Co-reporter:Yueting Wang;Qile Xu;Daiying Zuo;Haiqiu Tian;Jingwen Xu;Kai Bao;Yingliang Wu;Qi Guan;Maolin Sun;Weige Zhang
ACS Medicinal Chemistry Letters December 8, 2016 Volume 7(Issue 12) pp:1202-1206
Publication Date(Web):October 17, 2016
DOI:10.1021/acsmedchemlett.6b00252
A series of 3,6-diaryl-[1,2,4]triazolo[4,3-b]pyridazines were designed as a class of vinylogous CA-4 analogues. The easily isomerized (Z,E)-butadiene linker of vinylogous CA-4 was replaced by a rigid [1,2,4]triazolo[4,3-b]pyridazine scaffold. Twenty-one target compounds were synthesized and exhibited moderate to potent antiproliferative activity. The compound 4q with a 3-amino-4-methoxyphenyl moiety as the B-ring, comparable to CA-4 (IC50 = 0.009–0.012 μM), displayed the highly active antiproliferative activity against SGC-7901, A549, and HT-1080 cell lines with IC50 values of 0.014, 0.008, and 0.012 μM, respectively. Tubulin polymerization experiments indicated that 4q effectively inhibited tubulin polymerization, and immunostaining assay revealed that 4q significantly disrupted tubulin microtubule dynamics. Moreover, cell cycle studies revealed that compound 4q dramatically arrested cell cycle progression at G2/M phase in A549 cells. Molecular modeling studies showed that 4q could bind to the colchicine binding site on microtubules.Keywords: colchicine binding site; combretastatin A-4; molecular modeling; tubulin; [1,2,4]Triazolo[4,3-b]pyridazine;
Co-reporter:Dongjie Feng;Yue Wu;Hao Wang;Zhaoshi Bai;Defa Wang;Daiying Zuo;Kai Bao;Yingliang Wu;Weige Zhang
RSC Advances (2011-Present) 2017 vol. 7(Issue 46) pp:29103-29111
Publication Date(Web):2017/05/30
DOI:10.1039/C7RA02720F
A series of 2-aryl-4-(3,4,5-trimethoxybenzoyl)-1,2,3-triazols were designed as analogs of substituted methoxybenzoyl-aryl-thiazole (SMART) under the consideration of geometric features. The target compounds were synthesized via concise and efficient processes including microwave-assisted cyclization, and were evaluated for their antiproliferative activity against three human cancer cell lines. Most compounds exhibited moderate antiproliferative activity with IC50 values in the micromolar to sub-micromolar range. Tubulin polymerization and immunofluorescence studies demonstrated that (Z)-9a was a potent microtubule-destabilizing agent and disrupted the polymerization dynamics. Moreover, (Z)-9a significantly induced accumulation of cells in the G2/M phase and caused microtubule destabilization. Molecular modeling studies showed that (Z)-9a probably binds to the colchicine site of tubulin.
Co-reporter:Daiying Zuo, Xuewei Jiang, Mengting Han, Jiwei Shen, Binyue Lang, Qi Guan, Zhaoshi Bai, Chunming Han, Zengqiang Li, Weige Zhang, Yingliang Wu
Toxicology in Vitro 2017 Volume 42(Volume 42) pp:
Publication Date(Web):1 August 2017
DOI:10.1016/j.tiv.2017.04.019
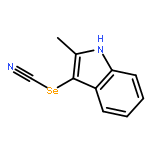
•M-24 is newly synthesized analogue of nocodazole.•M-24 behaves as a tubulin inhibitor.•M-24 exhibits potent anticancer properties against HeLa and MCF-7 cells.•M-24 induces G2/M phase arrest and apoptosis in HeLa and MCF-7 cells.•The mechanisms of M-24 induced apoptosis in two cell lines are different.Methyl 5-[(1H-indol-3-yl)selanyl]-1H-benzoimidazol-2-ylcarbamate (M-24) is a newly synthesized analogue of nocodazole by our group and has been found to be active for some cancer cells. However, its sensitivity to different cell lines and the underlying anticancer mechanism are still unclear. In this study, we proved that M-24 had strong time- and dose-dependent anti-proliferative effects on human cervical cancer HeLa cells and human breast carcinoma MCF-7 cells. We demonstrated that the growth inhibitory effects of M-24 in both cell lines were associated with microtubule depolymerization. Furthermore, M-24 treatment resulted in cell cycle arrest at the G2/M phase in a dose-dependent manner with subsequent apoptosis induction. Western blotting analysis revealed that up-regulation of cyclin B1 and cdc2 was related with G2/M arrest in both cell lines. In addition, M-24-induced HeLa cell apoptosis was mainly associated with mitochondria-dependent intrinsic pathway. However, M-24-induced MCF-7 cell apoptosis was associated with both mitochondrial and death receptor pathway. In conclusion, M-24 caused apoptosis through disrupting microtubule assembly and inducing cell cycle arrest in HeLa and MCF-7 cells. Therefore, the novel compound M-24 is a promising microtubule-destabilizing agent that has great potential for the therapy of various malignancies especially human cervical and breast cancers.Download high-res image (288KB)Download full-size image
Co-reporter:Zhaoshi Bai, Meiqi Gao, Huijuan Zhang, Qi Guan, Jingwen Xu, Yao Li, Huan Qi, Zhengqiang Li, Daiying Zuo, Weige Zhang, Yingliang Wu
Cancer Letters 2017 Volume 402(Volume 402) pp:
Publication Date(Web):28 August 2017
DOI:10.1016/j.canlet.2017.05.016
•BZML, as a novel CBSIs, has broad spectrum cytotoxic activity against various chemo-sensitive or -resistant cancer cells.•BZML is not a P-gp substrate and acts as an irreversible inhibitor of P-gp function in A549/Taxol cells.•BZML causes mitosis phase arrest by inhibiting tubulin polymerization in A549 and A549/Taxol cells.•BZML induces MC in A549/Taxol cells, which is different from apoptosis in A549 cells, to against apoptosis-resistance.Multidrug resistance (MDR) interferes with the efficiency of chemotherapy. Therefore, developing novel anti-cancer agents that can overcome MDR is necessary. Here, we screened a series of colchicine binding site inhibitors (CBSIs) and found that 5-(3, 4, 5-trimethoxybenzoyl)-4-methyl-2-(p-tolyl) imidazol (BZML) displayed potent cytotoxic activity against both A549 and A549/Taxol cells. We further explored the underlying mechanisms and found that BZML caused mitosis phase arrest by inhibiting tubulin polymerization in A549 and A549/Taxol cells. Importantly, BZML was a poor substrate for P-glycoprotein (P-gp) and inhibited P-gp function by decreasing P-gp expression at the protein and mRNA levels. Cell morphology changes and the expression of cycle- or apoptosis-related proteins indicated that BZML mainly drove A549/Taxol cells to die by mitotic catastrophe (MC), a p53-independent apoptotic-like cell death, whereas induced A549 cells to die by apoptosis. Taken together, our data suggest that BZML is a novel colchicine binding site inhibitor and overcomes MDR in A549/Taxol cells by inhibiting P-gp function and inducing MC. Our study also offers a new strategy to solve the problem of apoptosis-resistance.
Co-reporter:Qile Xu, Maolin Sun, Zhaoshi Bai, Yueting Wang, Yue Wu, Haiqiu Tian, Daiying Zuo, Qi Guan, Kai Bao, Yingliang Wu, Weige Zhang
European Journal of Medicinal Chemistry 2017 Volume 139(Volume 139) pp:
Publication Date(Web):20 October 2017
DOI:10.1016/j.ejmech.2017.05.065
•A series of antitubulin agents with a 5,5-fused-heterocycle scaffold were designed.•Compound 4f exhibited potent antiproliferative and antitubulin activities.•Compound 4f induced cell cycle arrest at G2/M phase.•The binding mode of 4f was determined by docking studies.A series of 3,6-diaryl-1H-pyrazolo[5,1-c][1,2,4]triazoles (I) and 3,6-diaryl-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazoles (II) as antitubulin agents were designed, synthesized and bioevaluated. Compounds (II) 4a, 4d, 4f, 4j, 4l and 4n showed potent antiproliferative activity at sub-micromolar or nanomolar concentrations against SGC-7901, A549 and HT-1080 cell lines, indicating that the bioisosteric replacement of the carbonyl group and B-ring of SMART and ABI with a 5,5-fused-heterocycle scaffold successfully maintained potent antiproliferative activity. Compound 4f exhibited the most excellent antiproliferative activity against the three cancer cell lines (IC50 = 0.022–0.029 μM). Consistent with its potent antiproliferative activity, 4f also displayed excellent antitubulin activity (IC50 = 0.77 μM). Furthermore, compound 4f could dramatically affect cell morphology and microtubule networking, while cell cycle studies demonstrated that 4f significantly induced SGC-7901 cells arrest in G2/M phase. In addition, molecular docking studies supported the biological assay data and suggested that 4f may be a potential antitubulin agent.Download high-res image (398KB)Download full-size image
Co-reporter:Zhiwei Wang, Huan Qi, Qirong Shen, Guodong Lu, Mingyang Li, Kai Bao, Yingliang Wu, Weige Zhang
European Journal of Medicinal Chemistry 2016 Volume 122() pp:520-529
Publication Date(Web):21 October 2016
DOI:10.1016/j.ejmech.2016.06.055
•The combretastatin A-4/oltipraz hybrids were first introduced into CA-4 analogues.•A set of 4,5-diaryl-3H-1,2-dithiole-3-thiones and related compounds were synthesized.•4d and 5c showed potent antiproliferative activities at sub-micromolar IC50 values.•4d inhibited tubulin polymerization and caused cellular arrest in the G2/M phase.•4d was docked with a superimposed docked pose of CA-4.A new series of 4,5-diaryl-3H-1,2-dithiole-3-thiones and related compounds were designed and synthesised as combretastatin A-4/oltipraz hybrids. We evaluated the antiproliferative activities, inhibition of tubulin polymerization, and cell-cycle effects of these compounds. Several compounds in this series, such as 4d and 5c, displayed significant activity against SGC-7901, KB and HT-1080 cell lines, as determined using MTT assays. The most active compound, 4d, markedly inhibited tubulin polymerization, with an IC50 value of 4.44 μM being observed. In mechanistic studies, 4d caused cell arrest in G2/M phase, induced apoptotic cell death, and disrupted microtubule formation. Molecular docking studies revealed that 4d interacts and binds efficiently with the tubulin protein.
Co-reporter:Jun Sun;Lei Chen;Chunjiang Liu;Zhan Wang;Daiying Zuo;Jiatong Pan;Huan Qi;Kai Bao;Yingliang Wu;Weige Zhang
Chemical Biology & Drug Design 2015 Volume 86( Issue 6) pp:1541-1547
Publication Date(Web):
DOI:10.1111/cbdd.12617
A series of novel 1,2-diaryl pyrroles as analogues of combretastatin A-4 (CA-4, 1a) were synthesized and evaluated for their antitumour potential against three cancer cell lines. Most compounds exhibited growth inhibition against all of the cancer cell lines. Compound 7q not only exhibited prominent antitumour efficacy with IC50 values of 0.390 μm in SGC-7901, 0.070 μm in HT-1080 and 0.045 μm in KB cell lines but also showed low activity with IC50 values of 30.08 μm in normal L929 cell line. Moreover, compound 7q inhibited tubulin polymerization into microtubules and caused microtubule destabilization. A molecular docking study of 7q was performed to determine its binding mode at the colchicine site in the tubulin dimer.
Co-reporter:Qi Guan, Daiying Zuo, Nan Jiang, Huan Qi, Yanpeng Zhai, Zhaoshi Bai, Dongjie Feng, Lei Yang, Mingyang Jiang, Kai Bao, Chang Li, Yingliang Wu, Weige Zhang
Bioorganic & Medicinal Chemistry Letters 2015 Volume 25(Issue 3) pp:631-634
Publication Date(Web):1 February 2015
DOI:10.1016/j.bmcl.2014.12.004
A series of new CA-4 analogues bearing maleic anhydride/N-substituted maleimide moiety were synthesized via a microwave-assisted process. They were evaluated for the anti-proliferative activities against three tumor cell lines (SGC-7901, HT-1080 and KB). Most compounds showed moderate potencies in micromolar range, with the most promising analogue 6f showing active at submicromolar concentration against HT-1080 cancer cells which was selected to investigate the antitumor mechanisms. In addition, molecular docking studies within the colchicine binding site of tubulin were also in good agreement with the tubulin polymerization inhibitory data and provided a basis for further structure-guided design of novel CA-4 analogues.
Co-reporter:Qi Guan, Dongjie Feng, Zhaoshi Bai, Yuanhang Cui, Daiying Zuo, Min'an Zhai, Xuewei Jiang, Wenbo Zhou, Kai Bao, Yingliang Wu and Weige Zhang
MedChemComm 2015 vol. 6(Issue 8) pp:1484-1493
Publication Date(Web):12 Jun 2015
DOI:10.1039/C5MD00150A
A series of (3/5-aryl-1,2,4-oxadiazole-5/3-yl)(3,4,5-trimethoxyphenyl)methanone oxime derivatives were synthesized via a rapid and facile microwave-assisted synthesis method of building a 1,2,4-oxadiazole skeleton using mandelic acid as the starting material. Twenty-four target compounds were evaluated for their in vitro antiproliferative activities against three human cancer cell lines (SGC-7901, A549 and HT-1080). Among them, 16b exhibited the highest potency against different tumour cell lines, especially the A549 cell line (IC50 = 87 nM). Structure–activity relationship (SAR) studies revealed that the aryl substituent at the C-5 position on the 1,2,4-oxadiazole ring is superior to that at the C-3 position. An oxime as a connector can obviously increase the potency, contrary to that in SMART derivatives. Moreover, 16b significantly induced a cell cycle arrest in the G2/M phase and caused microtubule destabilization. Molecular docking studies provided a theoretical binding mode of 16b at the colchicine site in the tubulin dimer. Our work laid the foundation for further structure-guided design of novel tubulin polymerization inhibitors.
Co-reporter:Zhan Wang, Qingkun Yang, Zhaoshi Bai, Jun Sun, Xuewei Jiang, Hongrui Song, Yingliang Wu and Weige Zhang
MedChemComm 2015 vol. 6(Issue 5) pp:971-976
Publication Date(Web):02 Apr 2015
DOI:10.1039/C5MD00028A
