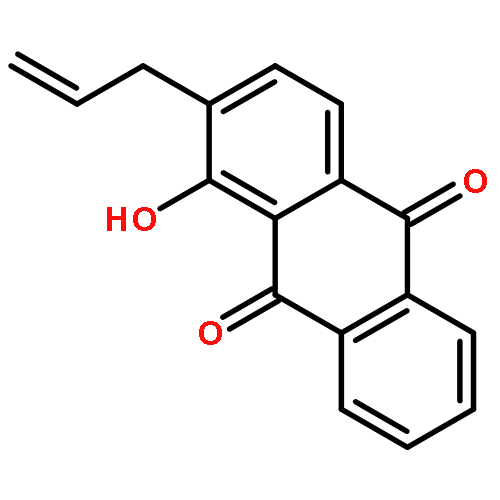
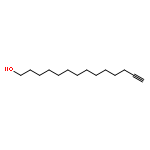
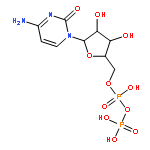
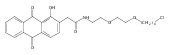
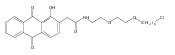
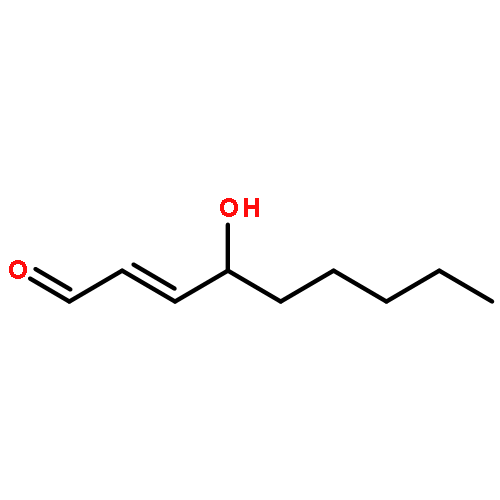
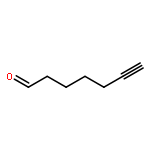
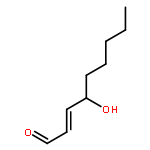
Co-reporter:Marcus J. C. Long, Jesse R. Poganik, Souradyuti Ghosh, and Yimon Aye
ACS Chemical Biology March 17, 2017 Volume 12(Issue 3) pp:586-586
Publication Date(Web):January 9, 2017
DOI:10.1021/acschembio.6b01148
Networks of redox sensor proteins within discrete microdomains regulate the flow of redox signaling. Yet, the inherent reactivity of redox signals complicates the study of specific redox events and pathways by traditional methods. Herein, we review designer chemistries capable of measuring flux and/or mimicking subcellular redox signaling at the cellular and organismal level. Such efforts have begun to decipher the logic underlying organelle-, site-, and target-specific redox signaling in vitro and in vivo. These data highlight chemical biology as a perfect gateway to interrogate how nature choreographs subcellular redox chemistry to drive precision redox biology.
Co-reporter:Marcus John Curtis Long, Yimon Aye
Cell Chemical Biology 2017 Volume 24, Issue 7(Volume 24, Issue 7) pp:
Publication Date(Web):20 July 2017
DOI:10.1016/j.chembiol.2017.05.023
This Perspective delineates how redox signaling affects the activity of specific enzyme isoforms and how this property may be harnessed for rational drug design. Covalent drugs have resurged in recent years and several reports have extolled the general virtues of developing irreversible inhibitors. Indeed, many modern pharmaceuticals contain electrophilic appendages. Several invoke a warhead that hijacks active-site nucleophiles whereas others take advantage of spectator nucleophilic side chains that do not participate in enzymatic chemistry, but are poised to bind/react with electrophiles. The latest data suggest that innate electrophile sensing—which enables rapid reaction with an endogenous signaling electrophile—is a quintessential resource for the development of covalent drugs. For instance, based on recent work documenting isoform-specific electrophile sensing, isozyme non-specific drugs may be converted to isozyme-specific analogs by hijacking privileged first-responder electrophile-sensing cysteines. Because this approach targets functionally relevant cysteines, we can simultaneously harness previously untapped moonlighting roles of enzymes linked to redox sensing.
