Co-reporter:Long Chen, Zhuanling Bai, Lin Zhu, Linjuan Zhang, Yawen Cai, Yuxiang Li, Wei Liu, Yanlong Wang, Lanhua Chen, Juan Diwu, Jianqiang Wang, Zhifang Chai, and Shuao Wang
ACS Applied Materials & Interfaces September 27, 2017 Volume 9(Issue 38) pp:32446-32446
Publication Date(Web):September 14, 2017
DOI:10.1021/acsami.7b12396
Enrichment of uranyl from seawater is crucial for the sustainable development of nuclear energy, but current uranium extraction technology suffers from multiple drawbacks of low sorption efficiency, slow uptake kinetics, or poor extraction selectivity. Herein, we prepared the first example of amidoxime appended metal–organic framework UiO-66-AO by a postsynthetic modification method for rapid and efficient extraction of uranium from seawater. UiO-66-AO can remove 94.8% of uranyl ion from Bohai seawater within 120 min and 99% of uranyl ion from Bohai seawater containing extra 500 ppb uranium within 10 min. The uranyl sorption capacity in a real seawater sample was determined to be 2.68 mg/g. In addition, the recyclability of the UiO-66-AO framework was demonstrated for at least three adsorption/desorption cycles. The origin for the superior sorption capability was further probed by extended X-ray absorption fine structure (EXAFS) analysis on the uranium-sorbed sample, suggesting multiple amidoxime ligands are able to chelate uranyl(VI) ions, forming a hexagonal bipyramid coordination geometry.Keywords: adsorption; amidoxime; metal−organic frameworks; seawater; uranyl;
Co-reporter:Lin Zhu, Daopeng Sheng, Chao Xu, Xing Dai, Mark A. Silver, Jie Li, Peng Li, Yaxing Wang, Yanlong Wang, Lanhua Chen, Chengliang Xiao, Jing Chen, Ruhong Zhou, Chao Zhang, Omar K. Farha, Zhifang Chai, Thomas E. Albrecht-Schmitt, and Shuao Wang
Journal of the American Chemical Society October 25, 2017 Volume 139(Issue 42) pp:14873-14873
Publication Date(Web):October 6, 2017
DOI:10.1021/jacs.7b08632
Effective and selective removal of 99TcO4– from aqueous solution is highly desirable for both waste partitioning and contamination remediation purposes in the modern nuclear fuel cycle, but is of significant challenge. We report here a hydrolytically stable and radiation-resistant cationic metal–organic framework (MOF), SCU-101, exhibiting extremely fast removal kinetics, exceptional distribution coefficient, and high sorption capacity toward TcO4–. More importantly, this material can selectively remove TcO4– in the presence of large excesses of NO3– and SO42–, as even 6000 times of SO42– in excess does not significantly affect the sorption of TcO4–. These superior features endow that SCU-101 is capable of effectively separating TcO4– from Hanford low-level waste melter off-gas scrubber simulant stream. The sorption mechanism is directly unraveled by the single crystal structure of TcO4–-incorporated SCU-101, as the first reported crystal structure to display TcO4– trapped in a sorbent material. A recognition site for the accommodation of TcO4– is visualized and is consistent with the DFT analysis results, while no such site can be resolved for other anions.
Co-reporter:Yaxing Wang, Tao Duan, Zhehui Weng, Jie Ling, Xuemiao Yin, Lanhua Chen, Daopeng Sheng, Juan Diwu, Zhifang Chai, Ning Liu, and Shuao Wang
Inorganic Chemistry November 6, 2017 Volume 56(Issue 21) pp:13041-13041
Publication Date(Web):October 9, 2017
DOI:10.1021/acs.inorgchem.7b01855
f-element-bearing iodate compounds are a large family mostly synthesized by hydrothermal reactions starting with actinide/lanthanide ions and iodic acid or iodate salt. In this work, we introduce melting periodic acid flux as a new reaction medium and provide a safe way for single-crystal growth of a series of new f-element iodate compounds including UO2(IO3)2·H2O (1), UO2(IO3)2(H2O)·HIO3 (2), α-Th(IO3)2(NO3)(OH) (3), β-Th(IO3)2(NO3)(OH) (4), and (H3O)9Nd9(IO3)36·3HIO3 (5). The structures of these compounds deviate from those afforded from hydrothermal reactions. Specifically, compounds 1 and 2 exhibit pillared structures consisting of uranyl pentagonal bipyramids and iodate trigonal pyramids. Compounds 3 and 4 represent two new thorium iodate compounds that are constructed from subunits of thorium dimers. Compound 5 exhibits a flower-shaped trivalent lanthanide iodate structure with HIO3 molecules and H3O+ cations filled in the channels. The aliovalent replacement of f elements in 5 is available from a hydrothermal process, further generating compounds of Th2(IO3)8(H2O) (6) and Ce2(IO3)8(H2O) (7). The distinct absorption features are observed in isotypic compounds 5–7, where 7 shows typical semiconductor behavior with a band gap of 2.43 eV. Remarkably, noncentrosymmetric 1, 6, and 7 exhibit strong second-harmonic-generation efficiencies of 1.3, 3.2, and 9.2 times, respectively, that of the commercial material KH2PO4. Additionally, the temperature-dependent emission spectra of 1 and 2 were also collected showing typical emission features of uranyl units and a negative correlation between the intensities of the emissions with temperature. Clearly, the presented low-temperature melting inorganic acid flux synthesis would provide a facile and effective strategy to produce a large new family of structurally versatile and multifunctional f-element inorganic compounds.
Co-reporter:Daopeng Sheng, Lin Zhu, Chao Xu, Chengliang Xiao, Yanlong Wang, Yaxing Wang, Lanhua Chen, Juan Diwu, Jing Chen, Zhifang Chai, Thomas E. Albrecht-Schmitt, and Shuao Wang
Environmental Science & Technology March 21, 2017 Volume 51(Issue 6) pp:3471-3471
Publication Date(Web):February 17, 2017
DOI:10.1021/acs.est.7b00339
99Tc is one of the most problematic radioisotopes in used nuclear fuel owing to its combined features of high fission yield, long half-life, and high environmental mobility. There are only a handful of functional materials that can remove TcO4– anion from aqueous solution and identifying for new, stable materials with high anion-exchange capacities, fast kinetics, and good selectivity remains a challenge. We report here an 8-fold interpenetrated three-dimensional cationic metal–organic framework material, SCU-100, which is assembled from a tetradentate neutral nitrogen-donor ligand and two-coordinate Ag+ cations as potential open metal sites. The structure also contains a series of 1D channels filled with unbound nitrate anions. SCU-100 maintains its crystallinity in aqueous solution over a wide pH range from 1 to 13 and exhibits excellent β and γ radiation-resistance. Initial anion exchange studies show that SCU-100 is able to both quantitatively and rapidly remove TcO4– from water within 30 min. The exchange capacity for the surrogate ReO4– reaches up to 541 mg/g and the distribution coefficient Kd is up to 1.9 × 105 mL/g, which are significantly higher than all previously tested inorganic anion sorbent materials. More importantly, SCU-100 can selectively capture TcO4– in the presence of large excess of competitive anions (NO3–, SO42–, CO32–, and PO43–) and remove as much as 87% of TcO4– from the Hanford low-level waste melter off-gas scrubber simulant stream within 2 h. The sorption mechanism is well elucidated by single crystal X-ray diffraction, showing that the sorbed ReO4– anion is able to selectively coordinate to the open Ag+ sites forming Ag–O–Re bonds and a series of hydrogen bonds. This further leads to a single-crystal-to-single-crystal transformation from an 8-fold interpenetrated framework with disordered nitrate anions to a 4-fold interpenetrated framework with fully ordered ReO4– anions. This work represents a practical case of TcO4– removal by a MOF material and demonstrates the promise of using this type of material as a scavenger for treating anionic radioactive contaminants during the nuclear waste partitioning and remediation processes.