A series of novel 2,3-diarylthiophene analogues of combretastatin A-4 (CA-4) were designed with a rigid thiophene moiety to retain the cis-olefin configuration of CA-4 and were subsequently synthesised. All of the target compounds were evaluated for their in vitro anti-proliferative activities. Among these compounds, 5f and 8d exhibited superior potency against different tumour cell lines with IC50 values at sub-micromolar levels. Moreover, compound 8d significantly inhibited tubulin polymerisation to microtubules and caused microtubule destabilisation. In addition, a molecular modelling study of compound 8d was performed to clarify its binding mode at the colchicine site in the tubulin dimer and to provide a basis for further structure-guided design of novel CA-4 analogues.
Co-reporter:Zhiyong Wen, Jingwen Xu, Zhiwei Wang, Huan Qi, Qile Xu, Zhaoshi Bai, Qian Zhang, Kai Bao, Yingling Wu, Weige Zhang
European Journal of Medicinal Chemistry 2015 90() pp: 184-194
Publication Date(Web):
DOI:10.1016/j.ejmech.2014.11.024
Co-reporter:Qi Guan, Zengjin Cheng, Xiaoxue Ma, Lijie Wang, Dongjie Feng, Yuanhang Cui, Kai Bao, Lan Wu, Weige Zhang
European Journal of Medicinal Chemistry 2014 Volume 85() pp:508-516
Publication Date(Web):6 October 2014
DOI:10.1016/j.ejmech.2014.08.014
•A series of new non-purine XO inhibitors were designed and synthesized.•The selenium-containing heterocycle was first introduced into XO inhibitors.•Inhibitory activity of compound 9e was 3-fold more potent than that of febuxostat.•Steady-state kinetics data demonstrated that 9e was a mixed-type XO inhibitor.•Molecular docking studies gained an insight into binding mode of 9e with XO.A series of 2-phenyl-4-methyl-1,3-selenazole-5-carboxylic acid derivatives (8a–f, 9a–m) were synthesized and evaluated for inhibitory activity against xanthine oxidase in vitro. Structure–activity relationship analyses have also been presented. Most of the target compounds exhibited potency levels in the nanomolar range. Compound 9e emerged as the most potent xanthine oxidase inhibitor (IC50 = 5.5 nM) in comparison to febuxostat (IC50 = 18.6 nM). Steady-state kinetics measurements with the bovine milk enzyme indicated a mixed type inhibition with Ki and Ki′ values of 0.9 and 2.3 nM, respectively. A molecular modeling study on compounds 9e was performed to gain an insight into its binding mode with xanthine oxidase, and to provide the basis for further structure-guided design of new non-purine xanthine oxidase inhibitors related with 2-phenyl-4-methyl-1,3-selenazole-5-carboxylic acid scaffold.
Co-reporter:Qi Guan, Fushan Yang, Dandan Guo, Jingwen Xu, Mingyang Jiang, Chunjiang Liu, Kai Bao, Yingliang Wu, Weige Zhang
European Journal of Medicinal Chemistry 2014 Volume 87() pp:1-9
Publication Date(Web):24 November 2014
DOI:10.1016/j.ejmech.2014.09.046
•The selenium-containing heterocycle was first introduced into CA-4 analogues.•A series of novel 3,4-diaryl-1,2,5-selenadiazol derivatives were synthesized.•Several compounds were more potent than the standard CA-4.•3n was selected to investigate the antitumour mechanisms.•The binding mode of 3n was determined by docking studies.A set of novel selenium-containing heterocyclic analogues of combretastatin A-4 (CA-4) have been designed and synthesised using a rigid 1,2,5-selenadiazole as a linker to fix the cis-orientation of ring-A and ring-B. All of the target compounds were evaluated for their in vitro anti-proliferative activities. Among these compounds, compounds 3a, 3i, 3n and 3q exhibited superior potency against different tumour cell lines with IC50 values at the nanomolar level. Moreover, compound 3n significantly induced cell cycle arrest in the G2/M phase, inhibited tubulin polymerisation into microtubules and caused microtubule destabilisation. A molecular modelling study of compound 3n was performed to elucidate its binding mode at the colchicine site in the tubulin dimer and to provide a basis for the further structure-guided design of novel CA-4 analogues.
Co-reporter:Qi Guan, Chunming Han, Daiying Zuo, Min'an Zhai, Zengqiang Li, Qian Zhang, Yanpeng Zhai, Xuewei Jiang, Kai Bao, Yingliang Wu, Weige Zhang
European Journal of Medicinal Chemistry 2014 Volume 87() pp:306-315
Publication Date(Web):24 November 2014
DOI:10.1016/j.ejmech.2014.09.071
•A series of benzimidazole carbamates bearing indole moieties were synthesized.•The target compounds were evaluated on different human cancer cell lines.•The antiproliferative activity of 10a was equivalent to nocodazole.•10a and 7e were taken for cellular tubulin polymerization assay.•The binding mode of 10a was determined by docking studies.A series of novel benzimidazole carbamates bearing indole moieties with sulphur or selenium atoms connecting the aromatic rings were synthesised and evaluated for their antiproliferative activities against three human cancer cell lines (SGC-7901, A-549 and HT-1080) using an MTT assay. Compounds 10a, 10b, 7a, 7b and 7f showed significant activities against these cell lines. The most potent compound in this series, 10a, was selected to investigate its antitumour mechanism. In addition, molecular docking studies suggested that compound 10a interacts very closely with the nocodazole docking pose through hydrogen bonds at the colchicine binding site of tubulin.
Co-reporter:Qirong Shen, Yi Dai, Guanghui Wang, Fei Yao, Yinghui Duan, Haifeng Chen, Weige Zhang, Xiaokun Zhang, Xinsheng Yao
Bioorganic & Medicinal Chemistry 2014 Volume 22(Issue 9) pp:2671-2677
Publication Date(Web):1 May 2014
DOI:10.1016/j.bmc.2014.03.024
Co-reporter:Foxiao Qiao;Daiying Zuo;Yingliang Wu;Huan Qi;Weige Zhang;Haifeng Wang;Xueqi Shen
Cancer Chemotherapy and Pharmacology 2012 Volume 70( Issue 2) pp:259-270
Publication Date(Web):2012/08/01
DOI:10.1007/s00280-012-1907-x
The anti-mitotic agent, combretastatin A-4 (CA-4), is the lead compound of a new class of anti-cancer drugs that target tumor vasculature. 2-Methoxy-5-(2-(3, 4, 5-trimethoxyphenyl) thiophen-3-yl) aniline (DAT-230) is a structurally novel CA-4 analog with more stability. We investigated its anti-tumor activity and mechanisms in vitro and in vivo for the first time.Cytotoxicity was measured by MTT method. Apoptosis, mitochondria membrane potential (ΔΨm) and NO generation were measured by flow cytometry. Intracellular microtubule network was detected by immunofluorescence experiments. Protein expression was analyzed by Western blotting. In vivo, the anti-tumor activity was assessed using fibrosarcoma xenografts subcutaneously established in BALB/c nude mice. Vasculature perfusion was identified using fluorescent DNA-binding compound Hoechst 33342.DAT-230 exhibited potent anti-proliferative activity against various cancer cells. DAT-230-treatment in HT-1080 cells resulted in microtubule de-polymerization and G2/M phase arrest preceding apoptosis. Phosphor-cdc2 (thr14/tyr15) reduction, cyclin B1 accumulation and aberrant spindles denoted the cyclin B1-cdc2 complex active and M phase arrest in HT-1080 cells treated with DAT-230. Apoptosis induced by DAT-230 was related with the activation of caspase-9, caspase-3 and PARP cleavage, which were at the downstream of mitochondria. The decrease ratio of Bcl-2/Bax, elevation of NO and disruption of ΔΨm confirmed the causal relationship between DAT-230 and mitochondrial pathway. In vivo, DAT-230 delayed tumor growth, induced tumor perfusion decrease and extensive hemorrhagic-necrosis.DAT-230 is a promising microtubule inhibitor that has great potential for the treatment of fibrosarcoma in vitro and in vivo. Its potential to be a candidate of anti-cancer agent is worth being further investigated.
Co-reporter:Kai Bao, Yi Dai, Zhi-Bin Zhu, Feng-Juan Tu, Wei-Ge Zhang, Xin-Sheng Yao
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 18) pp:6708-6714
Publication Date(Web):15 September 2010
DOI:10.1016/j.bmc.2010.07.062
Two new series of biphenyls, analogs of aglycone of natural product fortuneanoside E, were prepared using Suzuki–Miyaura cross-coupling and selective magnesium iodide demethylation/debenzylation, and their mushroom tyrosinase inhibitory activity was evaluated. Most of the 4-hydroxy-3,5-dimethoxyphenyl biphenyl compounds (series II, 20–36) were in general more active than 3,4,5-trimethoxyphenyl biphenyl compounds (series I, 1–19). Structure–activity relationships study showed that monosaccharide substituents, such as glucose, were not necessary and the presence of 4-hydroxy-3,5-dimethoxyphenyl moiety was crucial for inhibitory activity. Among the compounds synthesised, compound 21 (IC50 = 0.02 mM) was found to be the most active one, which exhibited an activity that was 7 times higher than that of fortuneanoside E (IC50 = 0.14 mM) and 10 times higher than that of arbutin (IC50 = 0.21 mM), known as potent tyrosinase inhibitors. The inhibition kinetics analyzed by Lineweaver–Burk plots revealed that compound 21 was a competitive inhibitor (Ki = 0.015 mM).
Co-reporter:Kai Bao, Aixue Fan, Yi Dai, Liang Zhang, Weige Zhang, Maosheng Cheng and Xinsheng Yao
Organic & Biomolecular Chemistry 2009 vol. 7(Issue 24) pp:5084-5090
Publication Date(Web):15 Oct 2009
DOI:10.1039/B916969E
An efficient selective demethylation and debenzylation method for aryl methyl/benzyl ethers using magnesium iodide under solvent-free conditions has been developed and applied to the synthesis of natural flavone and biphenyl glycosides. A variety of functional groups including glycoside were tolerated under the reaction conditions. Experimental results indicated that the removal of an O-benzyl group was easier than that of an O-methyl group, regardless of wherever they were meta or para to the carbonyl. Thus selective debenzylation can be achieved for substrates bearing both benzyloxy and methoxy groups.
Co-reporter:Kai Bao, Weige Zhang, Xiujuan Bu, Zhichun Song, Liang Zhang and Maosheng Cheng
Chemical Communications 2008 (Issue 42) pp:5429-5431
Publication Date(Web):16 Sep 2008
DOI:10.1039/B810086A
A novel N-formylation and related reactions proceed from cyanides promoted by esters.
Co-reporter:Jinshun Lin, Weige Zhang, Nan Jiang, Zeyu Niu, Kai Bao, Liang Zhang, Dailin Liu, Chao Pan and Xinsheng Yao
Journal of Natural Products 2008 Volume 71(Issue 11) pp:1938-1941
Publication Date(Web):October 29, 2008
DOI:10.1021/np800226n
The first total synthesis of bulbophylol-B (1) has been achieved with the longest linear sequence of 12 steps and an overall yield of 17.9% via a new and practical approach to construct the dihydrodibenz[b,f]oxepin skeleton employing Wittig, selective reduction, and intramolecular Ullmann reactions as key steps.
Co-reporter:Liang Zhang;En-Long Ma;Lan Wu;Kai Bao;Xiao-Long Wang;Yu-Ling Wang;Hong-Rui Song
Archiv der Pharmazie 2007 Volume 340(Issue 12) pp:
Publication Date(Web):12 NOV 2007
DOI:10.1002/ardp.200700077
The total synthesis of natural flavan racemates (±) 1, (±) 2 and natural flavones 3, 4 had thus been achieved. A straightforward synthetic procedure of flavans via the Pd-C catalyzed hydrogenation/hydrogenolysis of corresponding flavones was developed. Furthermore, the antiproliferative activities of racemic flavans (±) 1, (±) 2, flavones 3, 4, and five synthetic intermediates toward human SGC-7901, BEL-7402, HeLa, and HL-60 cell lines in vitro were evaluated by MTT assay, and the racemic flavans (±) 1 were found to have significant antiproliferative activity against all four cell lines.
Co-reporter:Wei-Ge Zhang;Rui Zhao;Jian Ren;Li-Xiang Ren;Jin-Guang Lin;Dai-Lin Liu;Ying-Liang Wu;Xin-Sheng Yao
Archiv der Pharmazie 2007 Volume 340(Issue 5) pp:
Publication Date(Web):22 MAY 2007
DOI:10.1002/ardp.200600146
A total synthetic route for two natural dihydrostilbenes with significant cytotoxicity toward human cancer cell lines, (3-(2-(7-methoxybenzo[d][1,3]dioxol-5-yl)ethyl)phenol 1a and 6-(3-hydroxyphenethyl)benzo[d][1,3]dioxol-4-ol 1b), which were isolated from Bulbophyllum odoratissimum Lindl, was developed via Wittig–Horner reaction. The natural products 1a and 1b were obtained in 28% and 20% overall yield, respectively. Additionally, nine analogues, 1c-1k, of the two natural dihydrostilbenes were synthesized and evaluated for their anti-proliferative activity against human SGC-7901, KB and HT-1080 cell lines by MTT assay. The activities of 1c and 1d were in the same range as those of the natural products 1a and 1b.
Co-reporter:Kai Bao, Aixue Fan, Yi Dai, Liang Zhang, Weige Zhang, Maosheng Cheng and Xinsheng Yao
Organic & Biomolecular Chemistry 2009 - vol. 7(Issue 24) pp:NaN5090-5090
Publication Date(Web):2009/10/15
DOI:10.1039/B916969E
An efficient selective demethylation and debenzylation method for aryl methyl/benzyl ethers using magnesium iodide under solvent-free conditions has been developed and applied to the synthesis of natural flavone and biphenyl glycosides. A variety of functional groups including glycoside were tolerated under the reaction conditions. Experimental results indicated that the removal of an O-benzyl group was easier than that of an O-methyl group, regardless of wherever they were meta or para to the carbonyl. Thus selective debenzylation can be achieved for substrates bearing both benzyloxy and methoxy groups.
Co-reporter:Kai Bao, Weige Zhang, Xiujuan Bu, Zhichun Song, Liang Zhang and Maosheng Cheng
Chemical Communications 2008(Issue 42) pp:
Publication Date(Web):
DOI:10.1039/B810086A

