Co-reporter:Robert-André F. Rarig, John M. Nelson, and Edwin Vedejs
The Journal of Organic Chemistry December 1, 2017 Volume 82(Issue 23) pp:12757-12757
Publication Date(Web):November 7, 2017
DOI:10.1021/acs.joc.7b01904
Tetrahydro-1,4-azaborines were accessed by hydroboration of N,N-diprenyltoluenesulfonamide 4. Conversion to the methylborinates 11 and 12 followed by heating with l-alanine and crystallization afforded (R,R,S)-13 (27%). Reduction of borinic acid (R,R)-18 with Soderquist’s KH* gave (R,R)-19, and hydride abstraction by TMSCl in the presence of alkenes resulted in hydroboration, 84–86% ee for (Z)-alkenes, but (E)-alkenes or 1,1-disubstituted alkenes gave <5% ee.
Co-reporter:Gorka Peris and Edwin Vedejs
The Journal of Organic Chemistry 2015 Volume 80(Issue 6) pp:3050-3057
Publication Date(Web):February 23, 2015
DOI:10.1021/jo502939a
A chiral benzylic ether serves as an auxiliary for oxindole carboxylation (dr 5.2:1.0) that sets C10 configuration in a potential diazonamide precursor. The chiral substituent allows diastereomer separation and departs during a subsequent acid-catalyzed ring closure to form a tetracyclic aminal. With suitable N-protection, crystallization affords the aminal with 98–99% ee.
Co-reporter:Aleksandrs Prokofjevs ; Jeff W. Kampf ; Andrey Solovyev ; Dennis P. Curran
Journal of the American Chemical Society 2013 Volume 135(Issue 42) pp:15686-15689
Publication Date(Web):October 2, 2013
DOI:10.1021/ja407458k
Hydride abstraction from monocationic hydride bridged salts [H(H2B–L)2]+ [B(C6F5)4]¯ (L = Lewis base) generates an observable primary borenium cation for L = iPr2NEt, but with L = Me3N, Me2NPr, or several N-heterocyclic carbenes, highly reactive dicationic dimers are formed.
Co-reporter:Clément Cazorla, Timothy S. De Vries, and Edwin Vedejs
Organic Letters 2013 Volume 15(Issue 5) pp:984-987
Publication Date(Web):January 30, 2013
DOI:10.1021/ol303203m
Internal borylation occurs upon activation of aryl di-isopropylphosphinite boranes with HNTf2 to give heterocyclic intermediates that can be reductively quenched to afford 6 or treated with KHF2 to give the phenolic potassium aryl trifluoroborate salts 10. The latter salts are useful for Pd-catalyzed coupling with aryl iodides under Molander conditions, provided that precautions are taken to remove the KNTf2 byproduct from the preceding KHF2 step.
Co-reporter:Aleksandrs Prokofjevs, Janis Jermaks, Alina Borovika, Jeff W. Kampf, and Edwin Vedejs
Organometallics 2013 Volume 32(Issue 22) pp:6701-6711
Publication Date(Web):October 8, 2013
DOI:10.1021/om400651p
Catalytic procedures are described for the amine-directed borylation of aliphatic and aromatic tertiary amine–boranes. Sequential double borylation is observed in cases where two or more C–H bonds are available that allow 5-center or 6-center intramolecular borylation. The HNTf2-catalyzed borylation of benzylamine–boranes provides a practical means for the synthesis of ortho-substituted arylboronic acid derivatives, suitable for Suzuki–Miyaura cross-coupling applications.
Co-reporter:Naresh Theddu and Edwin Vedejs
The Journal of Organic Chemistry 2013 Volume 78(Issue 10) pp:5061-5066
Publication Date(Web):April 13, 2013
DOI:10.1021/jo4005052
An aziridinyl stannatrane 8 couples with aryl or alkenyl halides RX under modified Stille conditions to afford substituted aziridines. Efficient coupling at room temperature is possible in the best examples in the presence of (tBu3P)2Pd and CuOP(O)Ph2 (CuDPP).
Co-reporter:Timothy S. De Vries, Aleksandrs Prokofjevs, and Edwin Vedejs
Chemical Reviews 2012 Volume 112(Issue 7) pp:4246
Publication Date(Web):April 20, 2012
DOI:10.1021/cr200133c
Co-reporter:Aleksandrs Prokofjevs ; Anne Boussonnière ; Linfeng Li ; Hélène Bonin ; Emmanuel Lacôte ; Dennis P. Curran
Journal of the American Chemical Society 2012 Volume 134(Issue 29) pp:12281-12288
Publication Date(Web):June 19, 2012
DOI:10.1021/ja305061c
Treatment of alkenes such as 3-hexene, 3-octene, and 1-cyclohexyl-1-butene with the N-heterocyclic carbene (NHC)-derived borane 2 and catalytic HNTf2 (Tf = trifluoromethanesulfonyl (CF3SO2)) effects hydroboration at room temperature. With 3-hexene, surprisingly facile migration of the boron atom from C(3) of the hexyl group to C(2) was observed over a time scale of minutes to hours. Oxidative workup gave a mixture of alcohols containing 2-hexanol as the major product. A similar preference for the C(2) alcohol was observed after oxidative workup of the 3-octene and 1-cyclohexyl-1-butene hydroborations. NHC-borenium cations (or functional equivalents) are postulated as the species that accomplish the hydroborations, and the C(2) selective migrations are attributed to the four-center interconversion of borenium cations with cationic NHC-borane-olefin π-complexes.
Co-reporter:Timothy S. De Vries, Supriyo Majumder, Angela M. Sandelin, Guoqiang Wang, and Edwin Vedejs
Organic Letters 2012 Volume 14(Issue 3) pp:688-691
Publication Date(Web):January 17, 2012
DOI:10.1021/ol2031203
Internal hydride transfer occurs when tethered carbocations are generated from unsaturated phosphine or phosphinite boranes. 3-Methylenecyclohexyl-derived boranes 12 or 18 react with MsOH to give ionic hydrogenation products with high syn-selectivity. With unsaturated amine boranes, initial hydrogen evolution gives BH2(OMs) complexes, but IH occurs using excess MsOH in a slower second stage. A diastereoselective reaction occurs from 26b using camphorsulfonic acid (first stage) and MsOH (second stage), affording 33 (68% ee) after hydrolysis.
Co-reporter:Susan D. Wiedner and Edwin Vedejs
The Journal of Organic Chemistry 2012 Volume 77(Issue 2) pp:1045-1055
Publication Date(Web):December 29, 2011
DOI:10.1021/jo202286a
The syntheses and reactivity of N-TBDPS and N-trityl protected derivatives of an aziridinomitosene corresponding to FK317 are described. New reactivity patterns were observed for these highly sensitive and functionally dense heterocycles under mild nucleophilic conditions approaching the threshold for degradation. Thus, the silyl or trityl protected aziridinomitosene reacted with Cs2CO3/CD3OD to give isomeric products where substitution occurred at C(10) and C(9a) (mitomycin numbering) providing a CD3 ether and a CD3 hemiaminal, respectively. These findings show that heterolysis at C(10) is faster than at aziridine C(1), in contrast to the behavior of typical aziridinomitosenes in the mitomycin series. The labile N-TBDPS hemiaminal and the more stable N-trityl hemiaminal resemble the mitomycin K substitution pattern. A reagent consisting of CsF in CF3CH2OH/CH3CN desilylated a simple N-TBDPS aziridine but caused nucleophilic cleavage at C(1) as well as C(10) without cleavage of the N-TBPDS group in the fully functionalized penultimate aziridinomitosene. The high reactivity of the C(10) carbamate with nucleophiles precludes the use of deprotection methodology that requires N-protonation for fully functionalized aziridinomitosenes in the FK317 series.
Co-reporter:Aleksandrs Prokofjevs
Journal of the American Chemical Society 2011 Volume 133(Issue 50) pp:20056-20059
Publication Date(Web):November 14, 2011
DOI:10.1021/ja208093c
Highly electrophilic boron cations derived from hindered amine borane complexes have been shown to undergo intramolecular aliphatic C–H borylation.
Co-reporter:Aleksrs Prokofjevs;Dr. Jeff W. Kampf ;Dr. Edwin Vedejs