Co-reporter:Yaxing Wang, Yumin Wang, Linjuan Zhang, Lanhua Chen, Zhiyong Liu, Xuemiao Yin, Daopeng Sheng, Juan Diwu, Jianqiang Wang, Ning Liu, Zhifang Chai, and Shuao Wang
Inorganic Chemistry March 20, 2017 Volume 56(Issue 6) pp:3702-3702
Publication Date(Web):March 9, 2017
DOI:10.1021/acs.inorgchem.7b00236
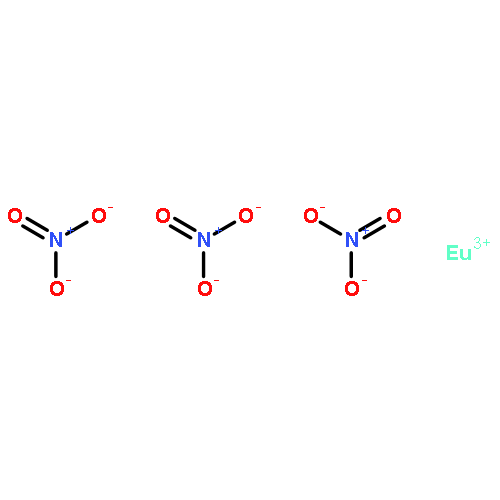
As the crucial soluble species of long-lived radionuclides 129I and 79Se, iodate and selenite anions commonly share similar geometry of the trigonal pyramid XO3 (X = I, Se) but in different valence states. Although large amounts of investigations have been performed aiming at understanding the environmental behavior of these two anions individually, studies on cases when they coexist are extremely scarce. Structurally well-characterized natural/synthetic crystalline solids simultaneously incorporating these two anions as potential solubility-limiting products at the nuclear waste geological depository remain elusive. We report here a crystalline solid Th(IO3)2(SeO3) representing the first example of aliovalent substitution between IO3– and SeO32– sharing the same structural site, as demonstrated by single crystal X-ray diffraction, laser-ablation inductively coupled plasma mass spectrometry analysis, and spectroscopic techniques including infrared, Raman, and X-ray absorption spectroscopies. Sequentially, in the Eu(IO3)3 solid matrix, we demonstrated that the IO3– site can be sufficiently substituted by SeO32– in the presence of Th4+ via simultaneous incorporation of Th4+ and SeO32– in a charge-balancing mechanism. The obtained results provide insights into the environmental behavior of fission products 79Se and 129I: they may cocrystallize in one solid matrix and may be efficiently immobilized by incorporation into each other’s solid phase through solid solution.
Co-reporter:Lanhua Chen, Juan Diwu, Daxiang Gui, Yaxing Wang, Zhehui Weng, Zhifang Chai, Thomas E. Albrecht-Schmitt, and Shuao Wang
Inorganic Chemistry June 19, 2017 Volume 56(Issue 12) pp:6952-6952
Publication Date(Web):May 26, 2017
DOI:10.1021/acs.inorgchem.7b00480
The oxidation state greatly affects the chemical behavior of uranium in the nuclear fuel cycle and in the environment. Phosphonate ligands, on the other hand, show strong complexation toward uranium at different oxidation states and are widely used in nuclear fuel reprocessing. Therefore, in this work, the reduction behavior of U(VI) with the presence of a phosphonate ligand is investigated under mild solvothermal conditions. By adjusting the reaction time, temperature, and counterion species, a series of uranium diphosphonates including two U(VI), seven U(IV), two mixed-valent U(IV/VI), and one distinct U(IV/V/VI) compounds were obtained. All these compounds were characterized by single crystal X-ray diffraction and UV–vis–NIR absorption and fluorescence spectroscopy. The structural diversity among those compounds not only illustrates the intrinsic structural complexity in this system, but also illuminates the in situ reduction pathways that are affected by the variation of reaction conditions. The UV–vis–NIR absorption spectra of the tetravalent uranium compounds show that the absorption features are closely related to the local coordination environments of the uranium centers as well as the bonding modes of the phosphonate ligand. The fluorescence spectra of mixed-valent uranium compounds show unique emission features of U(VI) luminescence that are partially quenched by the multiple electronic transitions of U(IV) centers in the visible and NIR regions.
Co-reporter:Zhuanling Bai, Yanlong Wang, Wei Liu, Yuxiang Li, Jian Xie, Lanhua Chen, Daopeng Sheng, Juan Diwu, Zhifang Chai, and Shuao Wang
Crystal Growth & Design July 5, 2017 Volume 17(Issue 7) pp:3847-3847
Publication Date(Web):May 15, 2017
DOI:10.1021/acs.cgd.7b00469
Searching for new host materials tailored for the high proton conductivity is highly desirable for the new generation of fuel cell system. We report here an anion-exchangeable cationic metal organic framework with the formula of [Ce(Ccbp)2]Br0.25Cl0.75·6H2O·2DMF (compound 1), which is constructed through the self-assembly of zwitterionic-based ligands H2CcbpBr (H2CcbpBr = 4-carboxy-1-(4-carboxybenzyl)pyridinium bromide) and (NH4)2Ce(NO3)6. During the investigation of humidity-dependent proton conduction behavior, we observed a rare case of rapid water-induced single-crystal-to-single-crystal phase transformation from compound 1 to a neutral chain [Ce(Ccbp)3(H2O)3]·8H2O (compound 2). This structural transformation originates from the coordination of water to Ce(III) metal centers, distortion of ligands, and the soft nature of the cationic framework 1, as probed and confirmed by a variety of investigations including color change, water vapor adsorption measurement, powder X-ray diffraction, single-crystal X-ray diffraction, humidity-dependent proton-conducting measurements, IR and UV–vis spectroscopies, and thermogravimetric analysis. As a consequence, this process introduces significant amounts of both coordinated and lattice water molecules into the structure, further giving rise to a decent water-assisted proton conductivity of 1.104 × 10–4 S cm–1 at 368 K and 95% relative humidity.
Co-reporter:Lin Zhu, Linjuan Zhang, Jie Li, Duo Zhang, Lanhua Chen, Daopeng Sheng, Shitong Yang, Chengliang Xiao, Jianqiang Wang, Zhifang Chai, Thomas E. Albrecht-Schmitt, and Shuao Wang
Environmental Science & Technology August 1, 2017 Volume 51(Issue 15) pp:8606-8606
Publication Date(Web):June 26, 2017
DOI:10.1021/acs.est.7b02006
Selenium is of great concern owing to its acutely toxic characteristic at elevated dosage and the long-term radiotoxicity of 79Se. The contents of selenium in industrial wastewater, agricultural runoff, and drinking water have to be constrained to a value of 50 μg/L as the maximum concentration limit. We reported here the selenium uptake using a structurally well-defined cationic layered rare earth hydroxide, Y2(OH)5Cl·1.5H2O. The sorption kinetics, isotherms, selectivity, and desorption of selenite and selenate on Y2(OH)5Cl·1.5H2O at pH 7 and 8.5 were systematically investigated using a batch method. The maximum sorption capacities of selenite and selenate are 207 and 124 mg/g, respectively, both representing the new records among those of inorganic sorbents. In the low concentration region, Y2(OH)5Cl·1.5H2O is able to almost completely remove selenium from aqueous solution even in the presence of competitive anions such as NO3–, Cl–, CO32–, SO42–, and HPO42–. The resulting concentration of selenium is below 10 μg/L, well meeting the strictest criterion for the drinking water. The selenate on loaded samples could be desorbed by rinsing with concentrated noncomplexing NaCl solutions whereas complexing ligands have to be employed to elute selenite for the material regeneration. After desorption, Y2(OH)5Cl·1.5H2O could be reused to remove selenate and selenite. In addition, the sorption mechanism was unraveled by the combination of EDS, FT-IR, Raman, PXRD, and EXAFS techniques. Specifically, the selenate ions were exchanged with chloride ions in the interlayer space, forming outer-sphere complexes. In comparison, besides anion exchange mechanism, the selenite ions were directly bound to the Y3+ center in the positively charged layer of [Y2(OH)5(H2O)]+ through strong bidentate binuclear inner-sphere complexation, consistent with the observation of the higher uptake of selenite over selenate. The results presented in this work confirm that the cationic layered rare earth hydroxide is an emerging and promising material for efficient removal of selenite and selenate as well as other anionic environmental pollutants.
