Co-reporter:Raquel Travieso-Puente, Simon Budzak, Juan Chen, Peter Stacko, Johann T. B. H. Jastrzebski, Denis Jacquemin, and Edwin Otten
Journal of the American Chemical Society March 8, 2017 Volume 139(Issue 9) pp:3328-3328
Publication Date(Web):February 20, 2017
DOI:10.1021/jacs.6b12585
A straightforward synthetic route to arylazoindazoles via nucleophilic aromatic substitution is presented. Upon deprotonation of the NH group, a C6F5-substituted formazan undergoes facile cyclization as a result of intermolecular nucleophilic substitution (SNAr). This new class of azo photoswitches containing an indazole five-membered heterocycle shows photochemical isomerization with high fatigue resistance. In addition, the Z-isomers have long thermal half-lives in the dark of up to several days at room temperature. The fluorinated indazole group offers a handle for further functionalization and tuning of its properties, as it is shown to be susceptible to a subsequent, highly selective nucleophilic displacement reaction.
Co-reporter:Ranajit Mondol;Daan A. Snoeken;Mu-Chieh Chang
Chemical Communications 2017 vol. 53(Issue 3) pp:513-516
Publication Date(Web):2017/01/03
DOI:10.1039/C6CC08166E
Redox-active formazanate ligands are emerging as tunable electron-reservoirs in coordination chemistry. Here we show that boron diphenyl complexes with formazanate ligands, despite their (formal) negative charge, can be further reduced by up to two electrons. A combined crystallographic, spectroscopic and computational study establishes that formazanate ligands are stable in mono-, di- and trianionic form.
Co-reporter:Raquel Travieso-Puente; J. O. P. Broekman; Mu-Chieh Chang; Serhiy Demeshko; Franc Meyer
Journal of the American Chemical Society 2016 Volume 138(Issue 17) pp:5503-5506
Publication Date(Web):April 14, 2016
DOI:10.1021/jacs.6b01552
Spin-crossover in a pseudo-tetrahedral bis(formazanate) iron(II) complex (1) is described. Structural, magnetic, and spectroscopic analyses indicate that this compound undergoes thermal switching between an S=0 and an S=2 state, which is very rare in four-coordinate complexes. The transition to the high-spin state is accompanied by an increase in Fe–N bond lengths and a concomitant contraction of intraligand N–N bonds. The latter suggests that stabilization of the low-spin state is due to the π-acceptor properties of the ligand. One-electron reduction of 1 leads to the formation of the corresponding anion, which contains a low-spin (S=1/2) Fe(I) center. The findings are rationalized by electronic structure calculations using density functional theory.
Co-reporter:M.-C. Chang, A. Chantzis, D. Jacquemin and E. Otten
Dalton Transactions 2016 vol. 45(Issue 23) pp:9477-9484
Publication Date(Web):10 May 2016
DOI:10.1039/C6DT01226D
The synthesis of a series of (formazanate)boron difluorides and their 1-electron reduction products is described. The neutral compounds are fluorescent with large Stokes shifts. DFT calculations suggest that a large structural reorganization accompanies photoexictation and accounts for the large Stokes shift. Reduction of the neutral boron difluorides occurs at the ligand and generates the corresponding radical anions. These complexes are non-fluorescent, allowing switching of the emission by changing the ligand oxidation state.
Co-reporter:Mu-Chieh Chang and Edwin Otten
Organometallics 2016 Volume 35(Issue 4) pp:534-542
Publication Date(Web):February 5, 2016
DOI:10.1021/acs.organomet.5b00968
The solution-phase thermolysis of (formazanate)boron dihydrides (LBH2; 2) results in the formation of aminoborane compounds (4) via a series of boron-to-ligand intramolecular hydride transfer reactions. Monitoring the reactions by NMR spectroscopy allowed identification of several intermediates, and a reaction mechanism is proposed. In the case of a ligand with an N-Mes substituent it was possible to characterize an intermediate (7b-i) during this transformation that shows an unexpected cyclohexadiene moiety, which results from hydride transfer to the ortho-position of the mesityl substituent. Two consecutive boron-to-ligand hydride transfers eventually result in reductive N–N bond cleavage to give triazaboroles as the final product.