Angewandte Chemie International Edition 2011 Volume 50( Issue 9) pp:2098-2101
Publication Date(Web):
DOI:10.1002/anie.201005663
Co-reporter:Aleksrs Prokofjevs;Dr. Jeff W. Kampf ;Dr. Edwin Vedejs
Angewandte Chemie 2011 Volume 123( Issue 9) pp:2146-2149
Publication Date(Web):
DOI:10.1002/ange.201005663
Co-reporter:Susan D. Wiedner and Edwin Vedejs
Organic Letters 2010 Volume 12(Issue 18) pp:4030-4033
Publication Date(Web):August 25, 2010
DOI:10.1021/ol101595u
Aziridinomitosane ketones 4 and 24 are accessed by internal acyl anion equivalent-lactam cyclization of 29 in a convergent route. The key aziridinolactam 6 is prepared by tin−lithium exchange via the lithiated aziridine 11.
Co-reporter:John M. Nelson and Edwin Vedejs
Organic Letters 2010 Volume 12(Issue 22) pp:5085-5087
Publication Date(Web):October 14, 2010
DOI:10.1021/ol101904a
The palladium-catalyzed coupling of an aziridinylzinc chloride intermediate with alkenyl and aryl halides has been demonstrated. The method provides products with retention of aziridine stereochemistry. The utility of the coupling procedure is illustrated in the synthesis of structures related to l-furanomycin.
Co-reporter:Timothy S. De Vries ; Aleksandrs Prokofjevs ; Jeremy N. Harvey
Journal of the American Chemical Society 2009 Volume 131(Issue 41) pp:14679-14687
Publication Date(Web):September 28, 2009
DOI:10.1021/ja905369n
The first examples of borylation under conditions of borenium ion generation from hydrogen-bridged boron cations are described. The observable H-bridged cations are generated by hydride abstraction from N,N-dimethylamine boranes Ar(CH2)nNMe2BH3 using Ph3C+ (C6F5)4B− (TrTPFPB) as the hydride acceptor. In the presence of excess TrTPFPB, the hydrogen-bridged cations undergo internal borylation to afford cyclic amine borane derivatives with n = 1−3. The products are formed as the corresponding cyclic borenium ions according to reductive quenching experiments and 11B and 1H NMR spectroscopy in the case with Ar = C6H5 and n = 1. The same cyclic borenium cation is also formed from the substrate with Ar = o-C6H4SiMe3 via desilylation, but the analogous system with Ar = o-C6H4CMe3 affords a unique cyclization product that retains the tert-butyl substituent. An ortho-deuterated substrate undergoes cyclization with a product-determining isotope effect of kH/kD 2.8. Potential cationic intermediates have been evaluated using B3LYP/6-31G* methods. The computations indicate that internal borylation from 14a occurs via a C−H insertion transition state that is accessible from either the borenium π complex or from a Wheland intermediate having nearly identical energy. The Ar = o-C6H4SiMe3 example strongly favors formation of the Wheland intermediate, and desilylation occurs via internal SiMe3 migration from carbon to one of the hydrides attached to boron.
Co-reporter:Edwin Vedejs ;Mara Jure
Angewandte Chemie International Edition 2005 Volume 44(Issue 26) pp:
Publication Date(Web):8 JUN 2005
DOI:10.1002/anie.200460842
The Walden memorial at the Technical University in Riga is pictured in the frontispiece to mark the recent centennial of the Walden inversion. This is a rare public monument to key events from the first era of exploration in stereocontrolled synthesis, and may be the only such monument to use the language of organic chemistry expressed at the molecular level. The reaction of racemic substrates with chiral nucleophiles is one of many methods currently known to achieve kinetic resolution, a phenomenon that ranks as the oldest and most general approach for the synthesis of highly enantioenriched substances. The first nonenzymatic kinetic resolutions as well as the original forms of the Walden inversion were studied in the 1890s. All of these investigations were conducted within the first generation following the demonstration that carbon is tetrahedral, and provided abundant evidence that the principles and importance of enantiocontrolled syntheses were understood. However, a reliable, rapid technique to quantify results and guide the optimization process was still lacking. Many decades passed before this problem was solved by the advent of HPLC and GLPC assays on chiral supports, which stimulated explosive growth in the synthesis of nonracemic substances by kinetic resolution. The Walden monument is accessible to passers-by for hands-on inspection as well as for contemplation and learning. In a similar way, kinetic resolution is experimentally accessible and can be thought-provoking at several levels. We follow the story of kinetic resolution from the early discoveries through fascinating historical milestones and conceptual developments, and close with a focus on modern techniques that maximize efficiency.
Co-reporter:Edwin Vedejs ;Mara Jure
Angewandte Chemie 2005 Volume 117(Issue 26) pp:
Publication Date(Web):8 JUN 2005
DOI:10.1002/ange.200460842
Das im Vortitel gezeigte Walden-Denkmal auf dem Campus der Technischen Universität Riga ist sicher eines der wenigen öffentlichen Monumente, die Schlüsselereignisse aus der Anfangszeit der stereokontrollierten Synthese thematisieren und vielleicht das einzige, das die “molekulare Sprache” der organischen Chemie gebraucht. Die ersten nichtenzymatischen kinetischen Racematspaltungen wurden, ebenso wie die ursprünglichen Varianten der Walden-Umkehr, in den 1890er Jahren untersucht – also noch in der ersten Forschergeneration nach Entdeckung der tetraedrischen Natur des Kohlenstoffs. Aus diesen frühen Arbeiten ist abzulesen, dass die Prinzipien und die Bedeutung enantiokontrollierter Synthesen bereits gut verstanden waren. Was noch fehlte war eine verlässliche und schnelle Methode zur Quantifizierung der Ergebnisse und zur Optimierung der Experimente. Viele Jahrzehnte sollten vergehen, bis das Problem mit Einführung von HPLC- und GPLC-Testreihen auf chiralen Trägern gelöst wurde. Das Walden-Denkmal lädt den Vorübergehenden zur spielerischen Betrachtung ein – auf ähnliche Weise, wie auch die kinetische Racematspaltung zum Experimentieren einlädt. Dieser Aufsatz verfolgt den Verlauf der Geschichte angefangen von den ersten Entdeckungen über die faszinierenden historischen Meilensteine und konzeptionellen Entwicklungen und schließt mit einem Blick auf moderne Techniken zur Effizienzmaximierung.
Co-reporter:Edwin Vedejs;Peter L. Steck
Angewandte Chemie International Edition 1999 Volume 38(Issue 18) pp:
Publication Date(Web):15 SEP 1999
DOI:10.1002/(SICI)1521-3773(19990917)38:18<2788::AID-ANIE2788>3.0.CO;2-P
A striking red color appears upon heating the colorless title compound 1 due to rearrangement to the oxaphosphorane 3. The initiating event in this transformation may involve intramolecular O-to-P acyl transfer to give 2, followed by P-to-C acyl transfer, or an unprecedented concerted addition of acetyl and phosphanyl groups to the triple bond.
Co-reporter:Edwin Vedejs;Peter L. Steck
Angewandte Chemie 1999 Volume 111(Issue 18) pp:
Publication Date(Web):15 SEP 1999
DOI:10.1002/(SICI)1521-3757(19990917)111:18<2958::AID-ANGE2958>3.0.CO;2-Q
Kräftig rot werden Lösungen der farblosen Titelverbindung 1 beim Erhitzen, da sich diese zum Oxaphosphoran 3 umlagert. Der erste Reaktionsschritt könnte ein intramolekularer OP-Acyltransfer unter Bildung von 2 sein, auf den ein PC-Acyltransfer folgt, oder eine bislang beispiellose konzertierte Addition von Acetyl- und Phosphanylgruppen an die Dreifachbindung.