Co-reporter:Yawen Cai;Fang Yuan;Xiaomei Wang;Zhuang Sun;Yang Chen;Zhiyong Liu
Cellulose 2017 Volume 24( Issue 1) pp:175-190
Publication Date(Web):2017 January
DOI:10.1007/s10570-016-1094-8
In this work, a carboxymethyl cellulose (CMC)-modified Fe3O4 (denoted as Fe3O4@CMC) composite was synthesized via a simple co-precipitation approach. Fourier transform infrared spectroscopy, zeta potential and thermogravimetric analysis results indicated that CMC was successfully coated on the Fe3O4 surfaces with a weight percent of ~30 % (w/w). The prepared Fe3O4@CMC composite was stable in acidic solution and could be easily collected with the aid of an external magnet. A batch technique was adopted to check the ability of the Fe3O4@CMC composite to remove Eu(III) as a function of various environmental parameters such as contact time, solution pH, ionic strength, solid content and temperature. The sorption kinetics process achieved equilibrium within a contact time of 7 h. The sorption isotherms were well simulated by the Langmuir model, and the maximum sorption capacity at 293 K was calculated to be 2.78 × 10−4 mol/g, being higher than the series of adsorbent materials reported to date. The ionic strength-independent sorption behaviors, desorption experiments by using ammonium acetate and disodium ethylenediamine tetraacetate as well as the spectroscopic characterization suggested that Eu(III) was sequestrated on the hydroxyl and carboxyl sites of Fe3O4@CMC via inner-sphere complexation. Overall, the Fe3O4@CMC composite could be utilized as a cost-effective adsorbent for the removal of trivalent lanthanide/actinides (e.g., 152+154Eu, 241Am and 244Cm) from radioactive wastewater.
Co-reporter:Jian Xie;Yaxing Wang;Wei Liu;Xuemiao Yin;Lanhua Chen;Dr. Youming Zou; Juan Diwu; Zhifang Chai; Thomas E. Albrecht-Schmitt;Dr. Guokui Liu; Shuao Wang
Angewandte Chemie International Edition 2017 Volume 56(Issue 26) pp:7500-7504
Publication Date(Web):2017/06/19
DOI:10.1002/anie.201700919
AbstractPrecise detection of low-dose X- and γ-radiations remains a challenge and is particularly important for studying biological effects under low-dose ionizing radiation, safety control in medical radiation treatment, survey of environmental radiation background, and monitoring cosmic radiations. We report here a photoluminescent uranium organic framework, whose photoluminescence intensity can be accurately correlated with the exposure dose of X- or γ-radiations. This allows for precise and instant detection of ionizing radiations down to the level of 10−4 Gy, representing a significant improvement on the detection limit of approximately two orders of magnitude, compared to other chemical dosimeters reported up to now. The electron paramagnetic resonance analysis suggests that with the exposure to radiations, the carbonyl double bonds break affording oxo-radicals that can be stabilized within the conjugated uranium oxalate-carboxylate sheet. This gives rise to a substantially enhanced equatorial bonding of the uranyl(VI) ions as elucidated by the single-crystal structure of the γ-ray irradiated material, and subsequently leads to a very effective photoluminescence quenching through phonon-assisted relaxation. The quenched sample can be easily recovered by heating, enabling recycled detection for multiple runs.
Co-reporter:Yawen Cai;Chunfang Wu;Zhiyong Liu;Linjuan Zhang;Lanhua Chen;Jianqiang Wang;Xiangke Wang;Shitong Yang
Environmental Science: Nano 2017 vol. 4(Issue 9) pp:1876-1886
Publication Date(Web):2017/09/14
DOI:10.1039/C7EN00412E
Uranium is not only a strategic resource for nuclear power but also a highly toxic contaminant in the environment. Although a series of traditional capturing materials including zeolites, metal–organic frameworks, mesoporous silica, and carbon-based nanomaterials have been investigated and developed, the combined advantages of decent stability, ultrafast removal kinetics, high sorption capacity, great selectivity, and potential recyclability have yet to be integrated into a single material. Herein, a new synthesis strategy was developed to synthesize a novel phosphorylated graphene oxide (GO)–chitosan (CS) composite (denoted as GO–CS–P) for U(VI) removal. The crosslinking of GO with CS and the subsequent phosphorylation of the GO–CS composite were demonstrated by scanning electron microscopy (SEM), powder X-ray diffraction (PXRD), Fourier transform infrared spectroscopy (FTIR), zeta potential measurement, thermogravimetric (TG) analysis, and scanning transmission electron microscopy (STEM). Batch experiments and spectroscopic analysis were performed to explore the removal performance and mechanism of GO–CS–P towards U(VI). The results showed that the uptake of U(VI) was ultrafast as the sorption equilibrium could be reached within 15 min. The maximum sorption capacity of U(VI) at pH 5.0 and 293 K was calculated to be 779.44 mg g−1, one of the highest values among the currently reported adsorbents. GO–CS–P also exhibited an excellent selectivity for capturing U(VI) from a mixture containing multiple competing metal ions. According to the desorption experiments, FTIR, X-ray absorption spectroscopy (XAS) and X-ray photoelectron spectroscopy (XPS) analysis, the highly efficient immobilization of U(VI) in GO–CS–P was predominantly controlled by inner-sphere surface complexation with a minor contribution of surface reduction. The experimental findings demonstrated the feasibility of using GO–CS–P for used nuclear fuel partition and uranium-bearing wastewater remediation.
Co-reporter:Chengliang Xiao;Mark A. Silver
Dalton Transactions 2017 vol. 46(Issue 47) pp:16381-16386
Publication Date(Web):2017/12/06
DOI:10.1039/C7DT03670A
In this Frontier article, we pursue the sequestration of radionuclides from aqueous solution by using recently emerging metal–organic framework (MOF) materials. The design of MOF materials and their corresponding sorption properties towards radionuclides (137Cs, 90Sr, 238U, 79Se, and 99Tc) as well as their interaction mechanisms are highlighted. The present challenges and future prospects of removing radionulides with MOFs as sorbents are also demonstrated.