![BENZO[B]-1,3-DIOXOLO[4,5-I][1]BENZOXEPIN-9-OL, 6,7-DIHYDRO-4-METHOXY-](http://img.cochemist.com/ccimg/850600/850588-28-2.png)
![BENZO[B]-1,3-DIOXOLO[4,5-I][1]BENZOXEPIN-9-OL, 6,7-DIHYDRO-4-METHOXY-](http://img.cochemist.com/ccimg/850600/850588-28-2_b.png)
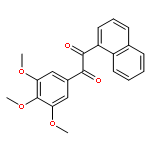
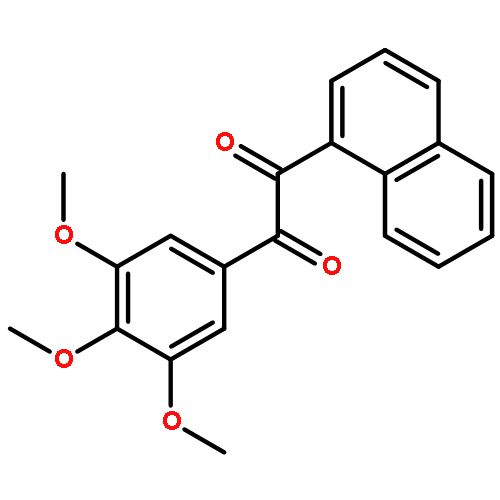
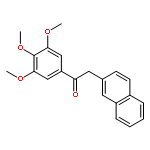
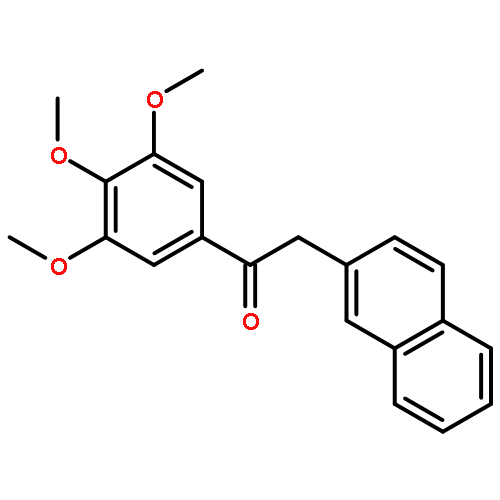