![Ethanone, 2-[(2,3-dihydroxypropyl)thio]-1-phenyl-](http://img.cochemist.com/ccimg/115200/115120-28-0.png)
![Ethanone, 2-[(2,3-dihydroxypropyl)thio]-1-phenyl-](http://img.cochemist.com/ccimg/115200/115120-28-0_b.png)
![Ethanone, 2-[[2-(acetyloxy)-3,3-dimethylbutyl]thio]-1-phenyl-](http://img.cochemist.com/ccimg/115200/115120-26-8.png)
![Ethanone, 2-[[2-(acetyloxy)-3,3-dimethylbutyl]thio]-1-phenyl-](http://img.cochemist.com/ccimg/115200/115120-26-8_b.png)
![Ethanone, 2-[[2-(acetyloxy)butyl]thio]-1-phenyl-](http://img.cochemist.com/ccimg/115200/115120-18-8.png)
![Ethanone, 2-[[2-(acetyloxy)butyl]thio]-1-phenyl-](http://img.cochemist.com/ccimg/115200/115120-18-8_b.png)
![Ethanethioic acid, S-[3-phenyl-1-(phenylsulfonyl)propyl] ester](http://img.cochemist.com/ccimg/115200/115120-16-6.png)
![Ethanethioic acid, S-[3-phenyl-1-(phenylsulfonyl)propyl] ester](http://img.cochemist.com/ccimg/115200/115120-16-6_b.png)
![Ethanethioic acid, S-[2-methyl-1-(phenylthio)propyl] ester](http://img.cochemist.com/ccimg/115200/115120-14-4.png)
![Ethanethioic acid, S-[2-methyl-1-(phenylthio)propyl] ester](http://img.cochemist.com/ccimg/115200/115120-14-4_b.png)
![ETHANETHIOIC ACID, S-[3-PHENYL-1-(PHENYLTHIO)PROPYL] ESTER](http://img.cochemist.com/ccimg/115200/115120-13-3.png)
![ETHANETHIOIC ACID, S-[3-PHENYL-1-(PHENYLTHIO)PROPYL] ESTER](http://img.cochemist.com/ccimg/115200/115120-13-3_b.png)