Co-reporter:Fang Yuan;Yawen Cai;Shitong Yang
Journal of Radioanalytical and Nuclear Chemistry 2017 Volume 311( Issue 1) pp:815-831
Publication Date(Web):28 October 2016
DOI:10.1007/s10967-016-5086-9
This study highlights the simultaneous sequestration of U(VI) and arsenate at the goethite/water interface. The uptake trends and speciation of these two components were related with molar arsenate/U(VI) ratio, solution pH, contact order and aging time. A metastable [UO2(H2AsO4)2·H2O] was observed after 3 days and then this solid completely transformed into Na2(UO2AsO4)2·3H2O after 7 days. The disodium ethylenediamine tetraacetate ligand gave rise to the complete dissolution of Na2(UO2AsO4)2·3H2O phase and the release of U(VI) and arsenate back into the solution. The experimental findings facilitated us better comprehend the migration and fate of coexisting U(VI) and arsenate in the aquatic environment.
Co-reporter:Daxiang Gui, Tao Zheng, Jian Xie, Yawen Cai, Yaxing Wang, Lanhua Chen, Juan Diwu, Zhifang Chai, and Shuao Wang
Inorganic Chemistry 2016 Volume 55(Issue 24) pp:12508-12511
Publication Date(Web):November 23, 2016
DOI:10.1021/acs.inorgchem.6b02308
A highly stable layered zirconium phosphate, (NH4)2[ZrF2(HPO4)2] (ZrP-1), was synthesized by an ionothermal method and contains an extremely dense two-dimensional hydrogen-bond network that is thermally stable up to 573 K, leading to combined ultrahigh water-assisted proton conductivities of 1.45 × 10–2 S cm–1 at 363 K/95% relative humidity and sustainable anhydrous proton conductivity of 1.1 × 10–5 S cm–1 at 503 K.
Co-reporter:Huangjie Lu, Yaxing Wang, Congzhi Wang, Lanhua Chen, Weiqun Shi, Juan Diwu, Zhifang Chai, Thomas E. Albrecht-Schmitt, and Shuao Wang
Inorganic Chemistry 2016 Volume 55(Issue 17) pp:8570-8575
Publication Date(Web):August 5, 2016
DOI:10.1021/acs.inorgchem.6b01110
A unique two-dimensional inorganic cationic network with the formula [Th3O2(IO3)5(OH)2]Cl was synthesized hydrothermally. Its crystal structure can best be described as positively charged slabs built with hexanuclear thorium clusters connected by iodate trigonal pyramids. Additional chloride anions are present in the interlayer spaces but surprisingly are not exchangeable, as demonstrated by a series of CrO42– uptake experiments. This is because all chloride anions are trapped by multiple strong halogen–halogen interactions with short Cl–I bond lengths ranging from 3.134 to 3.333 Å, forming a special Cl-centered trigonal-pyramidal polyhedron as a newly observed coordination mode for halogen bonds. Density functional theory calculations clarified that electrons transformed from central Cl atoms to I atoms, generating a halogen–halogen interaction energy with a value of about −8.3 kcal mol–1 per Cl···I pair as well as providing a total value of −57.9 kcal mol–1 among delocalized halogen–halogen bonds, which is a new record value reported for a single halogen atom. Additional hydrogen-bonding interaction is also present between Cl and OH, and the interaction energy is predicted to be −8.1 kcal mol–1, confirming the strong total interaction to lock the interlayer Cl anions.
Co-reporter:Lin Xu, Tao Zheng, Shitong Yang, Linjuan Zhang, Jianqiang Wang, Wei Liu, Lanhua Chen, Juan Diwu, Zhifang Chai, and Shuao Wang
Environmental Science & Technology 2016 Volume 50(Issue 7) pp:3852-3859
Publication Date(Web):March 10, 2016
DOI:10.1021/acs.est.5b05932
The permeable reactive barrier (PRB) technique has attracted an increasing level of attention for the in situ remediation of contaminated groundwater. In this study, the macroscopic uptake behaviors and microscopic speciation of Eu(III) on hydroxyapatite (HAP) were investigated by a combination of theoretical modeling, batch experiments, powder X-ray diffraction (PXRD) fitting, and X-ray absorption spectroscopy (XAS). The underlying removal mechanisms were identified to further assess the application potential of HAP as an effective PRB backfill material. The macroscopic analysis revealed that nearly all dissolved Eu(III) in solution was removed at pH 6.5 within an extremely short reaction time of 5 min. In addition, the thermodynamic calculations, desorption experiments, and PXRD and XAS analyses definitely confirmed the formation of the EuPO4·H2O(s) phase during the process of uptake of dissolved Eu(III) by HAP via the dissolution–precipitation mechanism. A detailed comparison of the present experimental findings and related HAP–metal systems suggests that the relative contribution of precipitation to the total Eu(III) removal increases as the P:Eu ratio decreases. The dosage of HAP-based PRB for the remediation of groundwater polluted by Eu(III) and analogous trivalent actinides [e.g., Am(III) and Cm(III)] should be strictly controlled depending on the dissolved Eu(III) concentration to obtain an optimal P:M (M represents Eu, Am, or Cm) ratio and treatment efficiency.
Co-reporter:Yaxing Wang, Linjuan Zhang, Lanhua Chen, Weifeng Li, Xing Dai, Juan Diwu, Jianqiang Wang, Zhifang Chai, and Shuao Wang
Inorganic Chemistry 2016 Volume 55(Issue 23) pp:12101-12104
Publication Date(Web):November 17, 2016
DOI:10.1021/acs.inorgchem.6b02010
Periodate is a strong oxidant and is often reduced to IO3– or I2 under hydrothermal conditions. Here, we present a rare case of a mixed-valent iodate(V)/periodate(VII) compound, Th(H2O)(IVO3)2[IVII0.6V1.76O7(OH)], prepared with a hydrothermal method starting from periodic acid. Crystallographic results demonstrate that heptavalent iodine adopts IVIIO6 distorted octahedral geometries, which are stabilized on the crystallographically compatible crystal lattice sites of VO6 octahedra through an aliovalent substitutional disorder mechanism. X-ray photoelectron and synchrotron radiation X-ray absorption spectroscopes both quantitatively confirm the presence of mixed valent iodine oxoanions with a molar ratio (IV/IVII) of 4:1, consistent with the single crystal X-ray analysis. The crystallization of mixed-valent products with compatible lattice site can be fancily utilized for stabilizing the uncommon oxidation states of other elements in general.
Co-reporter:Daxiang Gui, Tao Zheng, Lanhua Chen, Yanlong Wang, Yuxiang Li, Daopeng Sheng, Juan Diwu, Zhifang Chai, Thomas E. Albrecht-Schmitt, and Shuao Wang
Inorganic Chemistry 2016 Volume 55(Issue 8) pp:3721-3723
Publication Date(Web):March 25, 2016
DOI:10.1021/acs.inorgchem.6b00293
The first thorium framework compound with mixed-valent phosphorus-based (phosphite and pyrophosphate) ligands, [BMMim]2[Th3(PO3)4(H2P2O7)3] (ThP-1), was synthesized by ionothermal reactions concurrent with the partial oxidation of phosphoric acid. The overall structural topology of ThP-1 highly resembles that of MOF-5, containing only one type of three-dimensional channels with a window size of 11.32 Å × 11.32 Å. ThP-1 has a free void volume of 50.8%, making it one of the most porous purely inorganic actinide-based framework materials. More importantly, ThP-1 is highly stable in aqueous solutions over an extremely wide pH range from 1 to 14 and thus may find potential applications in selective ion exchange and catalysis.