Co-reporter:Marcus John Long, Hong-Yu Lin, Saba Parvez, Yi Zhao, ... Yimon Aye
Cell Chemical Biology 2017 Volume 24, Issue 8(Volume 24, Issue 8) pp:
Publication Date(Web):17 August 2017
DOI:10.1016/j.chembiol.2017.06.009
•Antioxidant response (AR) negatively regulates Wnt signaling•Loss of β-TRCP1 binding at β-catenin N terminus sensitizes Wnt to AR inhibition•β-catenin upregulates AR, irrespective of the extent to which β-TRCP binds β-catenin•The delicate balance between Wnt and AR may be exploitable for drug discoverySimultaneous hyperactivation of Wnt and antioxidant response (AR) are often observed during oncogenesis. However, it remains unclear how the β-catenin-driven Wnt and the Nrf2-driven AR mutually regulate each other. The situation is compounded because many players in these two pathways are redox sensors, rendering bolus redox signal-dosing methods uninformative. Herein we examine the ramifications of single-protein target-specific AR upregulation in various knockdown lines. Our data document that Nrf2/AR strongly inhibits β-catenin/Wnt. The magnitude and mechanism of this negative regulation are dependent on the direct interaction between β-catenin N terminus and β-TrCP1 (an antagonist of both Nrf2 and β-catenin), and independent of binding between Nrf2 and β-TrCP1. Intriguingly, β-catenin positively regulates AR. Because AR is a negative regulator of Wnt regardless of β-catenin N terminus, this switch of function is likely sufficient to establish a new Wnt/AR equilibrium during tumorigenesis.Download high-res image (102KB)Download full-size image
Co-reporter:Marcus J. C. Long; Jesse R. Poganik
Journal of the American Chemical Society 2016 Volume 138(Issue 11) pp:3610-3622
Publication Date(Web):February 23, 2016
DOI:10.1021/jacs.5b12608
Proximity enhancement is a central chemical tenet underpinning an exciting suite of small-molecule toolsets that have allowed us to unravel many biological complexities. The leitmotif of this opus is “tethering”—a strategy in which a multifunctional small molecule serves as a template to bring proteins/biomolecules together. Scaffolding approaches have been powerfully applied to control diverse biological outcomes such as protein–protein association, protein stability, activity, and improve imaging capabilities. A new twist on this strategy has recently appeared, in which the small-molecule probe is engineered to unleash controlled amounts of reactive chemical signals within the microenvironment of a target protein. Modification of a specific target elicits a precisely timed and spatially controlled gain-of-function (or dominant loss-of-function) signaling response. Presented herein is a unique personal outlook conceptualizing the powerful proximity-enhanced chemical biology toolsets into two paradigms: “multifunctional scaffolding” versus “on-demand targeting”. By addressing the latest advances and challenges in the established yet constantly evolving multifunctional scaffolding strategies as well as in the emerging on-demand precision targeting (and related) systems, this Perspective is aimed at choosing when it is best to employ each of the two strategies, with an emphasis toward further promoting novel applications and discoveries stemming from these innovative chemical biology platforms.
Co-reporter:Somsinee Wisitpitthaya, Yi Zhao, Marcus J. C. Long, Minxing Li, Elaine A. Fletcher, William A. Blessing, Robert S. Weiss, and Yimon Aye
ACS Chemical Biology 2016 Volume 11(Issue 7) pp:2021
Publication Date(Web):May 9, 2016
DOI:10.1021/acschembio.6b00303
The enzyme ribonucleotide reductase (RNR) is a major target of anticancer drugs. Until recently, suicide inactivation in which synthetic substrate analogs (nucleoside diphosphates) irreversibly inactivate the RNR-α2β2 heterodimeric complex was the only clinically proven inhibition pathway. For instance, this mechanism is deployed by the multifactorial anticancer agent gemcitabine diphosphate. Recently reversible targeting of RNR-α-alone coupled with ligand-induced RNR-α-persistent hexamerization has emerged to be of clinical significance. To date, clofarabine nucleotides are the only known example of this mechanism. Herein, chemoenzymatic syntheses of the active forms of two other drugs, phosphorylated cladribine (ClA) and fludarabine (FlU), allow us to establish that reversible inhibition is common to numerous drugs in clinical use. Enzyme inhibition and fluorescence anisotropy assays show that the di- and triphosphates of the two nucleosides function as reversible (i.e., nonmechanism-based) inhibitors of RNR and interact with the catalytic (C site) and the allosteric activity (A site) sites of RNR-α, respectively. Gel filtration, protease digestion, and FRET assays demonstrate that inhibition is coupled with formation of conformationally diverse hexamers. Studies in 293T cells capable of selectively inducing either wild-type or oligomerization-defective mutant RNR-α overexpression delineate the central role of RNR-α oligomerization in drug activity, and highlight a potential resistance mechanism to these drugs. These data set the stage for new interventions targeting RNR oligomeric regulation.