![9-Thia-2-azabicyclo[3.3.1]nonan-7-one, 2-methyl-](http://img.cochemist.com/ccimg/114400/114325-99-4.png)
![9-Thia-2-azabicyclo[3.3.1]nonan-7-one, 2-methyl-](http://img.cochemist.com/ccimg/114400/114325-99-4_b.png)


![2-Thiabicyclo[2.2.1]hept-5-ene-3-carboxylic acid, ethyl ester](http://img.cochemist.com/ccimg/112800/112751-83-4.png)
![2-Thiabicyclo[2.2.1]hept-5-ene-3-carboxylic acid, ethyl ester](http://img.cochemist.com/ccimg/112800/112751-83-4_b.png)






![2(3H)-Furanone,5-[[[(1,1-dimethylethyl)diphenylsilyl]oxy]methyl]dihydro-, (S)-](http://img.cochemist.com/ccimg/102800/102717-29-3.png)
![2(3H)-Furanone,5-[[[(1,1-dimethylethyl)diphenylsilyl]oxy]methyl]dihydro-, (S)-](http://img.cochemist.com/ccimg/102800/102717-29-3_b.png)














![2-Oxa-4-azabicyclo[3.2.0]hept-6-ene-6,7-dicarboxylic acid,1,3,4,5-tetramethyl-, dimethyl ester](http://img.cochemist.com/ccimg/102600/102536-97-0.png)
![2-Oxa-4-azabicyclo[3.2.0]hept-6-ene-6,7-dicarboxylic acid,1,3,4,5-tetramethyl-, dimethyl ester](http://img.cochemist.com/ccimg/102600/102536-97-0_b.png)
















![Xylitol, 1,5-dideoxy-2-O-[(1,1-dimethylethyl)dimethylsilyl]-](http://img.cochemist.com/ccimg/100500/100428-64-6.png)
![Xylitol, 1,5-dideoxy-2-O-[(1,1-dimethylethyl)dimethylsilyl]-](http://img.cochemist.com/ccimg/100500/100428-64-6_b.png)












![Ribitol, 1,5-dideoxy-2-O-[(1,1-dimethylethyl)dimethylsilyl]-](http://img.cochemist.com/ccimg/100400/100312-83-2.png)
![Ribitol, 1,5-dideoxy-2-O-[(1,1-dimethylethyl)dimethylsilyl]-](http://img.cochemist.com/ccimg/100400/100312-83-2_b.png)






![Silane, dimethyl[(2Z)-1-methyl-2-butenyl]phenyl-](http://img.cochemist.com/ccimg/100400/100312-73-0.png)
![Silane, dimethyl[(2Z)-1-methyl-2-butenyl]phenyl-](http://img.cochemist.com/ccimg/100400/100312-73-0_b.png)





![2-Pyrrolidinone, 3-[(2-nitrophenyl)methoxy]-1-[(trimethylsilyl)methyl]-](http://img.cochemist.com/ccimg/96300/96246-59-2.png)
![2-Pyrrolidinone, 3-[(2-nitrophenyl)methoxy]-1-[(trimethylsilyl)methyl]-](http://img.cochemist.com/ccimg/96300/96246-59-2_b.png)
![{[(4-Methylphenyl)sulfonyl]amino}(phenyl)acetic acid](http://img.cochemist.com/ccimg/92900/92851-65-5.png)
![{[(4-Methylphenyl)sulfonyl]amino}(phenyl)acetic acid](http://img.cochemist.com/ccimg/92900/92851-65-5_b.png)


![Benzene, 1-methoxy-4-[(methoxymethoxy)methyl]-](http://img.cochemist.com/ccimg/92600/92565-78-1.png)
![Benzene, 1-methoxy-4-[(methoxymethoxy)methyl]-](http://img.cochemist.com/ccimg/92600/92565-78-1_b.png)






![Ethanone, 2-[[2-(acetyloxy)-2-phenylethyl]thio]-1-phenyl-](http://img.cochemist.com/ccimg/88400/88358-43-4.png)
![Ethanone, 2-[[2-(acetyloxy)-2-phenylethyl]thio]-1-phenyl-](http://img.cochemist.com/ccimg/88400/88358-43-4_b.png)