Co-reporter:Zhuanling Bai, Yanlong Wang, Yuxiang Li, Wei Liu, Lanhua Chen, Daopeng Sheng, Juan Diwu, Zhifang Chai, Thomas E. Albrecht-Schmitt, and Shuao Wang
Inorganic Chemistry 2016 Volume 55(Issue 13) pp:6358-6360
Publication Date(Web):June 16, 2016
DOI:10.1021/acs.inorgchem.6b00930
By controlling the extent of hydrolysis during the self-assembly process of a zwitterionic-based ligand with uranyl cations, we observed a structural evolution from the neutral uranyl–organic framework [(UO2)2(TTTPC)(OH)O(COOH)]·1.5DMF·7H2O (SCU-6) to the first cationic uranyl–organic framework with the formula of [(UO2)(HTTTPC)(OH)]Br·1.5DMF·4H2O (SCU-7). The crystal structures of SCU-6 and SCU-7 are layers built with tetranuclear and dinuclear uranyl clusters, respectively. Exchangeable halide anions are present in the interlaminar spaces balancing the positive charge of layers in SCU-7. Therefore, SCU-7 is able to effectively remove perrhenate anions from aqueous solution. Meanwhile, the H2PO4–-exchanged SCU-7 material exhibits a moderate proton conductivity of 8.70 × 10–5 S cm–1 at 50 °C and 90% relative humidity, representing nearly 80 times enhancement compared to the original material.
Co-reporter:Tao Zheng, Yang Gao, Daxiang Gui, Lanhua Chen, Daopeng Sheng, Juan Diwu, Zhifang Chai, Thomas E. Albrecht-Schmitt and Shuao Wang
Dalton Transactions 2016 vol. 45(Issue 22) pp:9031-9035
Publication Date(Web):06 May 2016
DOI:10.1039/C6DT01011C
The example of phase transformation from a centrosymmetric space group at low temperature (LT) to a chiral space group at high temperature (HT) is reported, which was clearly resolved in a single-crystal-to-single-crystal manner in a 3D uranyl(VI) phosphonate compound [TMA][(UO2)2(1,3-pbpH)(1,3-pbpH2)] (1) (TMA+ = tetramethylammonium cation; 1,3-pbpH4 = 1,3-phenylenebis(phosphonic acid)).
Co-reporter:Yanlong Wang; Zhiyong Liu; Yuxiang Li; Zhuanling Bai; Wei Liu; Yaxing Wang; Xiaomei Xu; Chengliang Xiao; Daopeng Sheng; Juan Diwu; Jing Su; Zhifang Chai; Thomas E. Albrecht-Schmitt
Journal of the American Chemical Society 2015 Volume 137(Issue 19) pp:6144-6147
Publication Date(Web):May 5, 2015
DOI:10.1021/jacs.5b02480
Searching for new chemically durable and radiation-resistant absorbent materials for actinides and their fission products generated in the nuclear fuel cycle remain highly desirable, for both waste management and contamination remediation. Here we present a rare case of 3D uranyl organic framework material built through polycatenating of three sets of graphene-like layers, which exhibits significant umbellate distortions in the uranyl equatorial planes studied thoroughly by linear transit calculations. This unique structural arrangement leads to high β and γ radiation-resistance and chemical stability in aqueous solutions within a wide pH range from 3 to 12. Being equipped with the highest surface area among all actinide compounds known to date and completely exchangeable [(CH3)2NH2]+ cations in the structure, this material is able to selectively remove cesium from aqueous solutions while retaining the polycatenated framework structure.
Co-reporter:Tao Zheng, Qun-Yan Wu, Yang Gao, Daxiang Gui, Shiwen Qiu, Lanhua Chen, Daopeng Sheng, Juan Diwu, Wei-Qun Shi, Zhifang Chai, Thomas E. Albrecht-Schmitt, and Shuao Wang
Inorganic Chemistry 2015 Volume 54(Issue 8) pp:3864-3874
Publication Date(Web):March 27, 2015
DOI:10.1021/acs.inorgchem.5b00024
Systematic control of the reactions between U(VI) and 1,4-phenylenebis(methylene))bis(phosphonic acid) (pmbH4) allows for alterations in the bonding between these constituents and affords three uranyl phosphonate compounds with chiral one-dimensional (1D), two-dimensional (2D), and three-dimensional (3D) structures, namely, [TPA][UO2(pmbH3)(pmbH2)H2O]·2H2O (1), [NH4]2[UO2(pmb)] (2), UO2(pmbH2) (3), and the first uranyl mixed phosphite/phosphonate compound [TMA]2[(UO2)2(pmb)(HPO3)] (4) (TPA = NPr4+, TMA = NMe4+). These compounds crystallize in the space groups P212121, P1̅, P21/c, and Cmcm, respectively. Further investigation of the local uranyl coordination environment reveals that in 1 only oxygen atoms from P═O moieties ligate the uranium centers; whereas in 2 only P–O– oxygen atoms are involved in bonding and yield a layered topology. Compound 3 differs sharply from the first two in that conjugated P═O and P–O– oxygen atoms chelate the uranium centers resulting in a 3D framework. In compound 4, a phosphonate group bridges three uranyl centers further coordinated with a phosphite ligand HPO32–, which is a product of pmbH4 decomposing, forming a 2D layered structure. Compounds 3 and 4 also contain a different coordination environment for U(VI) than that found in 1 or 2. In this case, tetragonal bipyramidal UO6 units occur instead of the far more common UO7 pentagonal bipyramids found in 1 and 2. Interestingly, 1 converts to 3 at elevated reaction temperatures, indicating that the formation of 1 is likely under kinetic control. This is supported by thermal analysis, which reveals that 3 has higher thermal stability than 1 or 2. UV–vis–near-IR absorption and fluorescence spectroscopy show that the absorption and photoluminescence intensity increases from 1 to 4. Density functional theory electronic structure calculations provide insight into the nature of the interactions between U(VI) and the phosphonate ligands.
Co-reporter:Yaxing Wang; Xuemiao Yin; Yanyan Zhao; Yang Gao; Lanhua Chen; Zhiyong Liu; Daopeng Sheng; Juan Diwu; Zhifang Chai; Thomas E. Albrecht-Schmitt
Inorganic Chemistry 2015 Volume 54(Issue 17) pp:8449-8455
Publication Date(Web):August 20, 2015
DOI:10.1021/acs.inorgchem.5b01141
Two new uranyl vanadates have been prepared from hydrothermal reactions and structurally characterized by single-crystal X-ray diffraction. The structure of (H3O)UO2VO4 (UVO-1) consists of anionic layers containing UO22+ pentagonal bipyramids coordinated by edge-sharing VO5 square pyramids, with the charge balanced by interlaminar H3O+ cations. Vanadium in (UO2)3(VO4)2(H2O)3 (UVO-2) exists as monomeric VO4 tetrahedra coordinating to UO22+ pentagonal bipyramids, forming a 3D uranyl(VI) vanadate framework. Similar reactions with the addition of Ln(NO3)3 (Ln = Nd, Eu) afford the three heterobimetallic lanthanide uranyl vanadate frameworks Nd(UO2)3(VO4)3(H2O)11 (NdUVO-1), Eu(UO2)3(VO4)3(H2O)10 (EuUVO-1), and Eu2(UO2)12(VO4)10(H2O)24 (EuUVO-2). In NdUVO-1 and EuUVO-1, Ln3+ cations are inserted into the interlayer space of UVO-1 substituting for H3O+ and further bridging adjacent layers into 3D frameworks. Similarly, EuUVO-2 adopts the same sheet topology as UVO-2, with Eu3+ ions replacing some of the interlayer uranyl ions in UVO-2. Our work has demonstrated that uranyl vanadate extended structures are excellent hosts for further incorporation of trivalent lanthanide/actinide cations and has provided a new way to create new heterobimetallic 4f–5f and 5f–5f compounds.