Co-reporter:Marcus J. C. Long and Yimon Aye
Chemical Research in Toxicology 2016 Volume 29(Issue 10) pp:1575
Publication Date(Web):September 12, 2016
DOI:10.1021/acs.chemrestox.6b00261
This perspective sets out to critically evaluate the scope of reactive electrophilic small molecules as unique chemical signal carriers in biological information transfer cascades. We consider these electrophilic cues as a new volatile cellular currency and compare them to canonical signaling circulation such as phosphate in terms of chemical properties, biological specificity, sufficiency, and necessity. The fact that nonenzymatic redox sensing properties are found in proteins undertaking varied cellular tasks suggests that electrophile signaling is a moonlighting phenomenon manifested within a privileged set of sensor proteins. The latest interrogations into these on-target electrophilic responses set forth a new horizon in the molecular mechanism of redox signal propagation wherein direct low-occupancy electrophilic modifications on a single sensor target are biologically sufficient to drive functional redox responses with precision timing. We detail how the various mechanisms through which redox signals function could contribute to their interesting phenotypic responses, including hormesis.
Co-reporter:Hong-Yu Lin; Joseph A. Haegele; Michael T. Disare; Qishan Lin
Journal of the American Chemical Society 2015 Volume 137(Issue 19) pp:6232-6244
Publication Date(Web):April 24, 2015
DOI:10.1021/ja5132648
Despite the known propensity of small-molecule electrophiles to react with numerous cysteine-active proteins, biological actions of individual signal inducers have emerged to be chemotype-specific. To pinpoint and quantify the impacts of modifying one target out of the whole proteome, we develop a target-protein-personalized “electrophile toolbox” with which specific intracellular targets can be selectively modified at a precise time by specific reactive signals. This general methodology, T-REX (targetable reactive electrophiles and oxidants), is established by (1) constructing a platform that can deliver a range of electronic and sterically different bioactive lipid-derived signaling electrophiles to specific proteins in cells; (2) probing the kinetics of targeted delivery concept, which revealed that targeting efficiency in cells is largely driven by initial on-rate of alkylation; and (3) evaluating the consequences of protein-target- and small-molecule-signal-specific modifications on the strength of downstream signaling. These data show that T-REX allows quantitative interrogations into the extent to which the Nrf2 transcription factor-dependent antioxidant response element (ARE) signaling is activated by selective electrophilic modifications on Keap1 protein, one of several redox-sensitive regulators of the Nrf2–ARE axis. The results document Keap1 as a promiscuous electrophile-responsive sensor able to respond with similar efficiencies to discrete electrophilic signals, promoting comparable strength of Nrf2–ARE induction. T-REX is also able to elicit cell activation in cases in which whole-cell electrophile flooding fails to stimulate ARE induction prior to causing cytotoxicity. The platform presents a previously unavailable opportunity to elucidate the functional consequences of small-molecule-signal- and protein-target-specific electrophilic modifications in an otherwise unaffected cellular background.
Co-reporter:Y Aye;M Li;M J C Long;R S Weiss
Oncogene 2015 34(16) pp:2011-2021
Publication Date(Web):2014-06-09
DOI:10.1038/onc.2014.155
Accurate DNA replication and repair is essential for proper development, growth and tumor-free survival in all multicellular organisms. A key requirement for the maintenance of genomic integrity is the availability of adequate and balanced pools of deoxyribonucleoside triphosphates (dNTPs), the building blocks of DNA. Notably, dNTP pool alterations lead to genomic instability and have been linked to multiple human diseases, including mitochondrial disorders, susceptibility to viral infection and cancer. In this review, we discuss how a key regulator of dNTP biosynthesis in mammals, the enzyme ribonucleotide reductase (RNR), impacts cancer susceptibility and serves as a target for anti-cancer therapies. Because RNR-regulated dNTP production can influence DNA replication fidelity while also supporting genome-protecting DNA repair, RNR has complex and stage-specific roles in carcinogenesis. Nevertheless, cancer cells are dependent on RNR for de novo dNTP biosynthesis. Therefore, elevated RNR expression is a characteristic of many cancers, and an array of mechanistically distinct RNR inhibitors serve as effective agents for cancer treatment. The dNTP metabolism machinery, including RNR, has been exploited for therapeutic benefit for decades and remains an important target for cancer drug development.