Co-reporter:Mu-Chieh Chang; Peter Roewen; Raquel Travieso-Puente; Martin Lutz
Inorganic Chemistry 2015 Volume 54(Issue 1) pp:379-388
Publication Date(Web):December 10, 2014
DOI:10.1021/ic5025873
A range of tetrahedral bis(formazanate)zinc complexes with different steric and electronic properties of the formazanate ligands were synthesized. The solid-state structures for several of these were determined by X-ray crystallography, which showed that complexes with symmetrical, unhindered ligands prefer coordination to the zinc center via the terminal N atoms of the NNCNN ligand backbone. Steric or electronic modifications can override this preference and give rise to solid-state structures in which the formazanate ligand forms a 5-membered chelate by binding to the metal center via an internal N atom. In solution, these compounds show dynamic equilibria that involve both 5- and 6-membered chelates. All compounds are intensely colored, and the effect of the ligand substitution pattern on the UV–vis absorption spectra was evaluated. In addition, their cyclic voltammetry is reported, which shows that all compounds may be electrochemically reduced to radical anionic (L2Zn–) and dianionic (L2Zn2–) forms. While unhindered NAr substituents lie in the plane of the ligand backbone (Ar = Ph), the introduction of sterically demanding substituents (Ar = Mes) favors a perpendicular orientation in which the NMes group is no longer in conjugation with the backbone, resulting in hypsochromic shifts in the absorption spectra. The redox potentials in the series of L2Zn compounds may be altered in a straightforward manner over a relatively wide range (∼700 mV) via the introduction of electron-donating or -withdrawing substituents on the formazanate framework.
Co-reporter:M.-C. Chang and E. Otten
Chemical Communications 2014 vol. 50(Issue 56) pp:7431-7433
Publication Date(Web):19 May 2014
DOI:10.1039/C4CC03244F
Mono(formazanate) boron difluoride complexes (LBF2), which show remarkably facile and reversible ligand-based redox-chemistry, were synthesized by transmetallation of bis(formazanate) zinc complexes with boron trifluoride. The one-electron reduction product [LBF2]−[Cp2Co]+ and a key intermediate for the transmetallation reaction, the six-coordinate zinc complex (L(BF3))2Zn were isolated and fully characterized.
Co-reporter:Raquel Travieso-Puente, Mu-Chieh Chang and Edwin Otten
Dalton Transactions 2014 vol. 43(Issue 48) pp:18035-18041
Publication Date(Web):03 Oct 2014
DOI:10.1039/C4DT02578D
Alkali metal salts of redox-active formazanate ligands were prepared, and their structures in the solid-state and in solution are determined. The nitrogen-rich [NNCNN] backbone of formazanates results in a varied coordination chemistry, with both the internal and terminal nitrogen atoms available for bonding with the alkali metal. The potassium salt K[PhNNC(p-tol)NNPh]·2THF (1-K) is dimeric in the solid state and even in THF solution, as a result of the K atom bridging via interaction with a terminal N atom and the aromatic ring of a second unit. Conversely, for the compounds Na[MesNNC(CN)NNMes]·2THF (2-Na) and Na[PhNNC(tBu)NNPh] (3-Na) polymeric and hexameric structures are found in the solid state respectively. The preference for binding the alkali metal through internal N atoms (1-K and 2-Na) to give a 4-membered chelate, or via internal/external N atoms (5-membered chelate in 3-Na), contrasts with the 6-membered chelate mode observed in our recently reported formazanate zinc complexes.
Co-reporter:Mu-Chieh Chang;Thomas Dann;Dr. David P. Day;Dr. Martin Lutz;Dr. Gregory G. Wildgoose;Dr. Edwin Otten
Angewandte Chemie 2014 Volume 126( Issue 16) pp:4202-4206
Publication Date(Web):
DOI:10.1002/ange.201309948
Abstract
The synthesis of bis(formazanate) zinc complexes is described. These complexes have well-behaved redox-chemistry, with the ligands functioning as a reversible electron reservoir. This allows the synthesis of bis(formazanate) zinc compounds in three redox states in which the formazanate ligands are reduced to “metallaverdazyl” radicals. The stability of these ligand-based radicals is a result of the delocalization of the unpaired electron over four nitrogen atoms in the ligand backbone. The neutral, anionic, and dianionic compounds (L2Zn0/−1/−2) were fully characterized by single-crystal X-ray crystallography, spectroscopic methods, and DFT calculations. In these complexes, the structural features of the formazanate ligands are very similar to well-known β-diketiminates, but the nitrogen-rich (NNCNN) backbone of formazanates opens the door to redox-chemistry that is otherwise not easily accessible.
