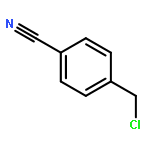
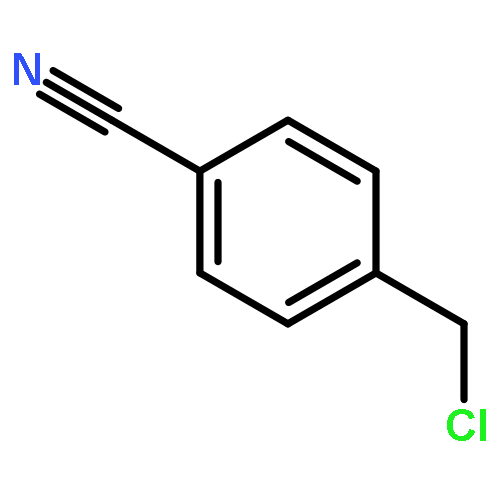
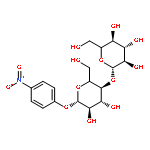
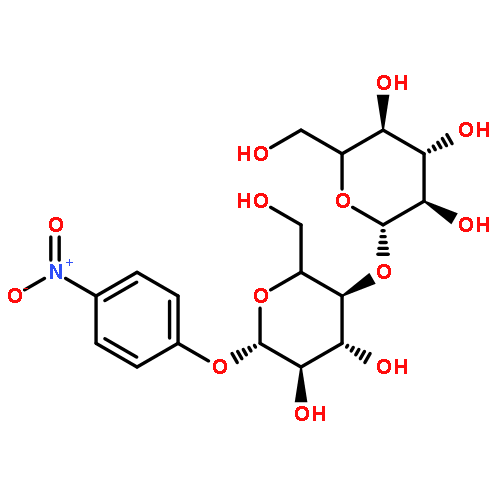
Co-reporter:Xukai JiangWen Li, Guanjun Chen, Lushan Wang
Journal of Chemical Information and Modeling 2017 Volume 57(Issue 2) pp:
Publication Date(Web):February 1, 2017
DOI:10.1021/acs.jcim.6b00692
The temperature dependence of enzyme catalysis is highly debated. Specifically, how high temperatures induce enzyme inactivation has broad implications for both fundamental and applied science. Here, we explored the mechanism of the reversible thermal inactivation in glycoside hydrolase family 12 (GH12) using comparative molecular dynamics simulations. First, we investigated the distribution of structural flexibility over the enzyme and found that the active site was the general thermal-sensitive region in GH12 cellulases. The dynamic perturbation of the active site before enzyme denaturation was explored through principal-component analysis, which indicated that variations in the collective motion and conformational ensemble of the active site may precisely correspond to enzyme transition from its active form to the inactive form. Furthermore, the degree of dynamic perturbation of the active site was found to be negatively correlated with the melting temperatures of GH12 enzymes, further proving the importance of the dynamic stability of the active site. Additionally, analysis of the residue–interaction network revealed that the active site in thermophilic enzyme was capable of forming additional contacts with other amino acids than those observed in the mesophilic enzyme. These interactions are likely the key mechanisms underlying the differences in rigidity of the active site. These findings provide further biophysical insights into the reversible thermal inactivation of enzymes and potential applications in future protein engineering.