Co-reporter:Saba Parvez; Yuan Fu; Jiayang Li; Marcus J. C. Long; Hong-Yu Lin; Dustin K. Lee; Gene S. Hu
Journal of the American Chemical Society 2014 Volume 137(Issue 1) pp:10-13
Publication Date(Web):December 25, 2014
DOI:10.1021/ja5084249
Lipid-derived electrophiles (LDEs) that can directly modify proteins have emerged as important small-molecule cues in cellular decision-making. However, because these diffusible LDEs can modify many targets [e.g., >700 cysteines are modified by the well-known LDE 4-hydroxynonenal (HNE)], establishing the functional consequences of LDE modification on individual targets remains devilishly difficult. Whether LDE modifications on a single protein are biologically sufficient to activate discrete redox signaling response downstream also remains untested. Herein, using T-REX (targetable reactive electrophiles and oxidants), an approach aimed at selectively flipping a single redox switch in cells at a precise time, we show that a modest level (∼34%) of HNEylation on a single target is sufficient to elicit the pharmaceutically important antioxidant response element (ARE) activation, and the resultant strength of ARE induction recapitulates that observed from whole-cell electrophilic perturbation. These data provide the first evidence that single-target LDE modifications are important individual events in mammalian physiology.
Co-reporter:Dr. Yuan Fu;Dr. Hong-Yu Lin;Somsinee Wisitpitthaya;William A. Blessing; Yimon Aye
ChemBioChem 2014 Volume 15( Issue 17) pp:2598-2604
Publication Date(Web):
DOI:10.1002/cbic.201402368
Abstract
Human ribonucleotide reductase (hRNR) is a target of nucleotide chemotherapeutics in clinical use. The nucleotide-induced oligomeric regulation of hRNR subunit α is increasingly being recognized as an innate and drug-relevant mechanism for enzyme activity modulation. In the presence of negative feedback inhibitor dATP and leukemia drug clofarabine nucleotides, hRNR-α assembles into catalytically inert hexameric complexes, whereas nucleotide effectors that govern substrate specificity typically trigger α-dimerization. Currently, both knowledge of and tools to interrogate the oligomeric assembly pathway of RNR in any species in real time are lacking. We therefore developed a fluorimetric assay that reliably reports on oligomeric state changes of α with high sensitivity. The oligomerization-directed fluorescence quenching of hRNR-α, covalently labeled with two fluorophores, allows for direct readout of hRNR dimeric and hexameric states. We applied the newly developed platform to reveal the timescales of α self-assembly, driven by the feedback regulator dATP. This information is currently unavailable, despite the pharmaceutical relevance of hRNR oligomeric regulation.
Co-reporter:Xinqiang Fang ; Yuan Fu ; Marcus J. C. Long ; Joseph A. Haegele ; Eva J. Ge ; Saba Parvez
Journal of the American Chemical Society 2013 Volume 135(Issue 39) pp:14496-14499
Publication Date(Web):September 9, 2013
DOI:10.1021/ja405400k
In-depth chemical understanding of complex biological processes hinges upon the ability to systematically perturb individual systems. However, current approaches to study impacts of biologically relevant reactive small molecules involve bathing of the entire cell or isolated organelle with excess amounts, leading to off-target effects. The resultant lack of biochemical specificity has plagued our understanding of how biological electrophiles mediate signal transduction or regulate responses that confer defense mechanisms to cellular electrophilic stress. Here we introduce a target-specific electrophile delivery platform that will ultimately pave the way to interrogate effects of reactive electrophiles on specific target proteins in cells. The new methodology is demonstrated by photoinducible targeted delivery of 4-hydroxynonenal (HNE) to the proteins Keap1 and PTEN. Covalent conjugation of the HNE-precursor to HaloTag fused to the target proteins enables directed HNE delivery upon photoactivation. The strategy provides proof of concept of selective delivery of reactive electrophiles to individual electrophile-responsive proteins in mammalian cells. It opens a new avenue enabling more precise determination of the pathophysiological consequences of HNE-induced chemical modifications on specific target proteins in cells.