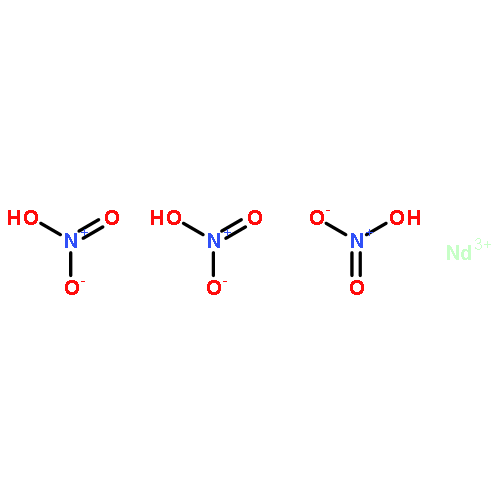
Co-reporter:Yaxing Wang, Zetian Tao, Xuemiao Yin, Jie Shu, Lanhua Chen, Daopeng Sheng, Zhifang Chai, Thomas E. Albrecht-Schmitt, and Shuao Wang
Inorganic Chemistry 2015 Volume 54(Issue 20) pp:10023-10029
Publication Date(Web):October 7, 2015
DOI:10.1021/acs.inorgchem.5b01801
The preparation of proton-conducting materials that are functional and stable at intermediate temperatures (393–573 K) is a focal point of fuel cell development. The purely inorganic material, HNd(IO3)4, which possesses a dense 3D framework structure, can reach a maximum of 4.6 × 10–4 S·cm–1 at 353 K and 95% relative humidity and exhibit a high conductivity of 8.0 × 10–5 S·cm–1 from 373 to 553 K under the flow of wet N2. HNd(IO3)4 exhibits a variety of improvements including high thermal stability, low solubility in water, and resistance to reducing atmosphere. The proton conductivity in such a wide temperature range originates from the intrinsic liberated protons in the structure and the resulting 1D hydrogen-bonding network confirmed by bond valence sum calculation and solid-state NMR analysis. Moreover, two different activation energies are observed in different temperature regions (0.23 eV below 373 K and 0.026 eV from 373 to 553 K), indicating that two types of proton motion are responsible for proton diffusion, as further domenstrated by temperature-dependent open-circuit voltage hysteresis in a tested fuel cell assembly as well as variable-temperature and double quantum filtered solid-state NMR measurements.
Co-reporter:Yuxiang Li, Zhehui Weng, Yanlong Wang, Lanhua Chen, Daopeng Sheng, Yunhai Liu, Juan Diwu, Zhifang Chai, Thomas E. Albrecht-Schmitt and Shuao Wang
Dalton Transactions 2015 vol. 44(Issue 48) pp:20867-20873
Publication Date(Web):29 Oct 2015
DOI:10.1039/C5DT03363B
The solvothermal reaction of thorium nitrate and tris-(4-carboxylphenyl)phosphine oxide in DMF affords a centrosymmetric porous thorium organic framework compound [Th(TPO)(OH)(H2O)]·8H2O (1). In contrast, the ionothermal reaction of the same reagents in the ionic liquid 1-butyl-2,3-dimethylimidazolium chloride results in the formation of a rare example of a chiral and porous thorium organic framework compound, [C9H17N2][Th(TPO)Cl2]·18H2O (2), which is derived solely from achiral starting materials. The geometries of the Th(IV) centers in compounds 1 and 2 are both atypical for low valent actinides, which can be best described as a ten-coordinate spherical sphenocorona and an irregular muffin, respectively. A large cavity of 17.5 Å (max. face to face) × 8 Å (min. face to face) with a BET surface area of 623 m2 g−1 in compound 2 is observed. The poor stability indicated by thermal gravimetric analysis and the water-resistance test for compound 2 may be due to the unique anisotropic coordination geometry for thorium. Temperature-dependent luminescence studies for both compounds indicate that the trends in the intensity vary as the Th–Th distance and the coordination environments of Th(IV) centers change.
Co-reporter:Tao Zheng, Yang Gao, Lanhua Chen, Zhiyong Liu, Juan Diwu, Zhifang Chai, Thomas E. Albrecht-Schmitt and Shuao Wang
Dalton Transactions 2015 vol. 44(Issue 41) pp:18158-18166
Publication Date(Web):14 Sep 2015
DOI:10.1039/C5DT02667A
The ionothermal reactions of uranyl nitrate and 1,3-pbpH4 (1,3-pbpH4 = 1,3-phenylenebis(phosphonic acid) ligand in ionic liquids of [C4mim][Dbp], [C4mpyr][Br], and [Etpy][Br], respectively, afforded three new uranyl phosphonates, namely [C4mim][(UO2)2(1,3-pbpH)(1,3-pbpH)·Hmim] (1), [UO2(1,3-pbpH2)H2O·mpr] (2), and [Etpy][UO2(1,3-pbpH2)F] (3). Compound 1 exhibits a rare example of a chiral uranyl phosphonate 3D framework structure built from achiral building units of tetragonal bipyramidal uranium polyhedra and 1,3-pbp ligands. The structure adopts a network with channels extending along the b axis, which are filled with C4mim+ and protonated 1-methylimidazole. In sharp contrast, compounds 2 & 3 both show pillared topology composed of uranyl pentagonal bipyramid polyhedra and phosphonate ligands. The layers are neutral in compound 2 with N-methylpyrrole molecules in the interlayer space, while compound 3 adopts anionic layer, and the charge is compensated with N-ethyl-pyridinium cations between the layers. Although compounds 1, 2, and 3 were synthesized under identical conditions with sole variation of the ionic liquid species, the resulting structures show a rich diversity in the local coordination environment of uranyl ions, the protonation of the phosphonate ligand, the conformation of ionic liquid ions, and the overall arrangement of the structure. All compounds were characterized by absorption, temperature dependent fluorescence, as well as infrared and Raman spectroscopies.
Co-reporter:Yanlong Wang, Yuxiang Li, Zhuanling Bai, Chengliang Xiao, Zhiyong Liu, Wei Liu, Lanhua Chen, Weiwei He, Juan Diwu, Zhifang Chai, Thomas E. Albrecht-Schmitt and Shuao Wang
Dalton Transactions 2015 vol. 44(Issue 43) pp:18810-18814
Publication Date(Web):29 Sep 2015
DOI:10.1039/C5DT02337H
The solvothermal reaction of [tris-(4-carboxylphenyl)phosphineoxide] (H3TPO) with UO2(NO3)2·6H2O in DMF affords a uranium-based chiral, microporous, metal–organic framework compound [(CH3)2NH2][UO2(TPO)]·4DMF·12.5H2O (SCU-3) that exhibits the highest void volume (68.8%) and second-harmonic generation (SHG) efficiency (1.54 KDP) for an actinide compound reported to date. The combination of large channels and the coordination capabilities of PO moieties in the structure enables SCU-3 to capture large amounts of Th(IV) from aqueous solutions, providing a new strategy for preparing heterobimetallic 5f/5f compounds, and may lead to applications in nuclear waste management.