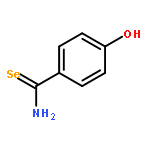
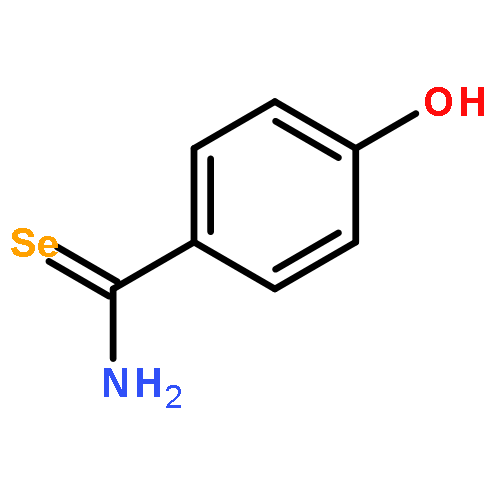




![1-([1,1'-Biphenyl]-4-yl)-3-((4-fluorophenyl)amino)propan-1-one](http://img.cochemist.com/ccimg/279700/279672-36-5.png)
![1-([1,1'-Biphenyl]-4-yl)-3-((4-fluorophenyl)amino)propan-1-one](http://img.cochemist.com/ccimg/279700/279672-36-5_b.png)
![1-([1,1'-Biphenyl]-4-yl)-3-((4-ethoxyphenyl)amino)propan-1-one](http://img.cochemist.com/ccimg/279700/279672-29-6.png)
![1-([1,1'-Biphenyl]-4-yl)-3-((4-ethoxyphenyl)amino)propan-1-one](http://img.cochemist.com/ccimg/279700/279672-29-6_b.png)


![1H-Indole, 1-[(4-fluorophenyl)methyl]-](http://img.cochemist.com/ccimg/204300/204205-77-6.png)
![1H-Indole, 1-[(4-fluorophenyl)methyl]-](http://img.cochemist.com/ccimg/204300/204205-77-6_b.png)




![1H-Benzimidazole, 2-[(4-chlorophenyl)methyl]-5-methyl-](http://img.cochemist.com/ccimg/200800/200799-58-2.png)
![1H-Benzimidazole, 2-[(4-chlorophenyl)methyl]-5-methyl-](http://img.cochemist.com/ccimg/200800/200799-58-2_b.png)
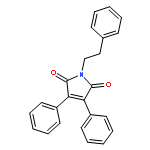
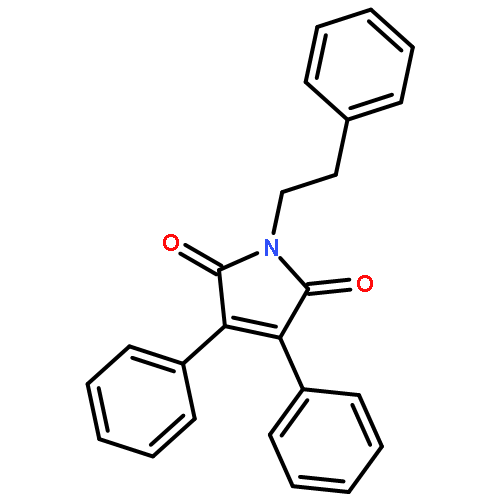






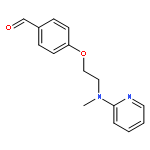
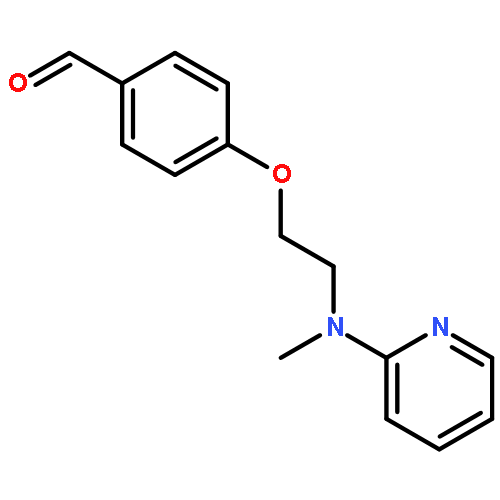


![1H-Benzimidazole, 5-methyl-2-[(4-nitrophenyl)methyl]-](http://img.cochemist.com/ccimg/119700/119691-82-6.png)
![1H-Benzimidazole, 5-methyl-2-[(4-nitrophenyl)methyl]-](http://img.cochemist.com/ccimg/119700/119691-82-6_b.png)
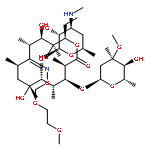
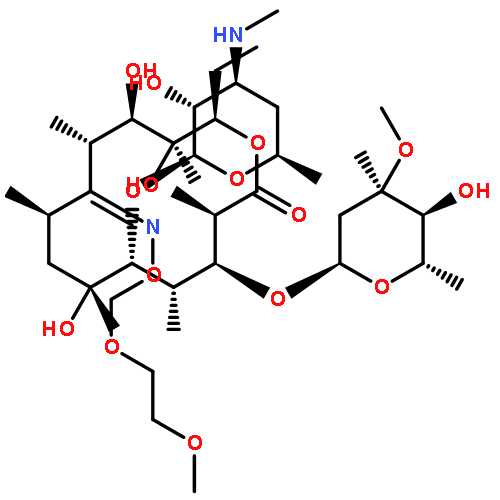





