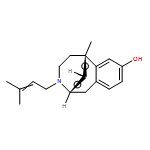
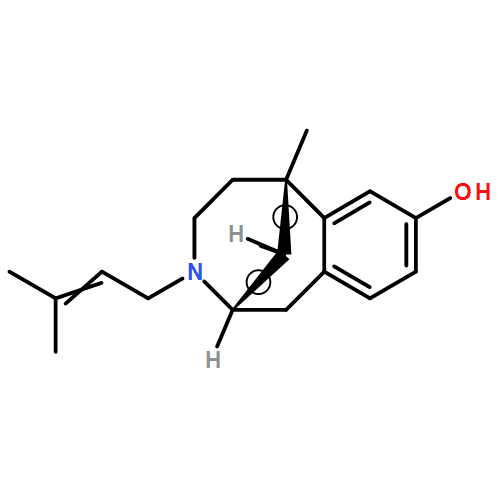
Co-reporter:Ahmed H. Abdelazeem, Shabana I. Khan, Stephen W. White, Kenneth J. Sufka, Christopher R. McCurdy
Bioorganic & Medicinal Chemistry 2015 23(13) pp: 3248-3259
Publication Date(Web):
DOI:10.1016/j.bmc.2015.04.057
Co-reporter:V. Blair Journigan ; Christophe Mésangeau ; Neha Vyas ; Shainnel O. Eans ; Stephen J. Cutler ; Jay P. McLaughlin ; Catherine Mollereau ;Christopher R. McCurdy
Journal of Medicinal Chemistry 2014 Volume 57(Issue 21) pp:8903-8927
Publication Date(Web):September 30, 2014
DOI:10.1021/jm500989n
Neuropeptide FF1 and FF2 receptors (NPFF1-R and NPFF2-R), and their endogenous ligand NPFF, are one of only several systems responsible for mediating opioid-induced hyperalgesia, tolerance, and dependence. Currently, no small molecules displaying good affinity or selectivity for either subtype have been reported, to decipher the role of NPFF2-R as it relates to opioid-mediated analgesia, for further exploration of NPFF1-R, or for medication development for either subtype. We report the first nonpeptide small molecule scaffold for NPFF1,2-R, the guanidino-piperidines, and SAR studies resulting in the discovery of a NPFF1 agonist (7b, Ki = 487 ± 117 nM), a NPFF1 antagonist (46, Ki = 81 ± 17 nM), and a NPFF2 partial antagonist (53a, Ki = 30 ± 5 nM), which serve as leads for the development of pharmacological probes and potential therapeutic agents. Testing of 46 alone was without effect in the mouse 48 °C warm-water tail-withdrawal test, but pretreatment with 46 prevented NPFF-induced hyperalgesia.
Co-reporter:Rohit Bhat, James A. Fishback, Rae R. Matsumoto, Jacques H. Poupaert, Christopher R. McCurdy
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 17) pp:5011-5013
Publication Date(Web):1 September 2013
DOI:10.1016/j.bmcl.2013.06.032
Herein we report the SAR study which involved structural modifications to the linker length, aryl substitution and alkylamine ring size of the benzo[d]thiazol-2(3H)one based sigma receptor (σ) ligands. Many compounds in this series displayed low nanomolar affinity for the σ receptor subtypes. In particular, 8a showed high affinity (σ-1 Ki = 4.5 nM) for σ-1 receptors and moderately high selectivity (483-fold) over σ-2 receptors.