Co-reporter:Yuan Fu, Marcus J. C. Long, Mike Rigney, Saba Parvez, William A. Blessing, and Yimon Aye
Biochemistry 2013 Volume 52(Issue 40) pp:
Publication Date(Web):September 11, 2013
DOI:10.1021/bi400781z
An N-terminal-domain (NTD) and adjacent catalytic body (CB) make up subunit-α of ribonucleotide reductase (RNR), the rate-limiting enzyme for de novo dNTP biosynthesis. A strong linkage exists between ligand binding at the NTD and oligomerization-coupled RNR inhibition, inducible by both dATP and nucleotide chemotherapeutics. These observations have distinguished the NTD as an oligomeric regulation domain dictating the assembly of inactive RNR oligomers. Inactive states of RNR differ between eukaryotes and prokaryotes (α6 in human versus α4β4 in Escherichia coli, wherein β is RNR’s other subunit); however, the NTD structurally interconnects individual α2 or α2 and β2 dimeric motifs within the respective α6 or α4β4 complexes. To elucidate the influence of NTD ligand binding on RNR allosteric and oligomeric regulation, we engineered a human–E. coli hybrid enzyme (HE) where human-NTD is fused to E. coli-CB. Both the NTD and the CB of the HE bind dATP. The HE specifically partners with E. coli-β to form an active holocomplex. However, although the NTD is the sole physical tether to support α2 and/or β2 associations in the dATP-bound α6 or α4β4 fully inhibited RNR complexes, the binding of dATP to the HE NTD only partially suppresses HE activity and fully precludes formation of higher-order HE oligomers. We postulate that oligomeric regulation is the ultimate mechanism for potent RNR inhibition, requiring species-specific NTD–CB interactions. Such interdomain cooperativity in RNR oligomerization is unexpected from structural studies alone or biochemical studies of point mutants.