Co-reporter:Mu-Chieh Chang;Thomas Dann;Dr. David P. Day;Dr. Martin Lutz;Dr. Gregory G. Wildgoose;Dr. Edwin Otten
Angewandte Chemie 2014 Volume 126( Issue 16) pp:
Publication Date(Web):
DOI:10.1002/ange.201401122
Co-reporter:Mu-Chieh Chang;Thomas Dann;Dr. David P. Day;Dr. Martin Lutz;Dr. Gregory G. Wildgoose;Dr. Edwin Otten
Angewandte Chemie International Edition 2014 Volume 53( Issue 16) pp:4118-4122
Publication Date(Web):
DOI:10.1002/anie.201309948
Abstract
The synthesis of bis(formazanate) zinc complexes is described. These complexes have well-behaved redox-chemistry, with the ligands functioning as a reversible electron reservoir. This allows the synthesis of bis(formazanate) zinc compounds in three redox states in which the formazanate ligands are reduced to “metallaverdazyl” radicals. The stability of these ligand-based radicals is a result of the delocalization of the unpaired electron over four nitrogen atoms in the ligand backbone. The neutral, anionic, and dianionic compounds (L2Zn0/−1/−2) were fully characterized by single-crystal X-ray crystallography, spectroscopic methods, and DFT calculations. In these complexes, the structural features of the formazanate ligands are very similar to well-known β-diketiminates, but the nitrogen-rich (NNCNN) backbone of formazanates opens the door to redox-chemistry that is otherwise not easily accessible.
Co-reporter:Mu-Chieh Chang;Thomas Dann;Dr. David P. Day;Dr. Martin Lutz;Dr. Gregory G. Wildgoose;Dr. Edwin Otten
Angewandte Chemie International Edition 2014 Volume 53( Issue 16) pp:
Publication Date(Web):
DOI:10.1002/anie.201401122
Co-reporter:Sébastien Perdriau;Mu-Chieh Chang;Dr. Edwin Otten;Dr. Hero J. Heeres;Dr. Johannes G. de Vries
Chemistry - A European Journal 2014 Volume 20( Issue 47) pp:15434-15442
Publication Date(Web):
DOI:10.1002/chem.201403236
Abstract
The [Ru(CO)H(PNN)] pincer complex based on a dearomatised PNN ligand (PNN: 2-di-tert-butylphosphinomethyl-6-diethylaminomethylpyridine) was examined for its ability to isomerise alkenes. The isomerisation reaction proceeded under mild conditions after activation of the complex with alcohols. Variable-temperature (VT) NMR experiments to investigate the role of the alcohol in the mechanism lend credence to the hypothesis that the first step involves the formation of a rearomatised alkoxide complex. In this complex, the hemilabile diethylamino side-arm can dissociate, allowing alkene binding cis to the hydride, enabling insertion of the alkene into the metal–hydride bond, whereas in the parent complex only trans binding is possible. During this study, a new uncommon Ru0 coordination complex was also characterised. The scope of the alkene isomerisation reaction was examined.
Co-reporter:Edwin Otten, Auke Meetsma, and Bart Hessen
Organometallics 2012 Volume 31(Issue 17) pp:6071-6079
Publication Date(Web):August 16, 2012
DOI:10.1021/om300421m
The cationic tantalum complex {[η6-Ar-CMe2-η5-C5H4]TaPr}[B(C6F5)4] (1; Ar = 3,5-Me2C6H3) serves as a starting material for a series of neutral, monocationic, and dicationic derivatives. The cationic hydride {[η6-Ar-CMe2-η5-C5H4]TaH}[B(C6F5)4] (2) that results from hydrogenolysis of 1 inserts the di- and trisubstituted olefins cyclopentene and 2-methyl-2-pentene; it reacts with styrene to give the 2,1-insertion product, for which the Ta–CH(Me)Ph group is bound in a σ3-allylic fashion. A neutral complex is obtained from 1 by reaction with Br–, and a dicationic derivative is available by hydride abstraction from 2 using the Lewis acidic trityl cation. All compounds described here retain the unusual ansa-(η5-cyclopentadienyl,η6-arene) coordination mode of the ligand that stabilizes the formally Ta(III) center. X-ray structures and DFT calculations show that the metal–arene interaction contains a significant π back-donation component (arene←Ta(III)) that differs only slightly in the series, despite the variation of charge at the metal center.
Co-reporter:Mu-Chieh Chang
Inorganic Chemistry () pp:
Publication Date(Web):
DOI:10.1021/acs.inorgchem.5b01287
Despite the current interest in structure and reactivity of sub-valent main group compounds, neutral boron analogues of N-heterocyclic carbenes have been elusive due to their high reactivity. Here we provide evidence that 2-electron reduction of a (formazanate)BF2 precursor leads to NaF elimination and formation of an N-heterocyclic boron carbenoid, and describe the formation of a series of unusual BN heterocycles that result from trapping of this fragment. Subsequent chemical oxidation by XeF2 demonstrates that the trapped (formazanate)B fragment retains carbenoid character and regenerates the boron difluoride starting material in good yield. These results indicate that the formazanate ligand framework provides a unique entry into sub-valent boron chemistry.