Co-reporter:Christophe Mésangeau, Emanuele Amata, Walid Alsharif, Michael J. Seminerio, Matthew J. Robson, Rae R. Matsumoto, Jacques H. Poupaert, Christopher R. McCurdy
European Journal of Medicinal Chemistry 2011 Volume 46(Issue 10) pp:5154-5161
Publication Date(Web):October 2011
DOI:10.1016/j.ejmech.2011.08.031
A series of novel indole-based analogs were prepared and their affinities for sigma receptors were determined using in vitro radioligand binding assays. The results of this study identified several compounds with nanomolar sigma-2 affinity and significant selectivity over sigma-1 receptors. In particular, 2-(4-(3-(4-fluorophenyl)indol-1-yl)butyl)-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline (9f) was found to display high affinity at sigma-2 receptors with good selectivity (σ-1/σ-2 = 395). The pharmacological binding profile for this compound was established with other relevant non-sigma sites.A series of novel indole-based analogs were prepared and their affinities for sigma receptors were determined.Highlights► Preparation and evaluation of a series of indoles as sigma receptor ligands. ► Identification of several sigma-2 selective derivatives. ► Extensive pharmacological profile characterization for the best compound.
Co-reporter:Christophe Mésangeau ; Sanju Narayanan ; Andrea M. Green ; Jamaluddin Shaikh ; Nidhi Kaushal ; Eddy Viard ; Yan-Tong Xu ; James A. Fishback ; Jacques H. Poupaert ; Rae R. Matsumoto ;Christopher R. McCurdy
Journal of Medicinal Chemistry 2008 Volume 51(Issue 5) pp:1482-1486
Publication Date(Web):February 16, 2008
DOI:10.1021/jm701357m
Cocaine’s toxicity can be mitigated by blocking its interaction with sigma-1 receptors. The involvement of sigma-2 receptors remains unclear. To investigate their potential role, we have designed compounds through a convergent synthesis utilizing a highly selective sigma-1 ligand and elements of a selective sigma-2 ligand. Among the synthesized compounds was produced a subnanomolar sigma-2 ligand with an 11-fold preference over sigma-1 receptors. These compounds may be useful in developing effective pharmacotherapies for cocaine toxicity.
Co-reporter:Nidhi Singh, Tammy L. Nolan, Christopher R. McCurdy
Journal of Molecular Graphics and Modelling 2008 Volume 27(Issue 2) pp:131-139
Publication Date(Web):September 2008
DOI:10.1016/j.jmgm.2008.03.007
In an effort to reduce or eliminate the centrally associated side effects produced by opioid analgesics there has been an interest in the preparation of peripherally acting opioid receptor agonists. These compounds would have very limited or no access to the central nervous system. As a first step towards developing peripheral kappa opioid receptor (KOP) agonists, we have developed a quantitatively predictive chemical function-based pharmacophore model of selective kappa opioid receptor agonists by using the HypoGen algorithm implemented in the Catalyst software. The input for HypoGen was a training set of 26 KOP agonists exhibiting Ki values ranging between 0.015 nM and 2300 nM. The best output hypothesis consists of four features: one hydrophobic (HYD), one ring aromatic (RA), one hydrogen bond acceptor (HBA), and one positive ionizable (PI) function. The predictive power of the model could be demonstrated by internal and external validation of the generated hypothesis. The resulting Catalyst pharmacophore can be used concurrently for rapid virtual screening of chemical databases to identify novel, selective KOP agonists that may be easily restricted to target tissues by synthetic modification. It is anticipated that such an approach will lead to the generation of novel selective KOP agonists that are clinically useful for the treatment of pain through peripheral mechanisms.
Co-reporter:Ashok E. Philip;Jacques H. Poupaert;Gwénaël Chevé
Medicinal Chemistry Research 2007 Volume 16( Issue 3) pp:130-135
Publication Date(Web):2007 December
DOI:10.1007/s00044-007-9016-9
Ameltolide shares with phenytoin and carbamazepine a common mode of action involving interaction with central voltage-dependent sodium channels. Ameltolide and structurally related benzanilides were subjected to molecular modeling studies using both molecular mechanics (MM2, Amber96, and OPLS) and semiempirical quantum mechanics (AM1, PM3, and PM3 Cosmo) to resolve a paradox: while compounds with a phenytoin-like pharmacological profile possess a CO-NH moiety in a cis-configuration, ameltolide was found via X-ray crystallography to exist in the trans-configuration. Results obtained both by molecular mechanics and semiempirical methods indicate that for ameltolide, the cis and trans forms have similar energy content. Additional ab initio calculations performed at 6–31G** gave a ΔE (Z – E) on the order of 3 kcal/mol. In view of this small energy difference between the cis and trans forms, it is conceivable that these benzanilides bind to their biological target in their cis configuration, therefore assuming a common structure–activity relationship with classical antiepileptic agents.