![4,13-Dioxabicyclo[8.2.1]tridec-12-en-5-one,7-[(2,6-dideoxy-3-C-methyl-3-O-methyl-a-L-ribo-hexopyranosyl)oxy]-3-[(1R,2R)-1,2-dihydroxy-1-methylbutyl]-2,6,8,10,12-pentamethyl-9-[[3,4,6-trideoxy-3-(dimethylamino)-b-D-xylo-hexopyranosyl]oxy]-,(2R,3R,6R,7S,8S,9R,10R)-](http://img.cochemist.com/ccimg/105900/105882-69-7.png)
![4,13-Dioxabicyclo[8.2.1]tridec-12-en-5-one,7-[(2,6-dideoxy-3-C-methyl-3-O-methyl-a-L-ribo-hexopyranosyl)oxy]-3-[(1R,2R)-1,2-dihydroxy-1-methylbutyl]-2,6,8,10,12-pentamethyl-9-[[3,4,6-trideoxy-3-(dimethylamino)-b-D-xylo-hexopyranosyl]oxy]-,(2R,3R,6R,7S,8S,9R,10R)-](http://img.cochemist.com/ccimg/105900/105882-69-7_b.png)
![Benzoxazole, 5-chloro-2-[(4-chlorophenyl)methyl]-](http://img.cochemist.com/ccimg/102400/102394-36-5.png)
![Benzoxazole, 5-chloro-2-[(4-chlorophenyl)methyl]-](http://img.cochemist.com/ccimg/102400/102394-36-5_b.png)
![Benzoxazole, 2-[(4-nitrophenyl)methyl]-](http://img.cochemist.com/ccimg/101600/101554-07-8.png)
![Benzoxazole, 2-[(4-nitrophenyl)methyl]-](http://img.cochemist.com/ccimg/101600/101554-07-8_b.png)


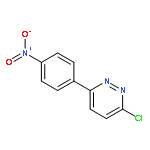
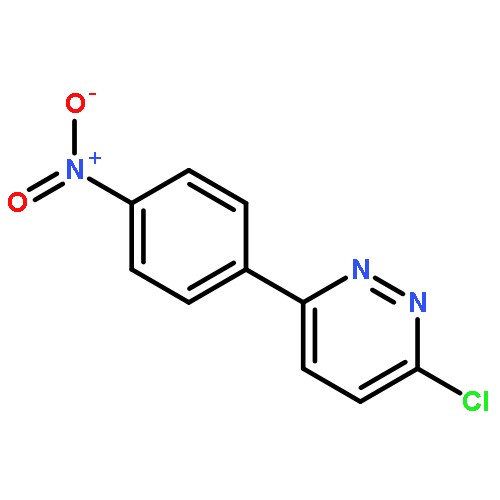
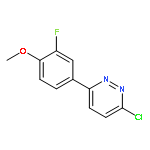





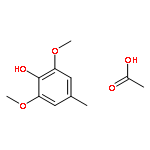
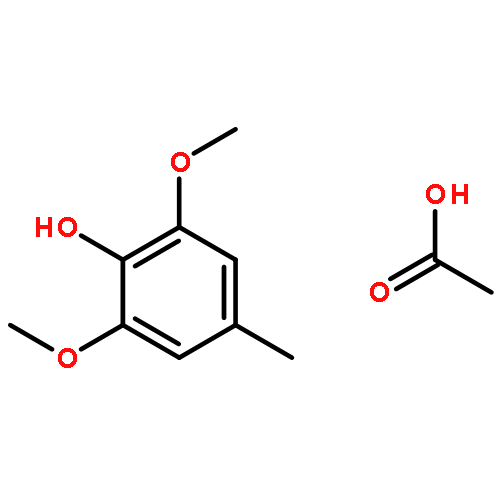
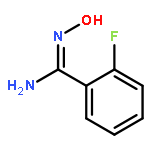


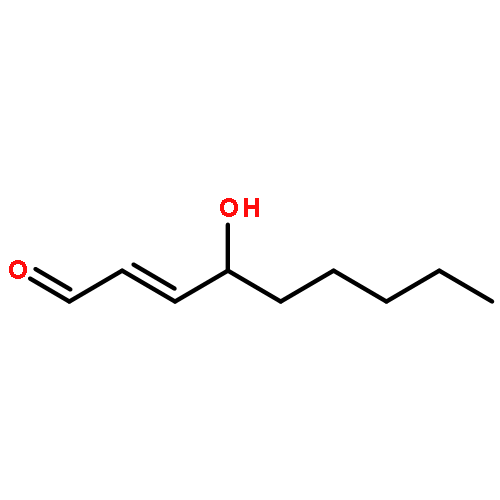
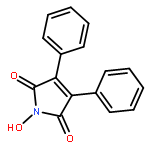
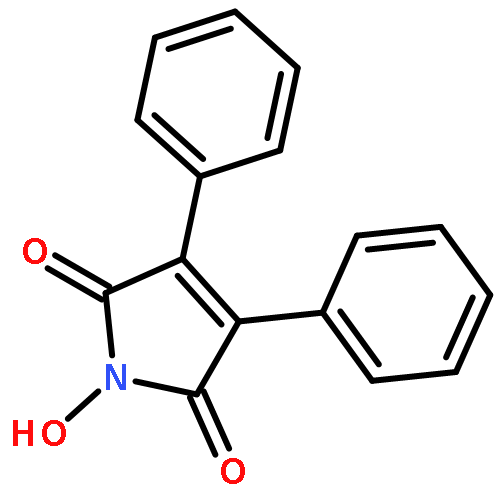
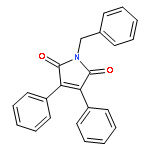
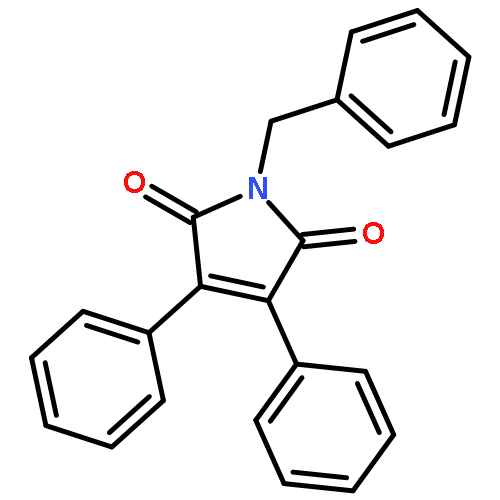
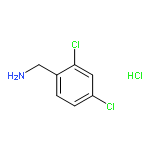
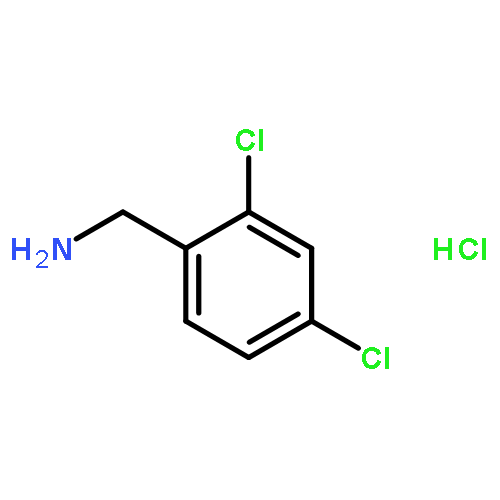
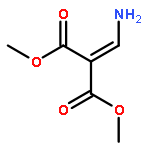
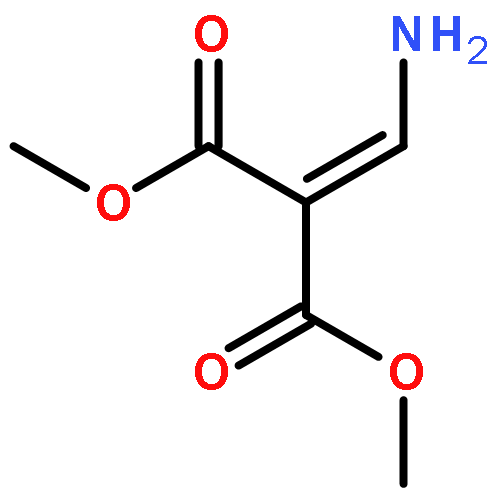
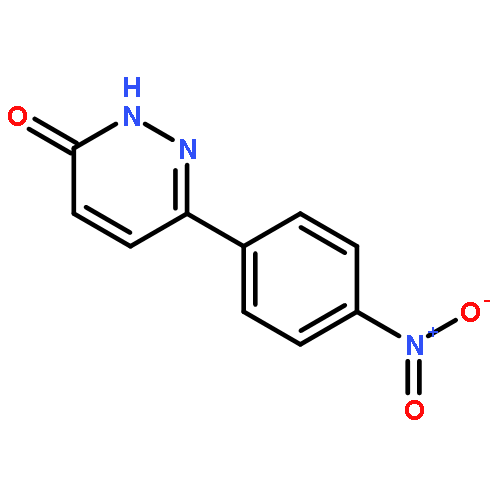
![Propanedioic acid,2-[[(3-chloro-4-fluorophenyl)amino]methylene]-, 1,3-diethyl ester](http://img.cochemist.com/ccimg/70100/70032-30-3.png)
![Propanedioic acid,2-[[(3-chloro-4-fluorophenyl)amino]methylene]-, 1,3-diethyl ester](http://img.cochemist.com/ccimg/70100/70032-30-3_b.png)