Co-reporter:M.-C. Chang, A. Chantzis, D. Jacquemin and E. Otten
Dalton Transactions 2016 - vol. 45(Issue 23) pp:NaN9484-9484
Publication Date(Web):2016/05/10
DOI:10.1039/C6DT01226D
The synthesis of a series of (formazanate)boron difluorides and their 1-electron reduction products is described. The neutral compounds are fluorescent with large Stokes shifts. DFT calculations suggest that a large structural reorganization accompanies photoexictation and accounts for the large Stokes shift. Reduction of the neutral boron difluorides occurs at the ligand and generates the corresponding radical anions. These complexes are non-fluorescent, allowing switching of the emission by changing the ligand oxidation state.
Co-reporter:Ranajit Mondol, Daan A. Snoeken, Mu-Chieh Chang and Edwin Otten
Chemical Communications 2017 - vol. 53(Issue 3) pp:NaN516-516
Publication Date(Web):2016/11/14
DOI:10.1039/C6CC08166E
Redox-active formazanate ligands are emerging as tunable electron-reservoirs in coordination chemistry. Here we show that boron diphenyl complexes with formazanate ligands, despite their (formal) negative charge, can be further reduced by up to two electrons. A combined crystallographic, spectroscopic and computational study establishes that formazanate ligands are stable in mono-, di- and trianionic form.
Co-reporter:Linda E. Eijsink, Sébastien C. P. Perdriau, Johannes G. de Vries and Edwin Otten
Dalton Transactions 2016 - vol. 45(Issue 40) pp:NaN16039-16039
Publication Date(Web):2016/08/25
DOI:10.1039/C6DT02478E
The pincer complex (PNN)RuH(CO), with a de-aromatized pyridine in the ligand backbone, is shown to react with nitriles in a metal–ligand cooperative manner. This leads to the formation of a series of complexes with new Ru–N(nitrile) and C(ligand)–C(nitrile) bonds. The initial nitrile cycloaddition products, the ketimido complexes 3, have a Brønsted basic (nitrile-derived) Ru–N fragment. This is able to deprotonate a CH2 side-arm of the pincer ligand to give ketimine complexes (4) with a de-aromatized pyridine backbone. Alternatively, the presence of a CH2 group adjacent to the nitrile functionality can lead to tautomerization to an enamido complex (5). Variable-temperature NMR studies and DFT calculations provide insight in the relative stability of these compounds and highlight the importance of their facile interconversion in the context of subsequent nitrile transformations.
Co-reporter:Raquel Travieso-Puente, Mu-Chieh Chang and Edwin Otten
Dalton Transactions 2014 - vol. 43(Issue 48) pp:NaN18041-18041
Publication Date(Web):2014/10/03
DOI:10.1039/C4DT02578D
Alkali metal salts of redox-active formazanate ligands were prepared, and their structures in the solid-state and in solution are determined. The nitrogen-rich [NNCNN] backbone of formazanates results in a varied coordination chemistry, with both the internal and terminal nitrogen atoms available for bonding with the alkali metal. The potassium salt K[PhNNC(p-tol)NNPh]·2THF (1-K) is dimeric in the solid state and even in THF solution, as a result of the K atom bridging via interaction with a terminal N atom and the aromatic ring of a second unit. Conversely, for the compounds Na[MesNNC(CN)NNMes]·2THF (2-Na) and Na[PhNNC(tBu)NNPh] (3-Na) polymeric and hexameric structures are found in the solid state respectively. The preference for binding the alkali metal through internal N atoms (1-K and 2-Na) to give a 4-membered chelate, or via internal/external N atoms (5-membered chelate in 3-Na), contrasts with the 6-membered chelate mode observed in our recently reported formazanate zinc complexes.
Co-reporter:M.-C. Chang and E. Otten
Chemical Communications 2014 - vol. 50(Issue 56) pp:NaN7433-7433
Publication Date(Web):2014/05/19
DOI:10.1039/C4CC03244F
Mono(formazanate) boron difluoride complexes (LBF2), which show remarkably facile and reversible ligand-based redox-chemistry, were synthesized by transmetallation of bis(formazanate) zinc complexes with boron trifluoride. The one-electron reduction product [LBF2]−[Cp2Co]+ and a key intermediate for the transmetallation reaction, the six-coordinate zinc complex (L(BF3))2Zn were isolated and fully characterized.