Co-reporter:Nidhi Singh;Mitchell A. Avery
Journal of Computer-Aided Molecular Design 2007 Volume 21( Issue 9) pp:
Publication Date(Web):2007 September
DOI:10.1007/s10822-007-9132-0
Mycobacterium tuberculosis 1-deoxy-d-xylulose-5-phosphate reductoisomerase (MtDXR) is a potential target for antitubercular chemotherapy. In the absence of its crystallographic structure, our aim was to develop a structural model of MtDXR. This will allow us to gain early insight into the structure and function of the enzyme and its likely binding to ligands and cofactors and thus, facilitate structure-based inhibitor design. To achieve this goal, initial models of MtDXR were generated using MODELER. The best quality model was refined using a series of minimizations and molecular dynamics simulations. A protein–ligand complex was also developed from the initial homology model of the target protein by including information about the known ligand as spatial restraints and optimizing the mutual interactions between the ligand and the binding site. The final model was evaluated on the basis of its ability to explain several site-directed mutagenesis data. Furthermore, a comparison of the homology model with the X-ray structure published in the final stages of the project shows excellent agreement and validates the approach. The knowledge gained from the current study should prove useful in the design and development of inhibitors as potential novel therapeutic agents against tuberculosis by either de novo drug design or virtual screening of large chemical databases.

![Tert-butyl N-[n'-(2-aminoethyl)-n-[(2-methylpropan-2-yl)oxycarbonyl]carbamimidoyl]carbamate](http://img.cochemist.com/ccimg/203300/203258-44-0.png)