![Ethanone, 2-[(2,2-dimethylpropyl)thio]-1-phenyl-](http://img.cochemist.com/ccimg/84900/84850-25-9.png)
![Ethanone, 2-[(2,2-dimethylpropyl)thio]-1-phenyl-](http://img.cochemist.com/ccimg/84900/84850-25-9_b.png)


![Propanoic acid, 2-[[(trifluoromethyl)sulfonyl]oxy]-, ethyl ester, (2S)-](http://img.cochemist.com/ccimg/84100/84028-88-6.png)
![Propanoic acid, 2-[[(trifluoromethyl)sulfonyl]oxy]-, ethyl ester, (2S)-](http://img.cochemist.com/ccimg/84100/84028-88-6_b.png)






![Ethane, [[(1,1-dimethylethyl)dimethylsilyl]-aci-nitro]-](http://img.cochemist.com/ccimg/77300/77242-15-0.png)
![Ethane, [[(1,1-dimethylethyl)dimethylsilyl]-aci-nitro]-](http://img.cochemist.com/ccimg/77300/77242-15-0_b.png)


![2-PYRROLIDINONE, 3-HYDROXY-1-[(TRIMETHYLSILYL)METHYL]-](http://img.cochemist.com/ccimg/76600/76596-20-8.png)
![2-PYRROLIDINONE, 3-HYDROXY-1-[(TRIMETHYLSILYL)METHYL]-](http://img.cochemist.com/ccimg/76600/76596-20-8_b.png)




















![SILANE, TRIMETHYL[(3-NITROPHENYL)METHYL]-](http://img.cochemist.com/ccimg/69400/69380-89-8.png)
![SILANE, TRIMETHYL[(3-NITROPHENYL)METHYL]-](http://img.cochemist.com/ccimg/69400/69380-89-8_b.png)


























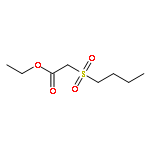
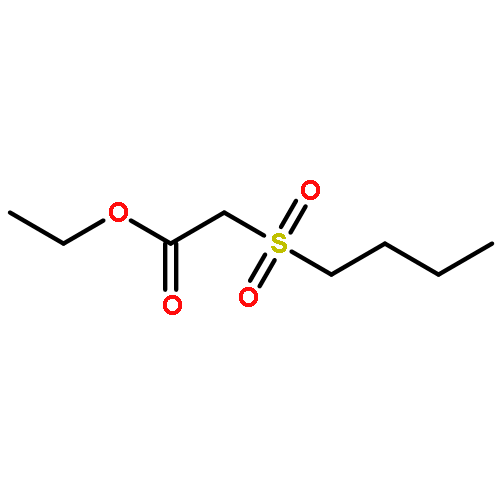


























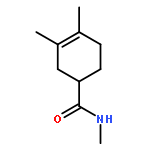














![Magnesium, [(1R,2S,5R)-5-methyl-2-(1-methylethyl)cyclohexyl]chloro-](http://img.cochemist.com/ccimg/63500/63474-24-8.png)
![Magnesium, [(1R,2S,5R)-5-methyl-2-(1-methylethyl)cyclohexyl]chloro-](http://img.cochemist.com/ccimg/63500/63474-24-8_b.png)













![PYRROLIDINE, 1-[(2E)-3-PHENYL-2-PROPENYL]-](http://img.cochemist.com/ccimg/58400/58325-60-3.png)
![PYRROLIDINE, 1-[(2E)-3-PHENYL-2-PROPENYL]-](http://img.cochemist.com/ccimg/58400/58325-60-3_b.png)


![Bicyclo[2.2.1]hept-2-ene, 5-methoxy-](http://img.cochemist.com/ccimg/56700/56663-04-8.png)
![Bicyclo[2.2.1]hept-2-ene, 5-methoxy-](http://img.cochemist.com/ccimg/56700/56663-04-8_b.png)



















![Ethanethioic acid, S-[(phenylthio)methyl] ester](http://img.cochemist.com/ccimg/42200/42177-11-7.png)
![Ethanethioic acid, S-[(phenylthio)methyl] ester](http://img.cochemist.com/ccimg/42200/42177-11-7_b.png)



![Benzene, [(1Z)-2-cyclohexylethenyl]-](http://img.cochemist.com/ccimg/40200/40132-69-2.png)
![Benzene, [(1Z)-2-cyclohexylethenyl]-](http://img.cochemist.com/ccimg/40200/40132-69-2_b.png)




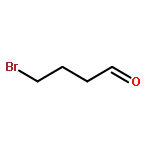
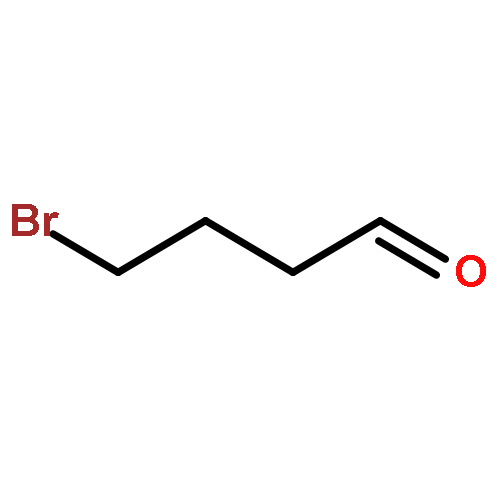
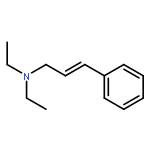
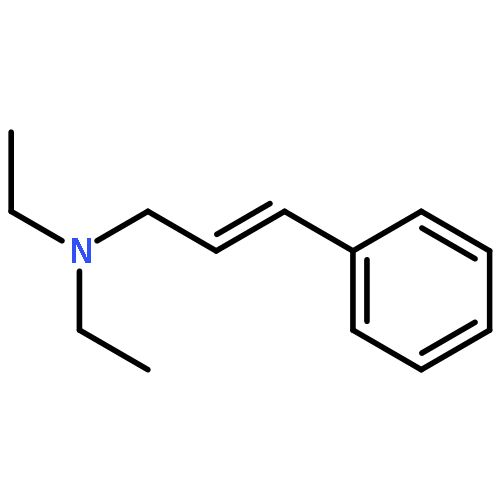




