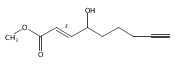
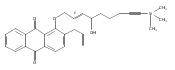
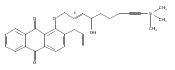
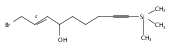


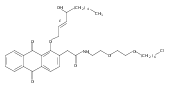


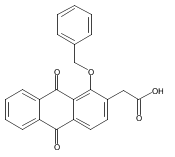
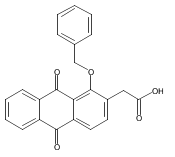
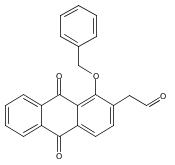
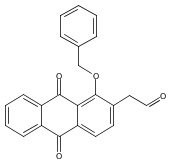
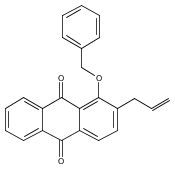
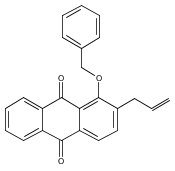



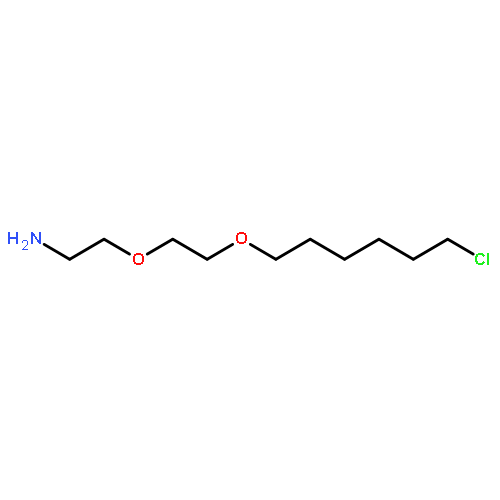




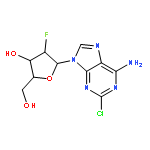
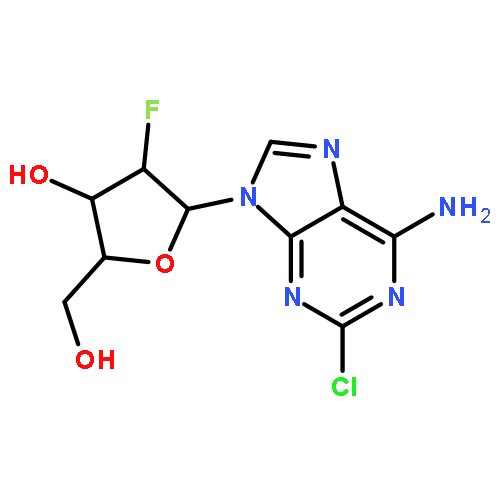
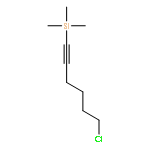
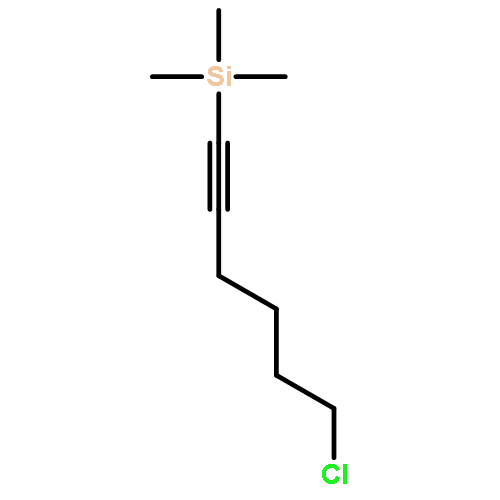
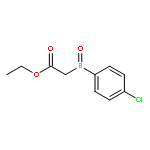
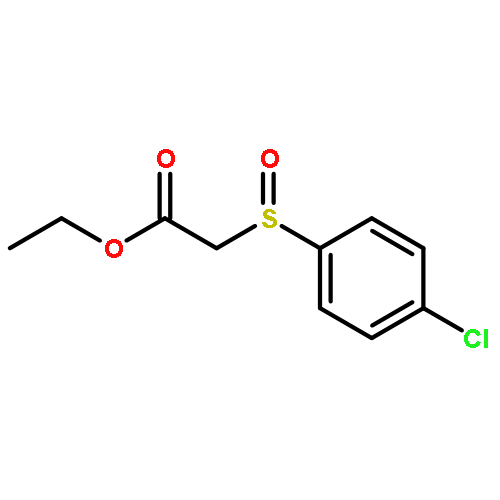
![Butanal, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/87200/87184-81-4.png)
![Butanal, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/87200/87184-81-4_b.png)


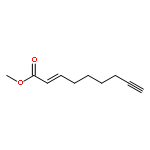
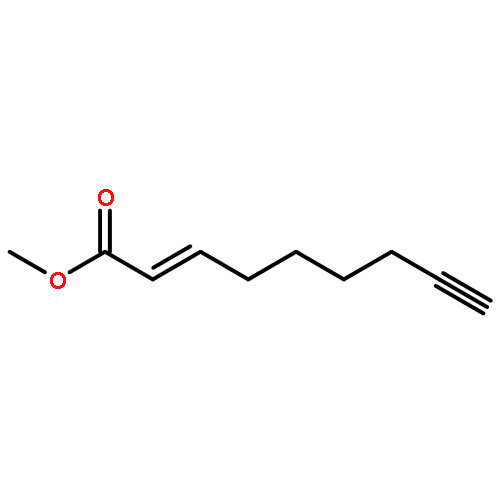
![9H-Purin-6-amine,2-fluoro-9-[5-O-[hydroxy[[hydroxy(phosphonooxy)phosphinyl]oxy]phosphinyl]-b-D-arabinofuranosyl]-](http://img.cochemist.com/ccimg/74900/74832-57-8.png)
![9H-Purin-6-amine,2-fluoro-9-[5-O-[hydroxy[[hydroxy(phosphonooxy)phosphinyl]oxy]phosphinyl]-b-D-arabinofuranosyl]-](http://img.cochemist.com/ccimg/74900/74832-57-8_b.png)