![Tert-butyl N-[n'-(2-aminoethyl)-n-[(2-methylpropan-2-yl)oxycarbonyl]carbamimidoyl]carbamate](http://img.cochemist.com/ccimg/203300/203258-44-0_b.png)
![1,4-Dioxa-8-azaspiro[4.5]decane, 8-(2-naphthalenylmethyl)-](http://img.cochemist.com/ccimg/898300/898222-58-7.png)
![1,4-Dioxa-8-azaspiro[4.5]decane, 8-(2-naphthalenylmethyl)-](http://img.cochemist.com/ccimg/898300/898222-58-7_b.png)
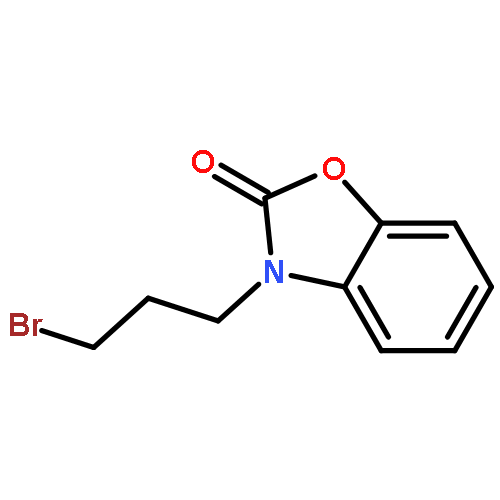
![Pentanoic acid,5-[[bis[[(1,1-dimethylethoxy)carbonyl]amino]methylene]amino]-](http://img.cochemist.com/ccimg/212600/212567-95-8.png)
![Pentanoic acid,5-[[bis[[(1,1-dimethylethoxy)carbonyl]amino]methylene]amino]-](http://img.cochemist.com/ccimg/212600/212567-95-8_b.png)






![Glycine,N-[bis[[(1,1-dimethylethoxy)carbonyl]amino]methylene]-](http://img.cochemist.com/ccimg/158500/158478-76-3.png)
![Glycine,N-[bis[[(1,1-dimethylethoxy)carbonyl]amino]methylene]-](http://img.cochemist.com/ccimg/158500/158478-76-3_b.png)
![2-Azabicyclo[3.3.1]nonan-7-one,5-(3-hydroxyphenyl)-2-methyl-8-(phenylmethylene)-, (1R,5R,8E)-](http://img.cochemist.com/ccimg/157800/157752-20-0.png)
![2-Azabicyclo[3.3.1]nonan-7-one,5-(3-hydroxyphenyl)-2-methyl-8-(phenylmethylene)-, (1R,5R,8E)-](http://img.cochemist.com/ccimg/157800/157752-20-0_b.png)


![1'-[4-[1-(4-fluorophenyl)indol-3-yl]butyl]spiro[1H-2-benzofuran-3,4'-piperidine]](http://img.cochemist.com/ccimg/147900/147817-50-3.png)
![1'-[4-[1-(4-fluorophenyl)indol-3-yl]butyl]spiro[1H-2-benzofuran-3,4'-piperidine]](http://img.cochemist.com/ccimg/147900/147817-50-3_b.png)




![L-Ornithine, N5-[bis[[(1,1-dimethylethoxy)carbonyl]amino]methylene]-N2-[(1,1-dimethylethoxy)carbonyl]-](/data/chemimg/122700/108787-91-3.png)
![L-Ornithine, N5-[bis[[(1,1-dimethylethoxy)carbonyl]amino]methylene]-N2-[(1,1-dimethylethoxy)carbonyl]-](/data/chemimg/122700/108787-91-3_b.png)