![Taniaphos SL-T001-1, (2S)-1-[(R)-(Dimethylamino)[2-(diphenylphosphino)phenyl]methyl]-2-(diphenylphosphino)ferrocene (acc to CAS)](http://img.cochemist.com/ccimg/1003100/1003012-96-1.png)
![Taniaphos SL-T001-1, (2S)-1-[(R)-(Dimethylamino)[2-(diphenylphosphino)phenyl]methyl]-2-(diphenylphosphino)ferrocene (acc to CAS)](http://img.cochemist.com/ccimg/1003100/1003012-96-1_b.png)

















![Benzene, 1,2-dimethoxy-5-methyl-3-[(1S)-1,2,2-trimethylcyclopentyl]-](http://img.cochemist.com/ccimg/222100/222022-93-7.png)
![Benzene, 1,2-dimethoxy-5-methyl-3-[(1S)-1,2,2-trimethylcyclopentyl]-](http://img.cochemist.com/ccimg/222100/222022-93-7_b.png)









![2-Oxazolidinone, 4-(1-methylethyl)-3-[(2E)-2-methyl-1-oxo-2-butenyl]-,(4S)-](http://img.cochemist.com/ccimg/136100/136057-71-1.png)
![2-Oxazolidinone, 4-(1-methylethyl)-3-[(2E)-2-methyl-1-oxo-2-butenyl]-,(4S)-](http://img.cochemist.com/ccimg/136100/136057-71-1_b.png)









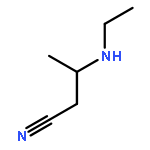
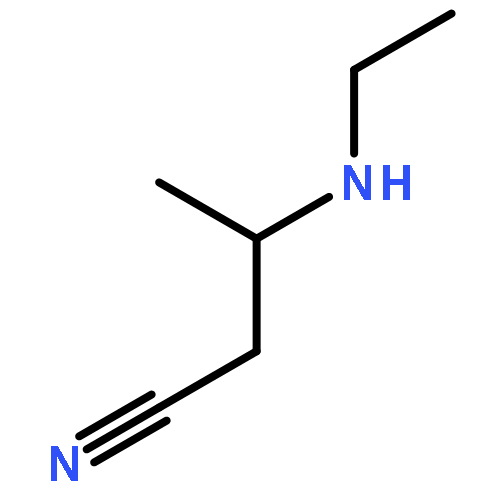
![BUTANENITRILE, 3-[(PHENYLMETHYL)AMINO]-](http://img.cochemist.com/ccimg/72700/72687-57-1.png)
![BUTANENITRILE, 3-[(PHENYLMETHYL)AMINO]-](http://img.cochemist.com/ccimg/72700/72687-57-1_b.png)



![PHOSPHINE OXIDE, DIPHENYL[(1E)-2-(TRIMETHYLSILYL)ETHENYL]-](http://img.cochemist.com/ccimg/57500/57415-83-5.png)
![PHOSPHINE OXIDE, DIPHENYL[(1E)-2-(TRIMETHYLSILYL)ETHENYL]-](http://img.cochemist.com/ccimg/57500/57415-83-5_b.png)





























![5-[(e)-prop-1-enyl]benzo[1,3]dioxole](http://img.cochemist.com/ccimg/4100/4043-71-4.png)
![5-[(e)-prop-1-enyl]benzo[1,3]dioxole](http://img.cochemist.com/ccimg/4100/4043-71-4_b.png)





















![5-Propylbenzo[d][1,3]dioxole](http://img.cochemist.com/ccimg/100/94-58-6.png)
![5-Propylbenzo[d][1,3]dioxole](http://img.cochemist.com/ccimg/100/94-58-6_b.png)


![Ruthenium,[6-[[bis(1,1-dimethylethyl)phosphino-kP]methylene]-N,N-diethyl-1,6-dihydro-2-pyridinemethanaminato-kN1,kN2]carbonylhydro-, (SP-5-52)-](http://img.cochemist.com/ccimg/864000/863971-63-5.png)
![Ruthenium,[6-[[bis(1,1-dimethylethyl)phosphino-kP]methylene]-N,N-diethyl-1,6-dihydro-2-pyridinemethanaminato-kN1,kN2]carbonylhydro-, (SP-5-52)-](http://img.cochemist.com/ccimg/864000/863971-63-5_b.png)