Co-reporter:Tao Zheng, Yang Gao, Lanhua Chen, Juan Diwu, Zhifang Chai, Thomas E. Albrecht-Schmitt, Shuao Wang
Inorganica Chimica Acta 2015 Volume 435() pp:131-136
Publication Date(Web):24 August 2015
DOI:10.1016/j.ica.2015.06.011
•Two uranyl phosphonates were obtained by ionothermal synthesis.•Oxalate ligand was obtained in-situ from acetic anion.•Temperature-dependent fluorescence spectra showed the intensity of the emission was in negative correlation with temperature.Two new uranyl phosphonates were synthesized by using p-xylenediphosphonic acid (pmbH4) to react with uranyl salts in Emim or BMMim ([EMim][PF6] = 1-ethyl-3-methylimidazolium hexafluorophosphate; [BMMim]Cl = 1-butyl-2,3-dimethylimidazolium chloride) at elevated temperature, namely [EMim][UO2(pmbH2)0.5(pmbH2)0.5(ox)0.5] (1) and [BMMim]2[(UO2)2(pmbH2)(pmb)] (2). Both compounds adopt two dimensional structures, consisting of an anionic layer of uranyl phosphonate, with cations filled in the interlayer spaces. The spectroscopic properties of the compounds were studied extensively using UV–Vis absorption, temperature dependent fluorescence, Raman and IR spectroscopies. The UV–Vis absorption spectrum contains typical peaks of uranyl complexes at around 320 nm and 420 nm, which are originated from the charge transfer transitions from O 2p orbitals to the U 5f/6d orbitals. Both compounds adopt five strong emission peaks, whose intensities increase with the decrease of temperature, as observed in the temperature dependent fluorescence spectra. In Raman and IR spectra, the peaks corresponding to the stretch of C–H/O–H, P–O/PO, and OUO bonds could all be located and assigned.Two novel layered uranyl phosphonate compounds were obtained by ionothermal method. These compounds were studied comprehensively using IR, Raman, UV–Vis absorption, and variable temperature fluorescence spectroscopy.
Co-reporter:Xiaowen Shu, Yingjie Wang, Shuang Zhang, Li Huang, Shuao Wang, Daoben Hua
Talanta 2015 Volume 131() pp:198-204
Publication Date(Web):January 2015
DOI:10.1016/j.talanta.2014.07.085
•A novel thermo-sensitive polymeric sensor was used to determine trace uranyl.•The fluorescent sensor exhibits a stable response for uranyl with high selectivity.•The determination of trace uranyl was achieved by centrifugation above 32 °C.Uranyl ion exists at trace levels in the environment and can cause severe adverse effects to human health. Therefore, it is desirable to develop analytical methods that can determine the trace uranyl ion in aqueous medium. We report here a new method using a thermo–responsive polymeric fluorescent sensor. Specifically, 5,10,15,20–tetrakis(4–carboxyphenyl)–porphyrin terminated poly(N–isopropylacrylamide) (TCPP–PNIPAM) was synthesized by controlled free radical polymerization for the detection of uranyl ion. The maximum fluorescence intensity at ~658 nm of TCPP–PNIPAM increases with molecular weights and is also closely related to the temperature. The polymeric sensor is sensitive to pH (1.0~5.0) with a fast responsive time (~3 min). Under optimized experimental conditions, the sensor exhibits a stable response for uranyl ion with high selectivity over a concentration range from 1.0×10–3 to 1.0×10–7 mol/L. For the trace uranyl ion (such as 1.0×10–8 or 10–9 mol/L), the determination could be successfully achieved after concentrating 100 times by centrifugation above 32 °C. The properties enable the polymeric sensor to have great potential for environmental application.
Co-reporter: Chengliang Xiao;Yaxing Wang;Lanhua Chen;Xuemiao Yin;Dr. Jie Shu;Daopeng Sheng; Zhifang Chai; Thomas E. Albrecht-Schmitt; Shuao Wang
Chemistry - A European Journal 2015 Volume 21( Issue 49) pp:17591-17595
Publication Date(Web):
DOI:10.1002/chem.201503733
Abstract
The limited long-term hydrolytic stability of rapidly emerging 3D-extended framework materials (MOFs, COFs, MOPs, etc.) is still one of major barriers for their practical applications as new solid-state electrolytes in fuel cells. To obtain hydrolytically stable materials, two H2PO4−-exchanged 3D inorganic cationic extended frameworks (CEFs) were successfully prepared by a facile anion-exchange method. Both anion-exchanged CEFs (YbO(OH)P and NDTBP) show significantly enhanced proton conductivity when compared with the original materials (YbO(OH)Cl and NDTB) with an increase of up to four orders-of-magnitude, reaching 2.36×10−3 and 1.96×10−2 S cm−1 at 98 % RH and 85 °C for YbO(OH)P and NDTBP, respectively. These values are comparable to the most efficient proton-conducting MOFs. In addition, these two anion-exchanged materials are stable in boiling water, which originates from the strong electrostatic interaction between the H2PO4− anion and the cationic host framework, showing a clear advance over all the acid-impregnated materials (H2SO4@MIL-101, H3PO4@MIL-101, and H3PO4@Tp-Azo) as practical solid-state fuel-cell electrolytes. This work offers a new general and efficient approach to functionalize 3D-extended frameworks through an anion-exchange process and achieves water-stability with ultra-high proton conductivity above 10−2 S cm−1.
Co-reporter:Tao Zheng, Yang Gao, Daxiang Gui, Lanhua Chen, Daopeng Sheng, Juan Diwu, Zhifang Chai, Thomas E. Albrecht-Schmitt and Shuao Wang
Dalton Transactions 2016 - vol. 45(Issue 22) pp:NaN9035-9035
Publication Date(Web):2016/05/06
DOI:10.1039/C6DT01011C
The example of phase transformation from a centrosymmetric space group at low temperature (LT) to a chiral space group at high temperature (HT) is reported, which was clearly resolved in a single-crystal-to-single-crystal manner in a 3D uranyl(VI) phosphonate compound [TMA][(UO2)2(1,3-pbpH)(1,3-pbpH2)] (1) (TMA+ = tetramethylammonium cation; 1,3-pbpH4 = 1,3-phenylenebis(phosphonic acid)).
Co-reporter:Yanlong Wang, Yuxiang Li, Zhuanling Bai, Chengliang Xiao, Zhiyong Liu, Wei Liu, Lanhua Chen, Weiwei He, Juan Diwu, Zhifang Chai, Thomas E. Albrecht-Schmitt and Shuao Wang
Dalton Transactions 2015 - vol. 44(Issue 43) pp:NaN18814-18814
Publication Date(Web):2015/09/29
DOI:10.1039/C5DT02337H
The solvothermal reaction of [tris-(4-carboxylphenyl)phosphineoxide] (H3TPO) with UO2(NO3)2·6H2O in DMF affords a uranium-based chiral, microporous, metal–organic framework compound [(CH3)2NH2][UO2(TPO)]·4DMF·12.5H2O (SCU-3) that exhibits the highest void volume (68.8%) and second-harmonic generation (SHG) efficiency (1.54 KDP) for an actinide compound reported to date. The combination of large channels and the coordination capabilities of PO moieties in the structure enables SCU-3 to capture large amounts of Th(IV) from aqueous solutions, providing a new strategy for preparing heterobimetallic 5f/5f compounds, and may lead to applications in nuclear waste management.