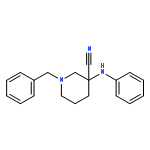
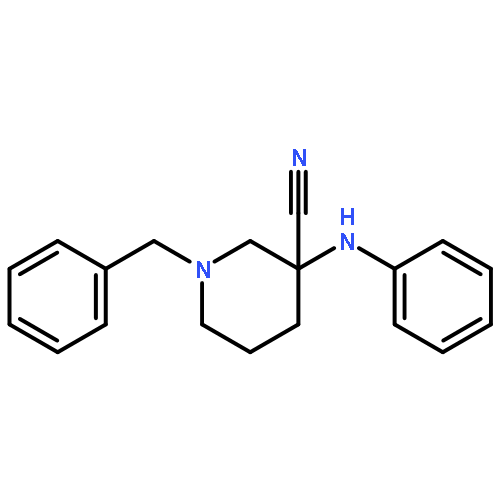
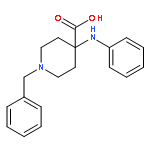
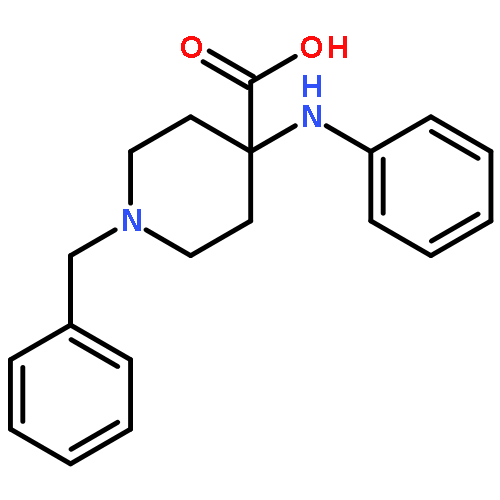
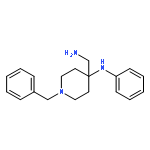
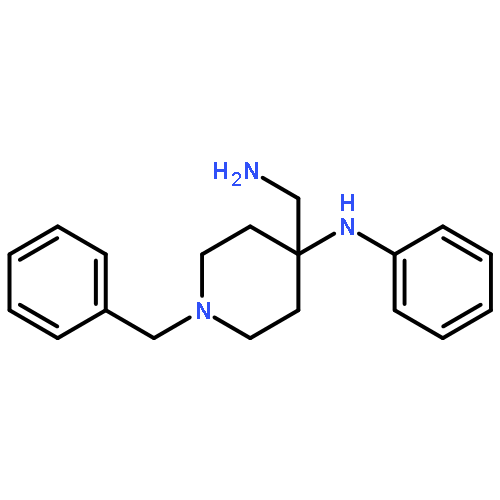
![(6aR,9S,10S,10aR)-6,6,9-trimethyl-3-pentyl-6a,7,8,9,10,10a-hexahydro-6H-benzo[c]chromene-1,9,10-triol](http://img.cochemist.com/ccimg/72300/72236-32-9.png)
![(6aR,9S,10S,10aR)-6,6,9-trimethyl-3-pentyl-6a,7,8,9,10,10a-hexahydro-6H-benzo[c]chromene-1,9,10-triol](http://img.cochemist.com/ccimg/72300/72236-32-9_b.png)
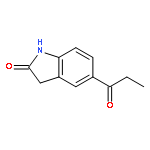
![6,6,9-trimethyl-3-pentyl-6H-benzo[c]chromene-1,8-diol](http://img.cochemist.com/ccimg/53900/53865-22-8.png)
![6,6,9-trimethyl-3-pentyl-6H-benzo[c]chromene-1,8-diol](http://img.cochemist.com/ccimg/53900/53865-22-8_b.png)



![6,6,9-TRIMETHYL-3-PROPYLBENZO[C]CHROMEN-1-OL](http://img.cochemist.com/ccimg/33800/33745-21-0.png)
![6,6,9-TRIMETHYL-3-PROPYLBENZO[C]CHROMEN-1-OL](http://img.cochemist.com/ccimg/33800/33745-21-0_b.png)
![6H-Dibenzo[b,d]pyran-1-ol,6a,7,8,10a-tetrahydro-6,6,9-trimethyl-3-propyl-, (6aR,10aR)-](http://img.cochemist.com/ccimg/31300/31262-37-0.png)
![6H-Dibenzo[b,d]pyran-1-ol,6a,7,8,10a-tetrahydro-6,6,9-trimethyl-3-propyl-, (6aR,10aR)-](http://img.cochemist.com/ccimg/31300/31262-37-0_b.png)
![Phenol, 2-[(2E)-3,7-dimethyl-2,6-octadienyl]-3-methoxy-5-pentyl-](http://img.cochemist.com/ccimg/29200/29106-17-0.png)
![Phenol, 2-[(2E)-3,7-dimethyl-2,6-octadienyl]-3-methoxy-5-pentyl-](http://img.cochemist.com/ccimg/29200/29106-17-0_b.png)
![3-(2-bromoethyl)benzo[d]oxazol-2(3H)-one](http://img.cochemist.com/ccimg/27200/27170-93-0.png)
![3-(2-bromoethyl)benzo[d]oxazol-2(3H)-one](http://img.cochemist.com/ccimg/27200/27170-93-0_b.png)