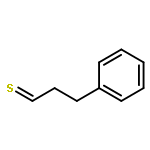
![Benzene,[(3-phenylpropyl)thio]-](http://img.cochemist.com/ccimg/30200/30134-12-4.png)
![Benzene,[(3-phenylpropyl)thio]-](http://img.cochemist.com/ccimg/30200/30134-12-4_b.png)

![(S)-2-[(Dimethylamino)methyl]pyrrolidine](http://img.cochemist.com/ccimg/29700/29618-57-3.png)
![(S)-2-[(Dimethylamino)methyl]pyrrolidine](http://img.cochemist.com/ccimg/29700/29618-57-3_b.png)







![1H-Cycloundec[d]isoindole-1,11(2H)-dione,15-(acetyloxy)-3,3a,4,5,6,6a,9,10,12,15-decahydro-6-hydroxy-4,10,12-trimethyl-5-methylene-3-(phenylmethyl)-,(3S,3aR,4S,6S,6aR,7E,10S,12S,13E,15R,15aR)-](http://img.cochemist.com/ccimg/26400/26399-27-9.png)
![1H-Cycloundec[d]isoindole-1,11(2H)-dione,15-(acetyloxy)-3,3a,4,5,6,6a,9,10,12,15-decahydro-6-hydroxy-4,10,12-trimethyl-5-methylene-3-(phenylmethyl)-,(3S,3aR,4S,6S,6aR,7E,10S,12S,13E,15R,15aR)-](http://img.cochemist.com/ccimg/26400/26399-27-9_b.png)
![Tetracyclo[3.3.1.02,8.04,6]nonan-3-ol](http://img.cochemist.com/ccimg/25300/25226-67-9.png)
![Tetracyclo[3.3.1.02,8.04,6]nonan-3-ol](http://img.cochemist.com/ccimg/25300/25226-67-9_b.png)






![1H-Cycloundec[d]isoindole-1,11(2H)-dione,15-(acetyloxy)-3,3a,4,5,6,6a,9,10,12,15-decahydro-6,12-dihydroxy-4,10,12-trimethyl-5-methylene-3-(phenylmethyl)-,(3S,3aR,4S,6S,6aR,7E,10S,12R,13E,15R,15aR)-](http://img.cochemist.com/ccimg/22200/22144-77-0.png)
![1H-Cycloundec[d]isoindole-1,11(2H)-dione,15-(acetyloxy)-3,3a,4,5,6,6a,9,10,12,15-decahydro-6,12-dihydroxy-4,10,12-trimethyl-5-methylene-3-(phenylmethyl)-,(3S,3aR,4S,6S,6aR,7E,10S,12R,13E,15R,15aR)-](http://img.cochemist.com/ccimg/22200/22144-77-0_b.png)

![Tricyclo[4.2.2.02,5]deca-3,7,9-triene](http://img.cochemist.com/ccimg/21700/21604-76-2.png)
![Tricyclo[4.2.2.02,5]deca-3,7,9-triene](http://img.cochemist.com/ccimg/21700/21604-76-2_b.png)
![TRICYCLO[2.2.1.02,6]HEPTANE, 3-METHOXY-](http://img.cochemist.com/ccimg/21600/21516-65-4.png)
![TRICYCLO[2.2.1.02,6]HEPTANE, 3-METHOXY-](http://img.cochemist.com/ccimg/21600/21516-65-4_b.png)





![1H-Pyrrolo[2,1-c][1,4]benzodiazepine-5,11(10H,11aH)-dione,2,3-dihydro-, (11aS)-](http://img.cochemist.com/ccimg/18900/18877-34-4.png)
![1H-Pyrrolo[2,1-c][1,4]benzodiazepine-5,11(10H,11aH)-dione,2,3-dihydro-, (11aS)-](http://img.cochemist.com/ccimg/18900/18877-34-4_b.png)
![Benzene, [(1E)-2-cyclohexylethenyl]-](http://img.cochemist.com/ccimg/18900/18869-27-7.png)
![Benzene, [(1E)-2-cyclohexylethenyl]-](http://img.cochemist.com/ccimg/18900/18869-27-7_b.png)












![1-methyl-9-oxabicyclo[6.1.0]nonane](http://img.cochemist.com/ccimg/16300/16240-40-7.png)
![1-methyl-9-oxabicyclo[6.1.0]nonane](http://img.cochemist.com/ccimg/16300/16240-40-7_b.png)




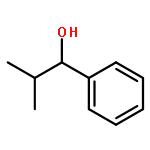
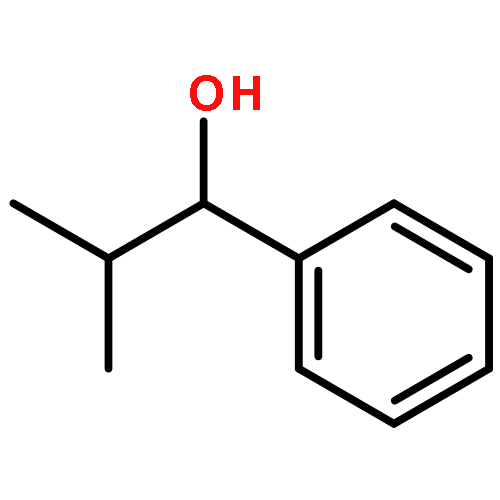
![Benzene,[(2-methylpropyl)thio]-](http://img.cochemist.com/ccimg/13400/13307-61-4.png)
![Benzene,[(2-methylpropyl)thio]-](http://img.cochemist.com/ccimg/13400/13307-61-4_b.png)