Co-reporter:Tao Zheng, Yang Gao, Lanhua Chen, Zhiyong Liu, Juan Diwu, Zhifang Chai, Thomas E. Albrecht-Schmitt and Shuao Wang
Dalton Transactions 2015 - vol. 44(Issue 41) pp:NaN18166-18166
Publication Date(Web):2015/09/14
DOI:10.1039/C5DT02667A
The ionothermal reactions of uranyl nitrate and 1,3-pbpH4 (1,3-pbpH4 = 1,3-phenylenebis(phosphonic acid) ligand in ionic liquids of [C4mim][Dbp], [C4mpyr][Br], and [Etpy][Br], respectively, afforded three new uranyl phosphonates, namely [C4mim][(UO2)2(1,3-pbpH)(1,3-pbpH)·Hmim] (1), [UO2(1,3-pbpH2)H2O·mpr] (2), and [Etpy][UO2(1,3-pbpH2)F] (3). Compound 1 exhibits a rare example of a chiral uranyl phosphonate 3D framework structure built from achiral building units of tetragonal bipyramidal uranium polyhedra and 1,3-pbp ligands. The structure adopts a network with channels extending along the b axis, which are filled with C4mim+ and protonated 1-methylimidazole. In sharp contrast, compounds 2 & 3 both show pillared topology composed of uranyl pentagonal bipyramid polyhedra and phosphonate ligands. The layers are neutral in compound 2 with N-methylpyrrole molecules in the interlayer space, while compound 3 adopts anionic layer, and the charge is compensated with N-ethyl-pyridinium cations between the layers. Although compounds 1, 2, and 3 were synthesized under identical conditions with sole variation of the ionic liquid species, the resulting structures show a rich diversity in the local coordination environment of uranyl ions, the protonation of the phosphonate ligand, the conformation of ionic liquid ions, and the overall arrangement of the structure. All compounds were characterized by absorption, temperature dependent fluorescence, as well as infrared and Raman spectroscopies.
Co-reporter:Yuxiang Li, Zhehui Weng, Yanlong Wang, Lanhua Chen, Daopeng Sheng, Yunhai Liu, Juan Diwu, Zhifang Chai, Thomas E. Albrecht-Schmitt and Shuao Wang
Dalton Transactions 2015 - vol. 44(Issue 48) pp:NaN20873-20873
Publication Date(Web):2015/10/29
DOI:10.1039/C5DT03363B
The solvothermal reaction of thorium nitrate and tris-(4-carboxylphenyl)phosphine oxide in DMF affords a centrosymmetric porous thorium organic framework compound [Th(TPO)(OH)(H2O)]·8H2O (1). In contrast, the ionothermal reaction of the same reagents in the ionic liquid 1-butyl-2,3-dimethylimidazolium chloride results in the formation of a rare example of a chiral and porous thorium organic framework compound, [C9H17N2][Th(TPO)Cl2]·18H2O (2), which is derived solely from achiral starting materials. The geometries of the Th(IV) centers in compounds 1 and 2 are both atypical for low valent actinides, which can be best described as a ten-coordinate spherical sphenocorona and an irregular muffin, respectively. A large cavity of 17.5 Å (max. face to face) × 8 Å (min. face to face) with a BET surface area of 623 m2 g−1 in compound 2 is observed. The poor stability indicated by thermal gravimetric analysis and the water-resistance test for compound 2 may be due to the unique anisotropic coordination geometry for thorium. Temperature-dependent luminescence studies for both compounds indicate that the trends in the intensity vary as the Th–Th distance and the coordination environments of Th(IV) centers change.
Co-reporter:Xiaomei Xu, Zhiyong Liu, Shitong Yang, Lanhua Chen, Juan Diwu, Evgeny V. Alekseev, Zhifang Chai, Thomas E. Albrecht-Schmitt and Shuao Wang
Dalton Transactions 2016 - vol. 45(Issue 39) pp:NaN15472-15472
Publication Date(Web):2016/08/29
DOI:10.1039/C6DT03299K
The equatorial coordination nature of the uranyl unit has resulted in only three uranyl borate 3D framework compounds reported so far formed from boric acid flux reactions conducted at 190 °C while all others are 2D layers. Here in this work, by increasing the reaction temperature to 250 °C, a new potassium uranyl borate K[(UO2)B6O10(OH)] (KUBO-4) framework compound is synthesized that shares the same layer topology with the previously reported 2D layered KUBO-1. The 3D structure of KUBO-4 is achieved by interlayer hydroborate condensation. The KUBO-4 was further characterized with single crystal XRD, SHG and fluorescence spectra, and TG/DSC measurements. A deep understanding regarding the dissolution behaviours of uranyl borate is achieved via solubility studies of the KUBO-1 and KUBO-4 performed using a combination of ICP-MS, powder XRD, and fluorescence spectroscopy techniques. The results confirm the lack of stability of borates in aqueous solutions with the presence of coordinating ligands in the environment regardless of the structure types.
Co-reporter:Yuxiang Li, Zhehui Weng, Yanlong Wang, Lanhua Chen, Daopeng Sheng, Juan Diwu, Zhifang Chai, Thomas E. Albrecht-Schmitt and Shuao Wang
Dalton Transactions 2016 - vol. 45(Issue 3) pp:NaN921-921
Publication Date(Web):2015/12/07
DOI:10.1039/C5DT04183J
Three thorium(IV)-based metal–organic hybrid compounds with 2D layered and 3D framework structures exhibiting graphene-like (6,3) sheet topologies were prepared with linkers with threefold symmetry. These compounds contain rare and relatively anisotropic coordination environments for low-valent actinides that are similar to those often observed for high-valent actinide ions.

![Benzoic acid, 4,4',4''-[20-[4-(7,7-dimethyl-1,6-dioxo-9-thioxo-2,5-dioxa-8,10-dithiadocos-1-yl)phenyl]-21H,23H-porphine-5,10,15-triyl]tris-](/data/chemimg/3723900/1624970-61-1.png)
![Benzoic acid, 4,4',4''-[20-[4-(7,7-dimethyl-1,6-dioxo-9-thioxo-2,5-dioxa-8,10-dithiadocos-1-yl)phenyl]-21H,23H-porphine-5,10,15-triyl]tris-](/data/chemimg/3723900/1624970-61-1_b.png)
![Pyridinium, 1,1',1''-[(2,4,6-trimethyl-1,3,5-benzenetriyl)tris(methylene)]tris[4-carboxy-, bromide (1:3)](/data/chemimg/3718700/1246090-62-9.png)
![Pyridinium, 1,1',1''-[(2,4,6-trimethyl-1,3,5-benzenetriyl)tris(methylene)]tris[4-carboxy-, bromide (1:3)](/data/chemimg/3718700/1246090-62-9_b.png)
![[1,1':3',1''-Terphenyl]-4,4'',5'-tricarboxylic acid](/data/chemimg/182600/1263218-51-4.png)
![[1,1':3',1''-Terphenyl]-4,4'',5'-tricarboxylic acid](/data/chemimg/182600/1263218-51-4_b.png)




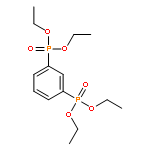
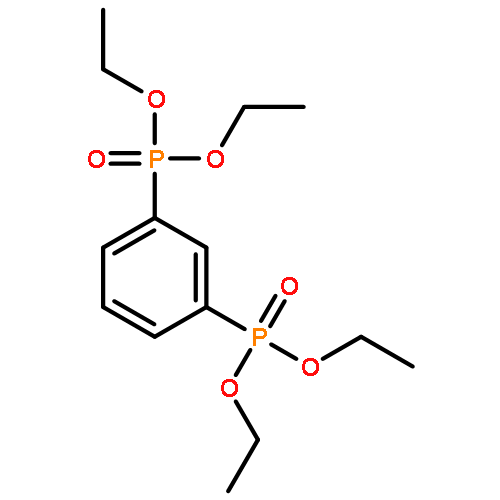

























![[4-(phosphonomethyl)phenyl]methylphosphonic Acid](http://img.cochemist.com/ccimg/4600/4546-06-9.png)
![[4-(phosphonomethyl)phenyl]methylphosphonic Acid](http://img.cochemist.com/ccimg/4600/4546-06-9_b.png)