![2-CHLORO-5-{(Z)-[(5Z)-5-{[5-(4,5-DIMETHYL-2-NITROPHENYL)-2-FURYL]<WBR />METHYLENE}-4-OXO-1,3-THIAZOLIDIN-2-YLIDENE]AMINO}BENZOIC ACID](http://img.cochemist.com/ccimg/25700/25654-31-3.png)
![2-CHLORO-5-{(Z)-[(5Z)-5-{[5-(4,5-DIMETHYL-2-NITROPHENYL)-2-FURYL]<WBR />METHYLENE}-4-OXO-1,3-THIAZOLIDIN-2-YLIDENE]AMINO}BENZOIC ACID](http://img.cochemist.com/ccimg/25700/25654-31-3_b.png)

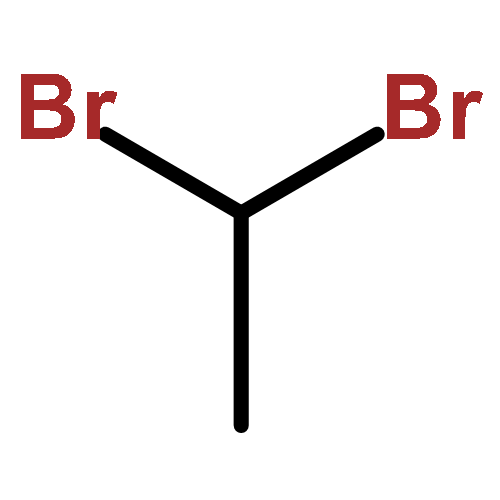
![3-[(2E)-3,7-dimethylocta-2,6-dien-1-yl]-2,4-dihydroxy-6-pentylbenzoic acid](http://img.cochemist.com/ccimg/25600/25555-57-1.png)
![3-[(2E)-3,7-dimethylocta-2,6-dien-1-yl]-2,4-dihydroxy-6-pentylbenzoic acid](http://img.cochemist.com/ccimg/25600/25555-57-1_b.png)
![2-[(1S,6S)-6-isopropenyl-3-methyl-1-cyclohex-2-enyl]-5-propyl-benzene-1,3-diol](http://img.cochemist.com/ccimg/24300/24274-48-4.png)
![2-[(1S,6S)-6-isopropenyl-3-methyl-1-cyclohex-2-enyl]-5-propyl-benzene-1,3-diol](http://img.cochemist.com/ccimg/24300/24274-48-4_b.png)


![6H-Dibenzo[b,d]pyran-2-carboxylicacid, 6a,7,8,10a-tetrahydro-1-hydroxy-6,6,9-trimethyl-3-pentyl-, (6aR,10aR)-](http://img.cochemist.com/ccimg/24000/23978-85-0.png)
![6H-Dibenzo[b,d]pyran-2-carboxylicacid, 6a,7,8,10a-tetrahydro-1-hydroxy-6,6,9-trimethyl-3-pentyl-, (6aR,10aR)-](http://img.cochemist.com/ccimg/24000/23978-85-0_b.png)


![2,4,6,8,9,10-Hexaoxa-1,3,5,7-tetraphosphatricyclo[3.3.1.13,7]decane,1,3,5,7-tetraoxide](http://img.cochemist.com/ccimg/16800/16752-60-6.png)
![2,4,6,8,9,10-Hexaoxa-1,3,5,7-tetraphosphatricyclo[3.3.1.13,7]decane,1,3,5,7-tetraoxide](http://img.cochemist.com/ccimg/16800/16752-60-6_b.png)
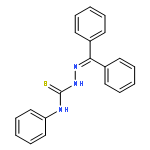
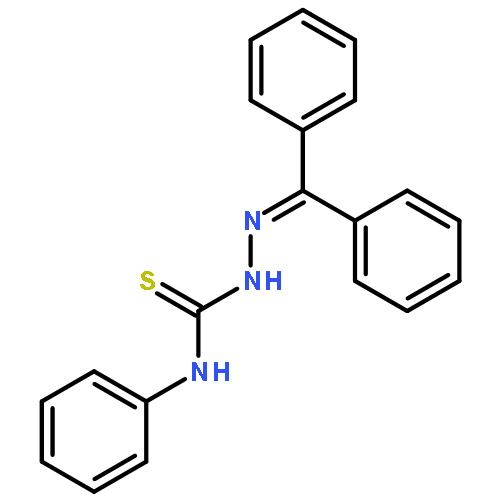
![1,3-Benzenediol,2-[(1R,6R)-3-methyl-6-(1-methylethenyl)-2-cyclohexen-1-yl]-5-pentyl-](http://img.cochemist.com/ccimg/14000/13956-29-1.png)