Co-reporter:Xukai Jiang, Yuying Wang, Limei Xu, Guanjun Chen, Lushan Wang
Biochemical and Biophysical Research Communications 2017 Volume 491, Issue 1(Issue 1) pp:
Publication Date(Web):9 September 2017
DOI:10.1016/j.bbrc.2017.07.086
•A hybrid mechanism for binding substrate in TfCel5A is found.•Active site residues can be classified based on conformational dynamics.•Conformational rebalance is more difficult than conformational selection.The role of protein dynamics in enzyme catalysis is one of the most active areas in current enzymological research. Here, using endoglucanase Cel5A from Thermobifida fusca (TfCel5A) as a model, we applied molecular dynamics simulations to explore the dynamic behavior of the enzyme upon substrate binding. The collective motions of the active site revealed that the mechanism of TfCel5A substrate binding can likely be described by the conformational-selection model; however, we observed that the conformations of active site residues changed differently along with substrate binding. Although most active site residues retained their native conformational ensemble, some (Tyr163 and Glu355) generated newly induced conformations, whereas others (Phe162 and Tyr189) exhibited shifts in the equilibration of their conformational distributions. These results showed that TfCel5A substrate binding relied on a hybrid mechanism involving induced fit and conformational selection. Interestingly, we found that TfCel5A active site could only partly rebalance its conformational dynamics upon substrate dissociation within the same simulation time, which implies that the conformational rebalance upon substrate dissociation is likely more difficult than the conformational selection upon substrate binding at least in the view of the time required. Our findings offer new insight into enzyme catalysis and potential applications for future protein engineering.
Co-reporter:Xukai Jiang, Guanjun Chen and Lushan Wang
Physical Chemistry Chemical Physics 2016 vol. 18(Issue 31) pp:21340-21350
Publication Date(Web):11 Jul 2016
DOI:10.1039/C6CP02998A
Understanding the molecular mechanism underlying protein thermostability is central to the process of efficiently engineering thermostable cellulases, which can provide potential advantages in accelerating the conversion of biomass into clean biofuels. Here, we explored the general factors that diversify enzyme thermostability in the glycoside hydrolase family 12 (GH12) using comparative molecular dynamics (MD) simulations coupled to a bioinformatics approach. The results indicated that protein stability is not equally distributed over the whole structure: the N-terminus is the most thermal-sensitive region of the enzymes with a β-sandwich architecture and it tends to lose its secondary structure during the course of protein unfolding. Furthermore, we found that the total interaction energy within the N-terminus is appreciably correlated with enzyme thermostability. Interestingly, the internal interactions within the N-terminus are organized in a special amphipathic pattern in which a hydrophobic packing cluster and a hydrogen bonding cluster lie at the two ends of the N-terminus. Finally, bioinformatics analysis demonstrated that the amphipathic pattern is highly conserved in GH12 and besides that, the evolution of the amino acids in the N-terminal region is an inherent mechanism underlying the diversity of enzyme thermostability. Taken together, our results demonstrate that the N-terminus is generally the structure that determines enzyme thermostability in GH12, and thereby it is also an ideal engineering target. The dynameomics study of a protein family can give a general view of protein functions, which will offer a wide range of applications in future protein engineering.
Co-reporter:Shijia Liu, Shangjin Shao, Linlin Li, Zhi Cheng, Li Tian, Peiji Gao, Lushan Wang
Carbohydrate Research 2015 Volume 418() pp:50-56
Publication Date(Web):11 December 2015
DOI:10.1016/j.carres.2015.10.002
•The sequence profiles of the chitosanase and chitinase active sites are constructed.•Substrate recognition is supported by hydrogen bonds with C2 functional groups.•CH–π interactions contribute to tighter binding and processivity.Chitinases and chitosanases, referred to as chitinolytic enzymes, are two important categories of glycoside hydrolases (GH) that play a key role in degrading chitin and chitosan, two naturally abundant polysaccharides. Here, we investigate the active site architecture of the major chitosanase (GH8, GH46) and chitinase families (GH18, GH19). Both charged (Glu, His, Arg, Asp) and aromatic amino acids (Tyr, Trp, Phe) are observed with higher frequency within chitinolytic active sites as compared to elsewhere in the enzyme structure, indicating significant roles related to enzyme function. Hydrogen bonds between chitinolytic enzymes and the substrate C2 functional groups, i.e. amino groups and N-acetyl groups, drive substrate recognition, while non-specific CH–π interactions between aromatic residues and substrate mainly contribute to tighter binding and enhanced processivity evident in GH8 and GH18 enzymes. For different families of chitinolytic enzymes, the number, type, and position of substrate atoms bound in the active site vary, resulting in different substrate-binding specificities. The data presented here explain the synergistic action of multiple enzyme families at a molecular level and provide a more reasonable method for functional annotation, which can be further applied toward the practical engineering of chitinases and chitosanases.