![Bicyclo[2.2.1]hept-2-ene, 5-methyl-, (1R,4R,5R)-rel-](/data/chemimg/1643800/10060-48-7.png)
![Bicyclo[2.2.1]hept-2-ene, 5-methyl-, (1R,4R,5R)-rel-](/data/chemimg/1643800/10060-48-7_b.png)



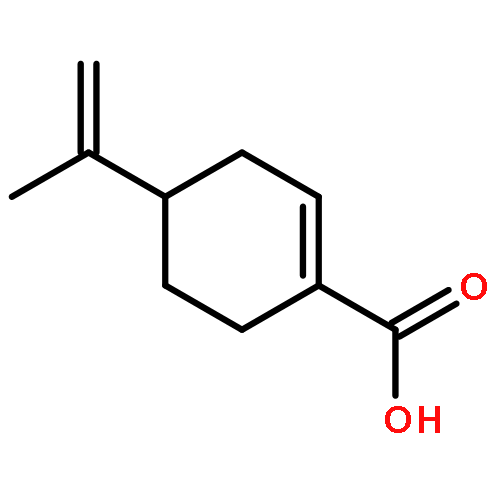




![2-THIABICYCLO[2.2.1]HEPT-5-ENE](http://img.cochemist.com/ccimg/6900/6841-59-4.png)
![2-THIABICYCLO[2.2.1]HEPT-5-ENE](http://img.cochemist.com/ccimg/6900/6841-59-4_b.png)


![tricyclo[2.2.1.0~2,6~]hept-3-yl acetate](http://img.cochemist.com/ccimg/6600/6555-48-2.png)
![tricyclo[2.2.1.0~2,6~]hept-3-yl acetate](http://img.cochemist.com/ccimg/6600/6555-48-2_b.png)




![Tricyclo[3.3.1.02,8]nona-3,6-dien-9-one](http://img.cochemist.com/ccimg/6100/6006-24-2.png)
![Tricyclo[3.3.1.02,8]nona-3,6-dien-9-one](http://img.cochemist.com/ccimg/6100/6006-24-2_b.png)
![Piperidine, 1-[(2E)-3-phenyl-2-propenyl]-](http://img.cochemist.com/ccimg/5900/5882-82-6.png)
![Piperidine, 1-[(2E)-3-phenyl-2-propenyl]-](http://img.cochemist.com/ccimg/5900/5882-82-6_b.png)

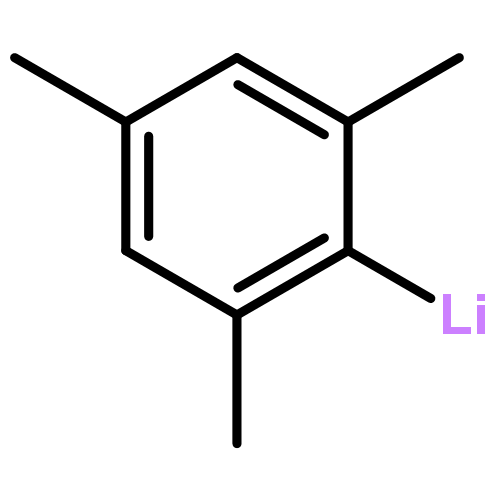





![1,4,5,6-tetrachloro-7,7-dimethoxybicyclo[2.2.1]hept-5-ene-2-carboxylic acid](http://img.cochemist.com/ccimg/5300/5259-71-2.png)
![1,4,5,6-tetrachloro-7,7-dimethoxybicyclo[2.2.1]hept-5-ene-2-carboxylic acid](http://img.cochemist.com/ccimg/5300/5259-71-2_b.png)






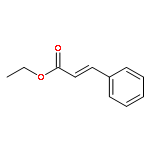
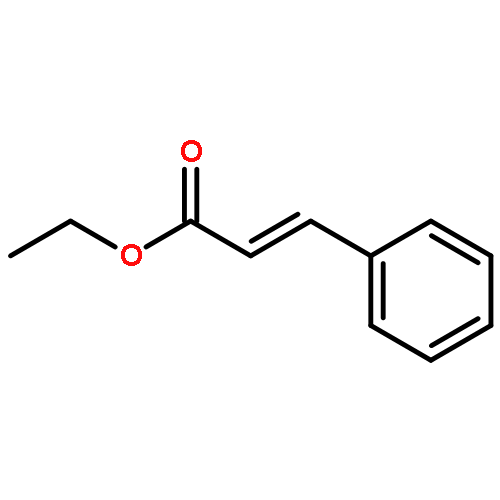