![1,3-Benzenediol,2-[(1R,6R)-3-methyl-6-(1-methylethenyl)-2-cyclohexen-1-yl]-5-pentyl-](http://img.cochemist.com/ccimg/14000/13956-29-1_b.png)
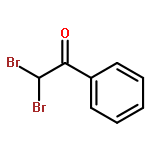
![(6aR)-1-methoxy-5,6,6a,7-tetrahydro-4H-dibenzo[de,g]quinolin-2-ol](http://img.cochemist.com/ccimg/6900/6871-21-2.png)
![(6aR)-1-methoxy-5,6,6a,7-tetrahydro-4H-dibenzo[de,g]quinolin-2-ol](http://img.cochemist.com/ccimg/6900/6871-21-2_b.png)
![4-Chloro-1-phenyl-1H-pyrazolo[3,4-d]pyrimidine](http://img.cochemist.com/ccimg/5400/5334-48-5.png)
![4-Chloro-1-phenyl-1H-pyrazolo[3,4-d]pyrimidine](http://img.cochemist.com/ccimg/5400/5334-48-5_b.png)
![2-{2-[4-(3,6-DIMETHYL-3-HEPTANYL)PHENOXY]ETHOXY}ETHANOL](http://img.cochemist.com/ccimg/3500/3423-07-2.png)
![2-{2-[4-(3,6-DIMETHYL-3-HEPTANYL)PHENOXY]ETHOXY}ETHANOL](http://img.cochemist.com/ccimg/3500/3423-07-2_b.png)
![4H-Dibenzo[de,g]quinolin-2-ol,5,6,6a,7-tetrahydro-1-methoxy-6-methyl-, (6aR)-](http://img.cochemist.com/ccimg/3200/3153-55-7.png)
![4H-Dibenzo[de,g]quinolin-2-ol,5,6,6a,7-tetrahydro-1-methoxy-6-methyl-, (6aR)-](http://img.cochemist.com/ccimg/3200/3153-55-7_b.png)





![4-Piperidinecarbonitrile, 4-[(3-methoxyphenyl)amino]-1-(phenylmethyl)-](http://img.cochemist.com/ccimg/1000/974-71-0.png)
![4-Piperidinecarbonitrile, 4-[(3-methoxyphenyl)amino]-1-(phenylmethyl)-](http://img.cochemist.com/ccimg/1000/974-71-0_b.png)
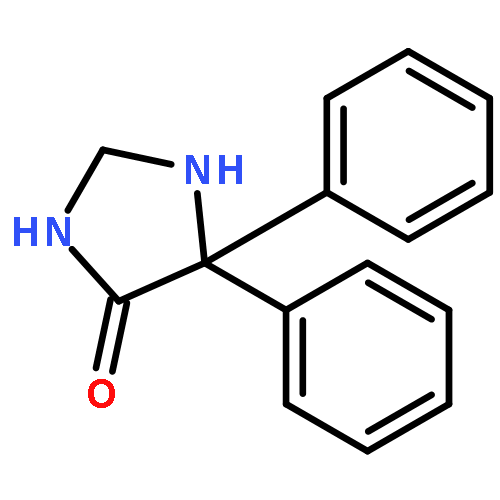
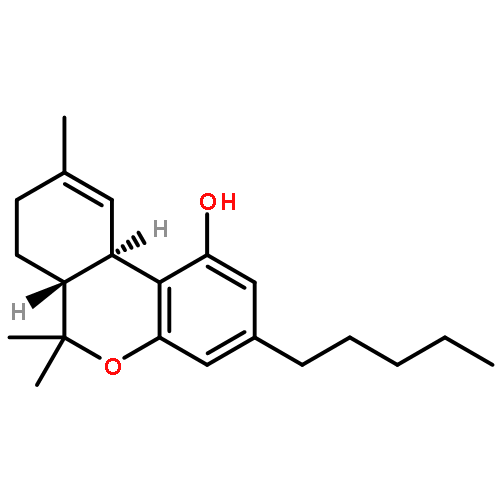
![7-Isoquinolinol,1,2,3,4-tetrahydro-1-[(4-hydroxyphenyl)methyl]-6-methoxy-, (1S)-](http://img.cochemist.com/ccimg/500/486-39-5.png)
![7-Isoquinolinol,1,2,3,4-tetrahydro-1-[(4-hydroxyphenyl)methyl]-6-methoxy-, (1S)-](http://img.cochemist.com/ccimg/500/486-39-5_b.png)