Co-reporter:Weili Gong;Huaiqiang Zhang;Shijia Liu
Applied Biochemistry and Biotechnology 2015 Volume 177( Issue 6) pp:1252-1271
Publication Date(Web):2015 November
DOI:10.1007/s12010-015-1811-z
Filamentous fungi such as Aspergillus spp., Trichoderma spp., and Penicillium spp. are frequently used to produce high concentrations of lignocellulosic enzymes. This study examined the discrepancies in the compositions and dynamic changes in the extracellular enzyme systems secreted by Aspergillus niger ATCC1015, Trichoderma reesei QM9414, and Penicillium oxalicum 114-2 cultured on corn stover and wheat bran. The results revealed different types and an abundance of monosaccharides and oligosaccharides were released during incubation, which induced the secretion of diverse glycoside hydrolases. Both the enzyme activities and isozyme numbers of the three fungal strains increased with time. A total of 279, 161, and 183 secretory proteins were detected in A. niger, T. reesei, and P. oxalicum secretomes, respectively. In the A. niger secretomes, more enzymes involved in the degradation of (galacto)mannan, xyloglucan, and the backbone of pectin distributed mostly in dicots were detected. In comparison, although P. oxalicum 114-2 hardly secreted any xyloglucanases, the diversities of enzymes involved in the degradation of xylan and β-(1,3;1,4)-d-glucan commonly found in monocots were higher. The cellulase system of P. oxalicum 114-2 was more balanced. The degradation preference provided a new perspective regarding the recomposition of lignocellulosic enzymes based on substrate types.
Co-reporter:Sheng Xing;Guoli Li;Xulu Sun;Su Ma
Applied Biochemistry and Biotechnology 2013 Volume 171( Issue 4) pp:832-846
Publication Date(Web):2013 October
DOI:10.1007/s12010-013-0402-0
Aspergillus niger is an effective secretor of glycoside hydrolases that facilitate the saprophytic lifestyle of the fungus by degrading plant cell wall polysaccharides. In the present study, a series of dynamic zymography assays were applied to quantify the secreted glycoside hydrolases of A. niger cultured in media containing different carbon sources. Differences in the diversity and concentrations of polysaccharide hydrolysates dynamically regulated the secretion of glycoside hydrolases. The secretion of β-1,4-endoglucanase isozymes was observed to lag at least 24 h behind, rather than coincide with, the secretion of xylanase isozymes. Low concentrations of xylose could induce many endoxylanases (such as Xyn1/XynA, Xyn2, and Xyn3/XynB). High concentrations of xylose could sustain the induction of Xyn2 and Xyn3/XynB but repress Xyn1/XynA (GH10 endoxylanase), which has a broad substrate specificity, and also triggers the low-level secretion of Egl3/EglA, which also has a broad substrate specificity. Mixed polysaccharide hydrolysates sustained the induction of Egl1, whereas the other β-1,4-endoglucanases were sustainably induced by the specific polysaccharide hydrolysates released during the hydrolysis process (such as Egl2 and Egl4). These results indicate that the secretion of glycoside hydrolases may be specifically regulated by the production of polysaccharide hydrolysates released during the process of biomass degradation.