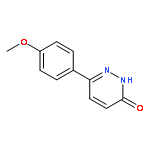
![Pyridazine, 3-chloro-6-[4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/66600/66548-88-7.png)
![Pyridazine, 3-chloro-6-[4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/66600/66548-88-7_b.png)
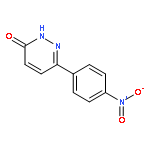
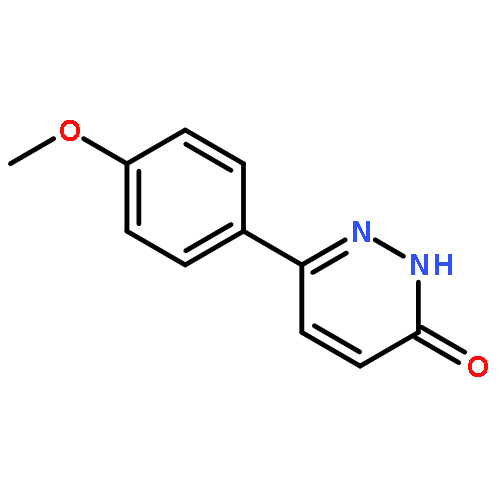
![3(2H)-Pyridazinone, 6-[4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/66600/66548-87-6.png)
![3(2H)-Pyridazinone, 6-[4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/66600/66548-87-6_b.png)








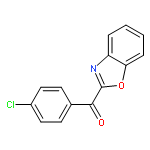
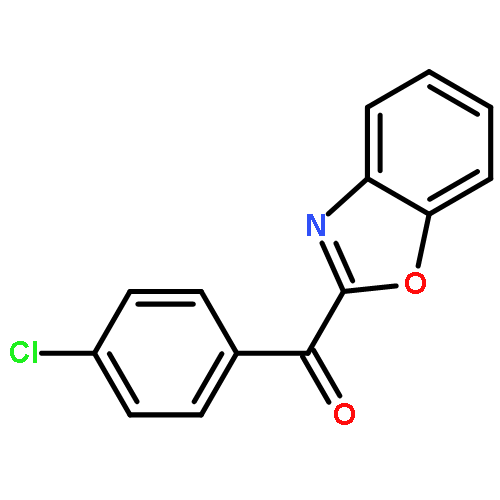
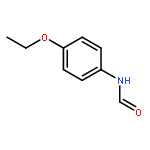
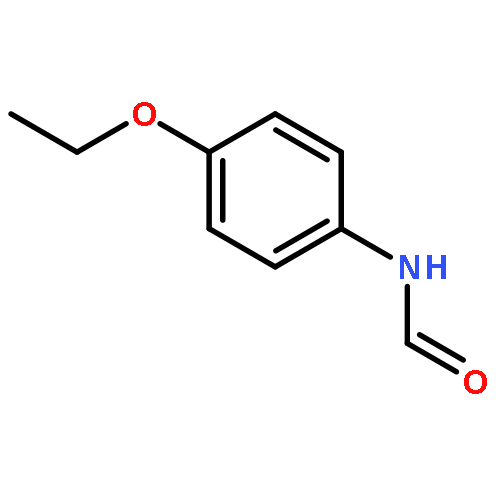

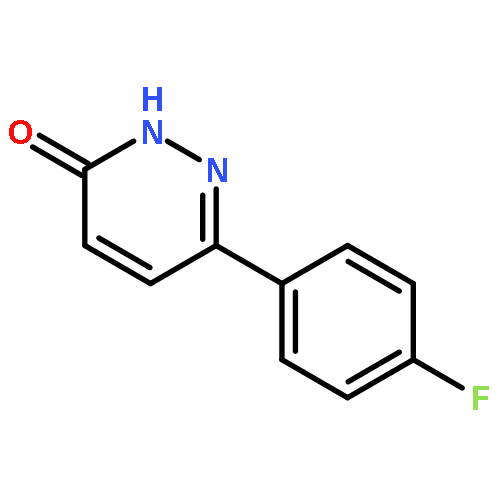
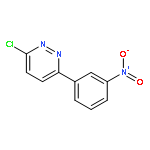
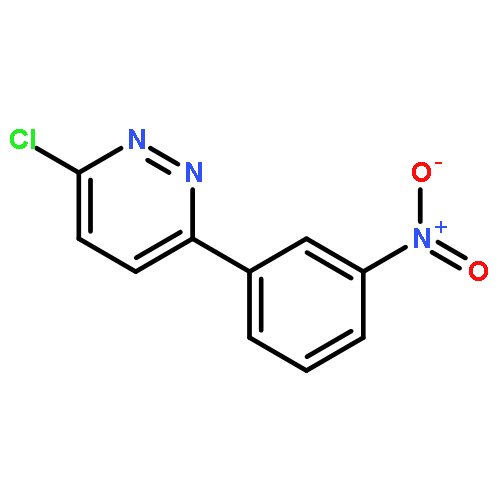



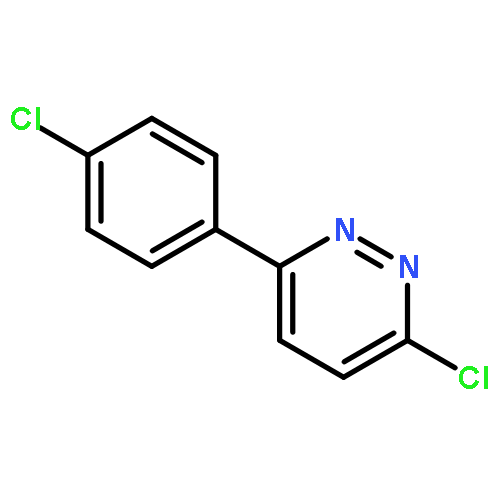







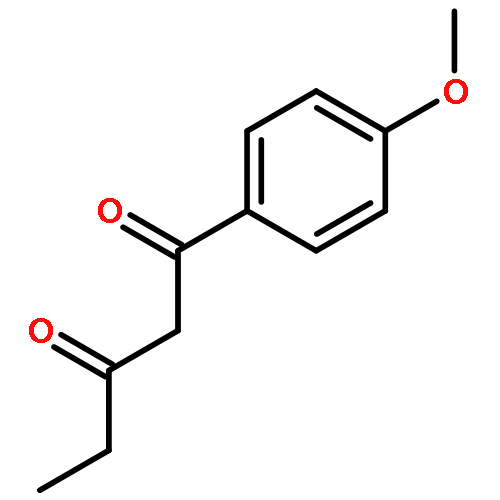




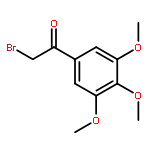

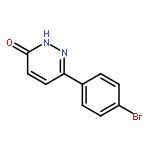

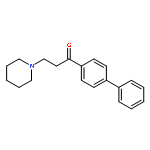
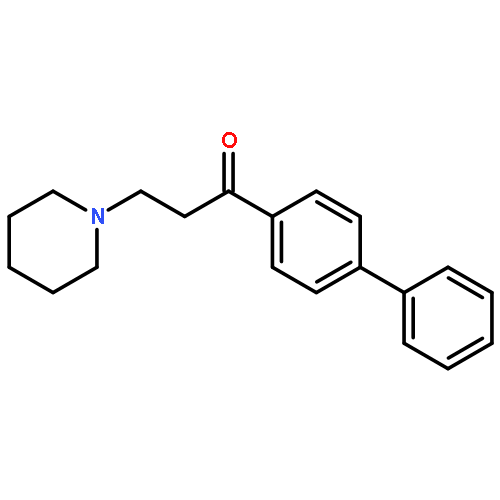
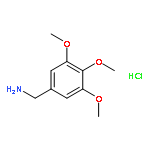
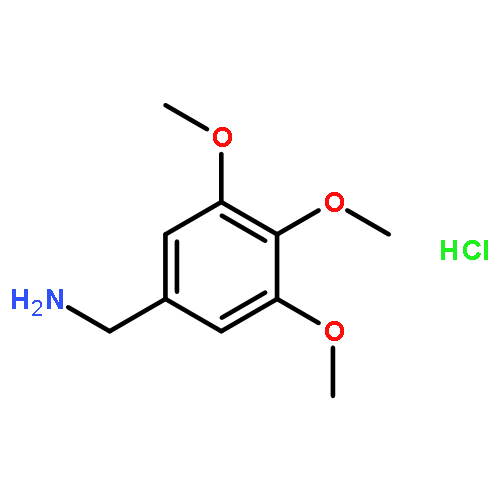


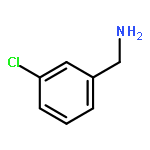
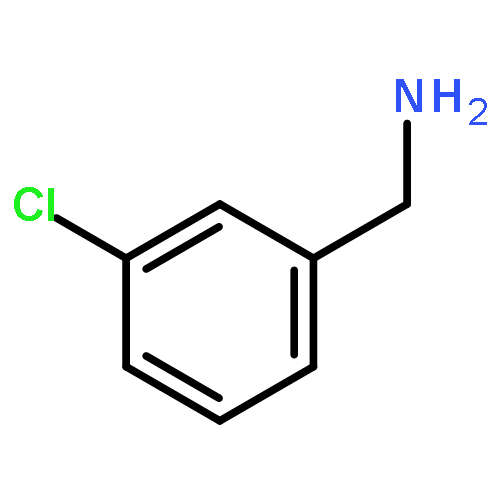