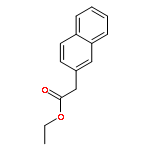















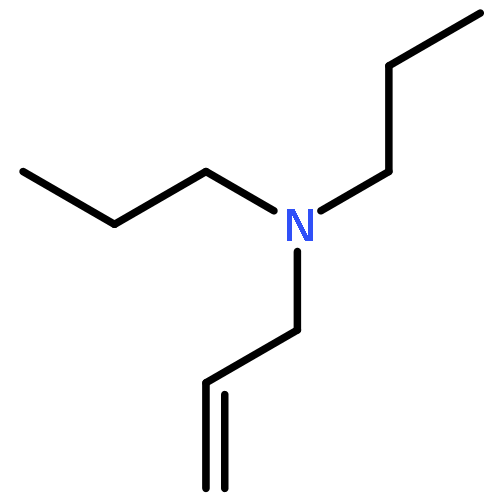
![7-Oxabicyclo[4.1.0]heptane, 1-methyl-4-(1-methylethenyl)-](http://img.cochemist.com/ccimg/1200/1195-92-2.png)
![7-Oxabicyclo[4.1.0]heptane, 1-methyl-4-(1-methylethenyl)-](http://img.cochemist.com/ccimg/1200/1195-92-2_b.png)
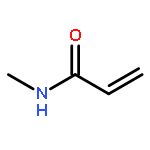
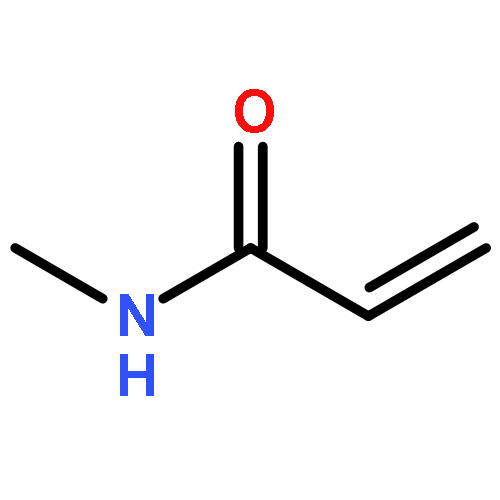
![5-phenylbenzo[b]phosphindole](http://img.cochemist.com/ccimg/1100/1088-00-2.png)
![5-phenylbenzo[b]phosphindole](http://img.cochemist.com/ccimg/1100/1088-00-2_b.png)
![Mercury, [(1R,2R,3S,4S)-3-(acetyloxy)bicyclo[2.2.1]hept-5-en-2-yl]chloro-](http://img.cochemist.com/ccimg/1100/1077-98-1.png)
![Mercury, [(1R,2R,3S,4S)-3-(acetyloxy)bicyclo[2.2.1]hept-5-en-2-yl]chloro-](http://img.cochemist.com/ccimg/1100/1077-98-1_b.png)


![Tricyclo[3.3.2.02,8]deca-3,6,9-triene](http://img.cochemist.com/ccimg/1100/1005-51-2.png)
![Tricyclo[3.3.2.02,8]deca-3,6,9-triene](http://img.cochemist.com/ccimg/1100/1005-51-2_b.png)


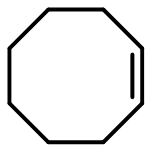
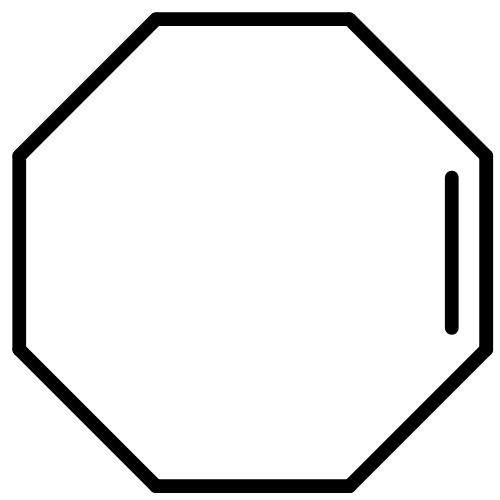








![9-Azabicyclo[6.1.0]nonane(6CI,7CI,8CI,9CI)](http://img.cochemist.com/ccimg/300/286-61-3.png)
![9-Azabicyclo[6.1.0]nonane(6CI,7CI,8CI,9CI)](http://img.cochemist.com/ccimg/300/286-61-3_b.png)
![5H-Benzo[b]phosphindole](http://img.cochemist.com/ccimg/300/244-87-1.png)
![5H-Benzo[b]phosphindole](http://img.cochemist.com/ccimg/300/244-87-1_b.png)




![1-(3,5-DITERT-BUTYLPHENYL)-3,6,6-TRIMETHYL-2,3,3A,4,5,6A-HEXAHYDROCYCLOPENTA[B]PHOSPHOLE](http://img.cochemist.com/ccimg/233000/232923-66-9.png)
![1-(3,5-DITERT-BUTYLPHENYL)-3,6,6-TRIMETHYL-2,3,3A,4,5,6A-HEXAHYDROCYCLOPENTA[B]PHOSPHOLE](http://img.cochemist.com/ccimg/233000/232923-66-9_b.png)
![Phosphine, [3,5-bis(1,1-dimethylethyl)phenyl]-](http://img.cochemist.com/ccimg/233000/232923-65-8.png)
![Phosphine, [3,5-bis(1,1-dimethylethyl)phenyl]-](http://img.cochemist.com/ccimg/233000/232923-65-8_b.png)










![Silane, (1,1-dimethylethyl)dimethyl[(1-methylene-2-propenyl)oxy]-](http://img.cochemist.com/ccimg/80800/80738-05-2.png)
![Silane, (1,1-dimethylethyl)dimethyl[(1-methylene-2-propenyl)oxy]-](http://img.cochemist.com/ccimg/80800/80738-05-2_b.png)






![Bicyclo[4.2.2]deca-2,4,7,9-tetraene](http://img.cochemist.com/ccimg/15700/15677-13-1.png)
![Bicyclo[4.2.2]deca-2,4,7,9-tetraene](http://img.cochemist.com/ccimg/15700/15677-13-1_b.png)