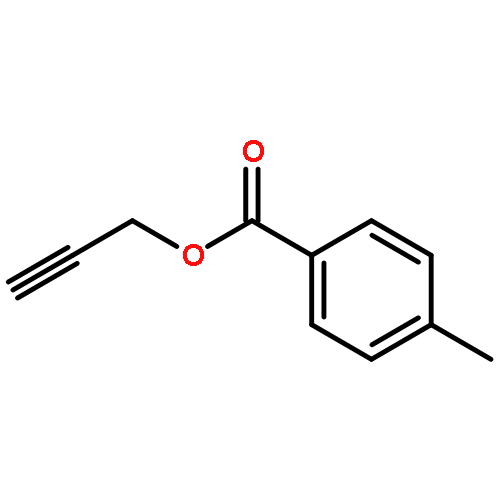




![Furan, 3-[(phenylmethoxy)methyl]-](http://img.cochemist.com/ccimg/89900/89858-76-4.png)
![Furan, 3-[(phenylmethoxy)methyl]-](http://img.cochemist.com/ccimg/89900/89858-76-4_b.png)