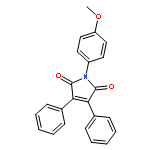
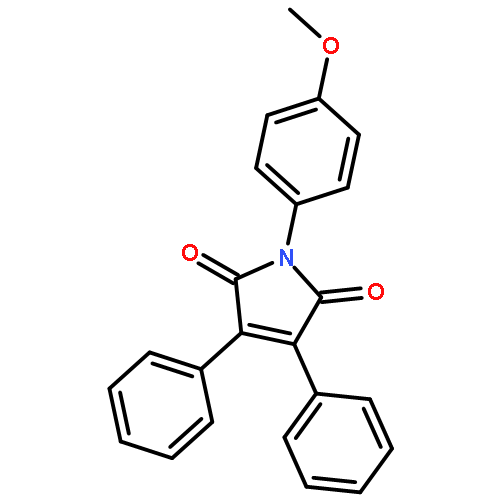


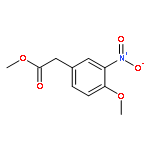


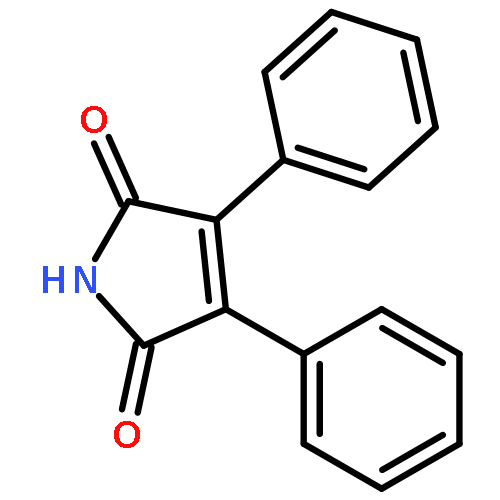









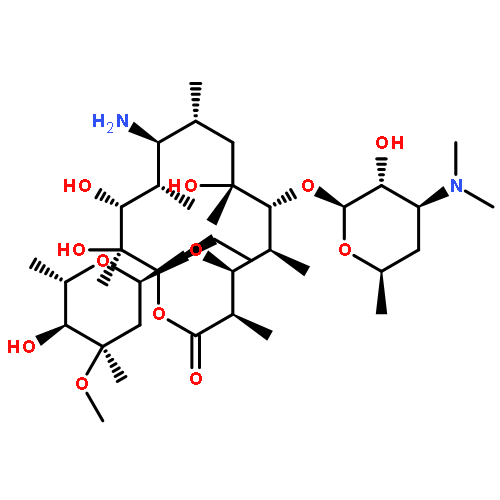
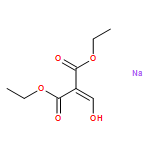
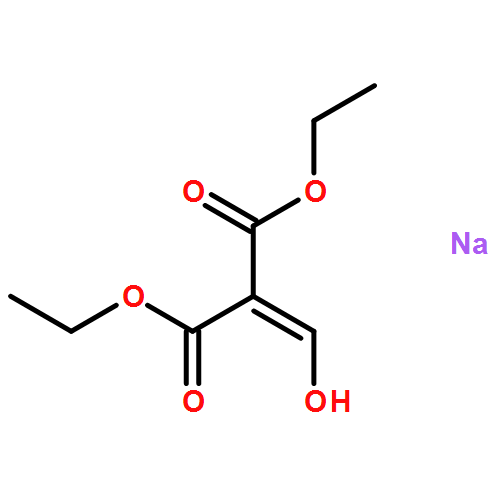
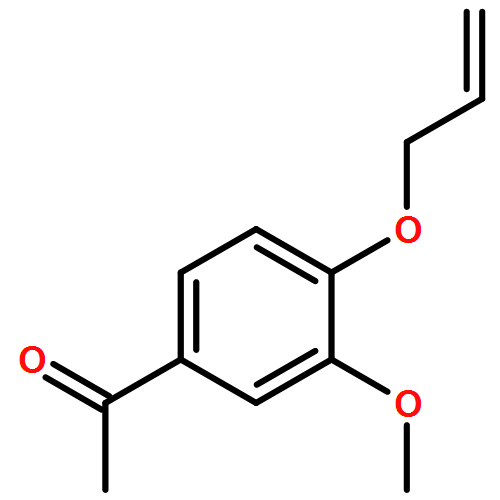
![Ethanone, 1-[4-methoxy-3-(2-propenyloxy)phenyl]-](http://img.cochemist.com/ccimg/24100/24064-42-4.png)
![Ethanone, 1-[4-methoxy-3-(2-propenyloxy)phenyl]-](http://img.cochemist.com/ccimg/24100/24064-42-4_b.png)
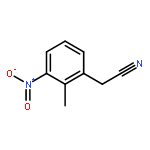
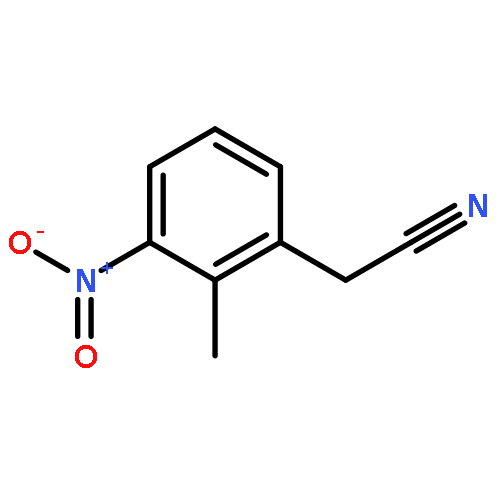


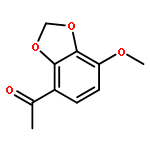
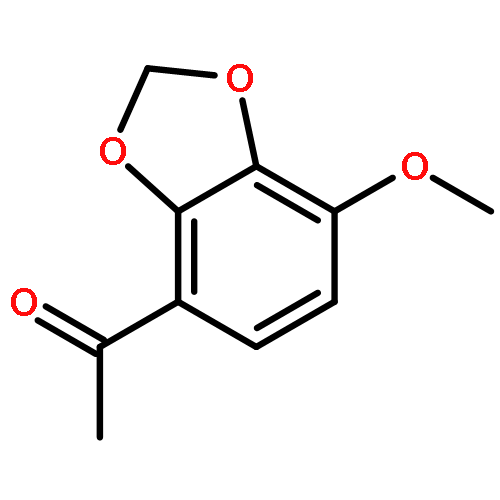



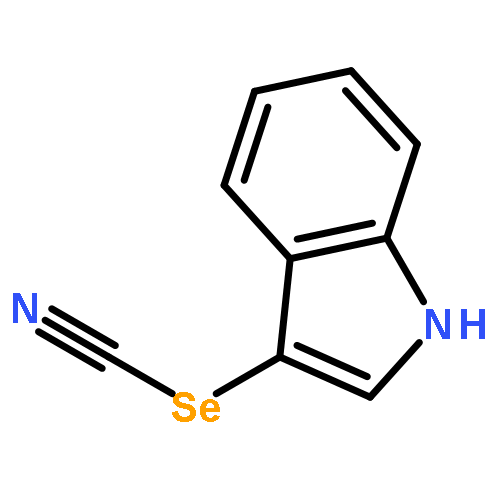
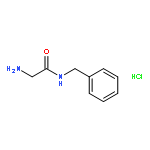
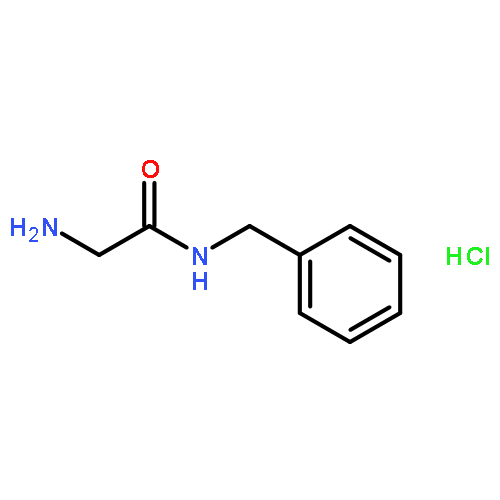
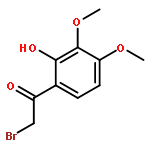

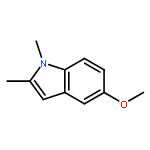
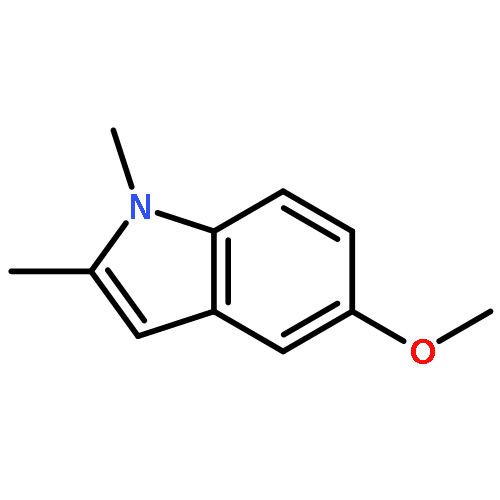
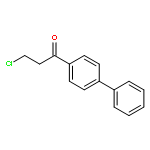