Co-reporter:Jinghua Li, Lushan Wang
Polymer Degradation and Stability 2011 Volume 96(Issue 5) pp:1009-1014
Publication Date(Web):May 2011
DOI:10.1016/j.polymdegradstab.2011.01.010
Xylanases from Bacillus circulans (BCX) are known as configuration-retaining glycoside hydrolases, which hydrolyze xylans with two glutamic acid residues (Glu78 and Glu172) serving as catalytic active residues according to a double displacement mechanism. Existing experimental researches show that mutating the asparagines (Asn) to aspartic acid (Asp) at position 35 next to Glu172 can obviously improve the catalytic activity of BCX. To better understand the inherent mechanism for the experimental finding, we performed quantum chemistry calculations on two model systems to mimic the catalyses of wild-type and mutant BCXs. Geometrical structures and relative energies of intermediates and transition states involved in the hydrolysis reactions are given in detail. It is found that in the wild-type model system Asn35 interacts with Glu172 via a loose hydrogen bond, while in the mutant model system Asp35 forms a very tight hydrogen bond with Glu172. The glycosidic bond cleavage is proposed to be the rate-determining step for the hydrolysis reaction, whose barrier varies from 98 to 65 kJ mol−1 when Asn35 is replaced by Asp35, showing the presence of Asp35 remarkably reduces the energy demand for the hydrolysis reaction. The present result provides a theoretical elucidation for why a single amino acid substitution can importantly influences catalytic activity of BCX.
Co-reporter:Jinghua Li, Likai Du, and Lushan Wang
The Journal of Physical Chemistry B 2010 Volume 114(Issue 46) pp:15261-15268
Publication Date(Web):October 28, 2010
DOI:10.1021/jp1064177
Cellulase Cel7A from Trichoderma reesei is one of the most abundant and effective cellulases. Structural studies have established that Cel7A is a retaining glycosidase and it can processively hydrolyze cellobiose units from the reducing end of a cellulose chain. Here, to elucidate the mechanism of enzymatic catalysis of cellulase Cel7A, we carried out a multisized level theoretical study by performing MD, QM, and QM/MM calculations. At the accurate level of theory, we showed the mechanism details of the catalytic cycle, which involves the configuration inversion of the anomeric center twice: the first results in the glycosidic bond cleavage and the formation of covalent glycosyl−enzyme intermediate, and the second restores the anomeric carbon to its original configuration. Calculated results have provided detailed structural and energetic information about these two processes, both of which proceed according to a SN2-type-like mechanism via loose transition state structures. It is clearly indicated that the glycosidic bond hydrolysis involves the formation of a covalent glycosyl−enzyme intermediate, which has been identified as the minimum on the potential energy surface. At the catalytic active region, hydrogen bond interactions exist throughout the whole process of the catalytic cycle, which are of special importance for stabilizing the glycosyl−enzyme intermediate. The present results provide a clear paradigm of the mechanisms of general glycosidases, which hydrolyze the glycosidic bonds with net retention of the anomeric configuration.
Co-reporter:Xukai Jiang, Guanjun Chen and Lushan Wang
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 31) pp:NaN21350-21350
Publication Date(Web):2016/07/11
DOI:10.1039/C6CP02998A
Understanding the molecular mechanism underlying protein thermostability is central to the process of efficiently engineering thermostable cellulases, which can provide potential advantages in accelerating the conversion of biomass into clean biofuels. Here, we explored the general factors that diversify enzyme thermostability in the glycoside hydrolase family 12 (GH12) using comparative molecular dynamics (MD) simulations coupled to a bioinformatics approach. The results indicated that protein stability is not equally distributed over the whole structure: the N-terminus is the most thermal-sensitive region of the enzymes with a β-sandwich architecture and it tends to lose its secondary structure during the course of protein unfolding. Furthermore, we found that the total interaction energy within the N-terminus is appreciably correlated with enzyme thermostability. Interestingly, the internal interactions within the N-terminus are organized in a special amphipathic pattern in which a hydrophobic packing cluster and a hydrogen bonding cluster lie at the two ends of the N-terminus. Finally, bioinformatics analysis demonstrated that the amphipathic pattern is highly conserved in GH12 and besides that, the evolution of the amino acids in the N-terminal region is an inherent mechanism underlying the diversity of enzyme thermostability. Taken together, our results demonstrate that the N-terminus is generally the structure that determines enzyme thermostability in GH12, and thereby it is also an ideal engineering target. The dynameomics study of a protein family can give a general view of protein functions, which will offer a wide range of applications in future protein engineering.