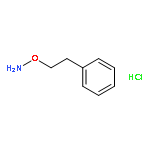
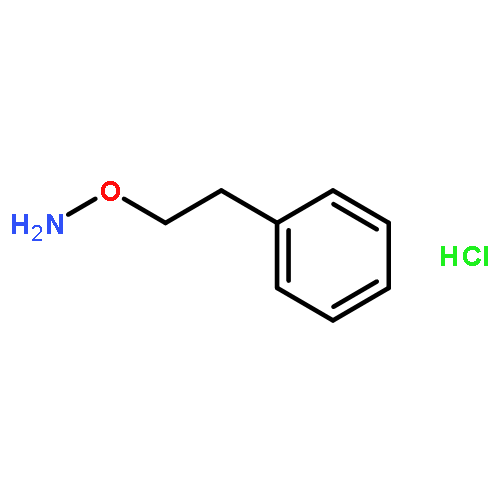
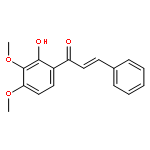
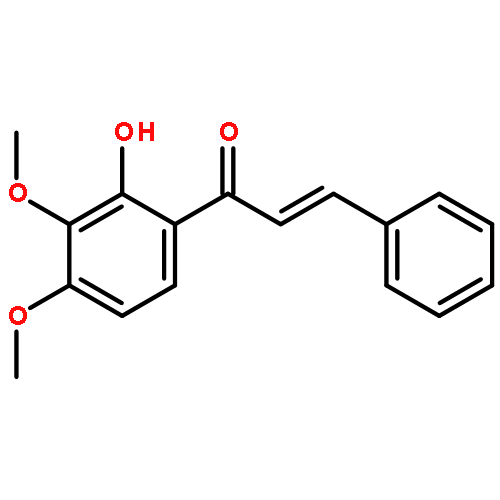





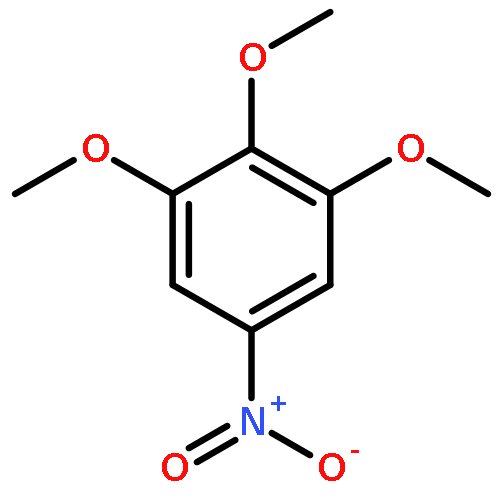





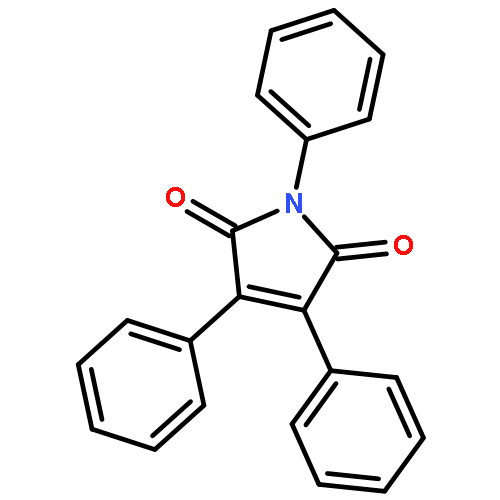
![(1H-Benzo[d]imidazol-2-yl)(4-chlorophenyl)methanol](http://img.cochemist.com/ccimg/5100/5028-38-6.png)
![(1H-Benzo[d]imidazol-2-yl)(4-chlorophenyl)methanol](http://img.cochemist.com/ccimg/5100/5028-38-6_b.png)
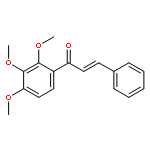
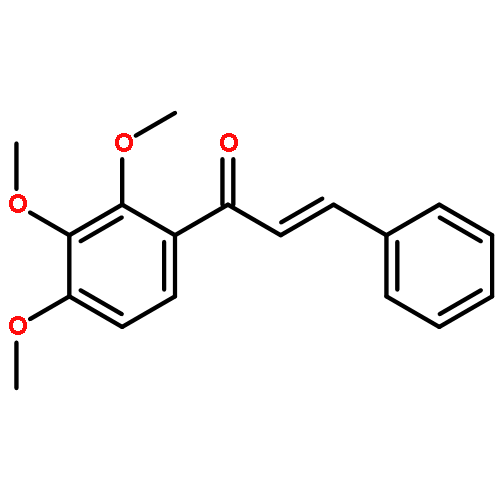
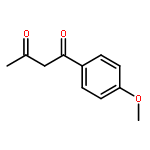
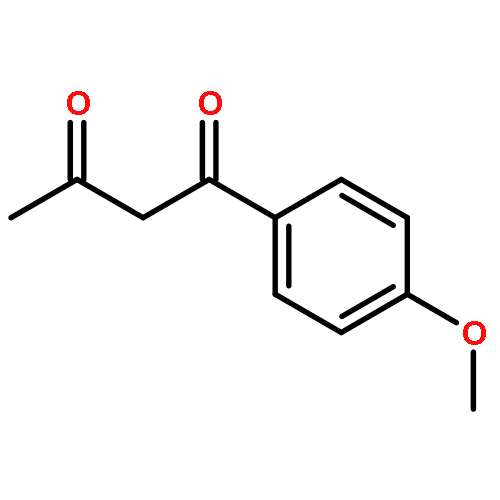
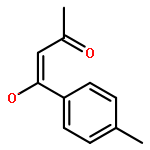
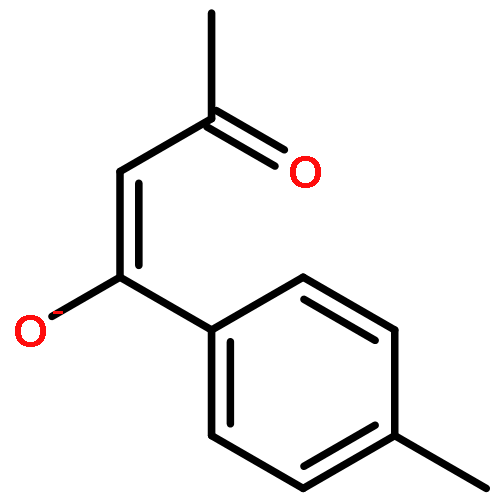

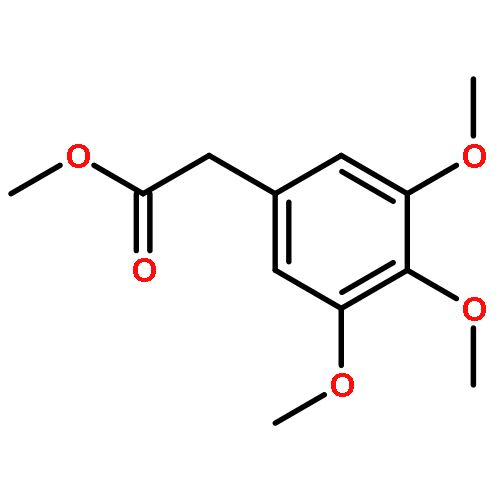
![5-[(aminooxy)methyl]-1,2,3-trimethoxybenzene hydrochloride (1:1)](http://img.cochemist.com/ccimg/2200/2173-27-5.png)
![5-[(aminooxy)methyl]-1,2,3-trimethoxybenzene hydrochloride (1:1)](http://img.cochemist.com/ccimg/2200/2173-27-5_b.png)





![2-Propanol, 1-[(1-methylethyl)amino]-3-(1-naphthalenyloxy)-](/data/chemimg/935900/525-66-6.png)
![2-Propanol, 1-[(1-methylethyl)amino]-3-(1-naphthalenyloxy)-](/data/chemimg/935900/525-66-6_b.png)














![2-methoxy-5-[(z)-2-(3,4,5-trimethoxyphenyl)ethenyl]phenol](http://img.cochemist.com/ccimg/117100/117048-59-6.png)
![2-methoxy-5-[(z)-2-(3,4,5-trimethoxyphenyl)ethenyl]phenol](http://img.cochemist.com/ccimg/117100/117048-59-6_b.png)





![5-[(2R)-2-hydroxy-2-(3,4,5-trimethoxyphenyl)ethyl]-2-methoxyphenol](http://img.cochemist.com/ccimg/82900/82855-09-2.png)
![5-[(2R)-2-hydroxy-2-(3,4,5-trimethoxyphenyl)ethyl]-2-methoxyphenol](http://img.cochemist.com/ccimg/82900/82855-09-2_b.png)





