Co-reporter:Qinghua Lyu;Jieyu Zhang;Koon Gee Neoh;Christina Li Lin Chai
Nanoscale (2009-Present) 2017 vol. 9(Issue 34) pp:12409-12415
Publication Date(Web):2017/08/31
DOI:10.1039/C7NR05293F
Biomimetic poly(catecholamine) coatings have gained much attention in recent years due to their versatility as functional materials. Despite this, only limited methods are available to modify the function and property of poly(catecholamine) coatings, primarily through post-modification methods. Our approach reported herein provides a simple approach to the fabrication of novel functionalized poly(catecholamine) coatings. The strategy employs the copolymerization of N-Ac-3,4-dihydroxyphenylalanine methyl ester (NADOPAMe) with nucleophilic additives, giving rise to nano-coatings on various surfaces including plastic, metal, glass and polymers. With the appropriate choice of nucleophilic additives, coatings with desired properties can be achieved. This is demonstrated through the fabrication of a redox responsive coating based on NADOPAMe with cysteamine as additive, which shows a concentration-dependent glutathione (GSH) responsive behavior. The ability to utilize this as a controlled release system is also demonstrated.
Co-reporter:Song Wei Benjamin Tan;Mark G. Moloney
Organic & Biomolecular Chemistry 2017 vol. 15(Issue 8) pp:1889-1912
Publication Date(Web):2017/02/22
DOI:10.1039/C6OB02828D
Mono and dihydroxypyrrolidinones are readily available by direct oxygenation of a pyroglutamate-derived bicyclic lactam with high diastereoselectivity, and these may be manipulated further in protected or unprotected form by Grignard addition to a pendant Weinreb amide to give acylhydroxypyrrolidinones, which are analogues of the natural product, pramanicin. Preliminary bioassay against S. aureus and E. coli indicated that some compounds exhibit selective Gram-negative antibacterial activity, and may offer promise for the development of novel systems suitable for antibacterial drug development.
Co-reporter:Jieyu Zhang, Yong Shung Cheah, Sridhar Santhanakrishnan, Koon Gee Neoh, Christina L.L. Chai
Polymer 2017 Volume 116(Volume 116) pp:
Publication Date(Web):5 May 2017
DOI:10.1016/j.polymer.2017.03.061
•An electron rich dopamine derivative, 5-methoxy dopamine (OMeDA), is synthesized.•The electron-donating group on OMeDA accelerates oxidative polymerization reactions.•For surface modification applications, OMeDA is superior to dopamine as monomer.•OMeDA can be used to efficiently fabricate Ag-containing antibacterial coatings.Deposition of polydopamine on substrates is a facile and effective method of surface modification and the deposited polydopamine can reduce silver ions to form silver nanoparticles (AgNPs) for antibacterial applications. However, polydopamine deposition is a time-consuming process that usually requires 24 h to produce a dense surface coating. Since oxidation of dopamine is critical for its polymerization, we hypothesize herein that substitution of an electron-donating group on the catechol ring of dopamine can enhance its oxidation potential and subsequently accelerate its polymerization. In this work, dopamine substituted with a 5-methoxy group (OMeDA) was prepared. OMeDA polymerized faster than dopamine under similar reaction conditions, resulting in a polymer coating of 13 nm thickness on a silicon surface after 8 h, compared to the 24 h required for dopamine to form a coating of similar thickness. A polymer layer with AgNPs can be directly formed on the silicon substrate after exposure to a solution containing OMeDA and silver nitrate. After 2 h exposure, the silver content on the modified surfaces prepared with OMeDA was 187% higher than that obtained with dopamine, and the antibacterial efficacy of the former against Staphylococcus aureus was correspondingly higher than that of the latter. This study demonstrates that OMeDA with an electron-donating group in the catechol ring offers improvements over dopamine for surface modification applications.Download high-res image (160KB)Download full-size image
Co-reporter:Quy T.N. Tran, W.S. Fred Wong, Christina L.L. Chai
Pharmacological Research 2017 Volume 124(Volume 124) pp:
Publication Date(Web):1 October 2017
DOI:10.1016/j.phrs.2017.07.019
The search for new anti-inflammatory agents is challenging due to the complexity of the inflammatory process and its role in host defense. Over the past few decades, a significant body of evidence has emerged, supporting the prominent role of labdane diterpenoids in therapeutic interventions of various inflammatory diseases. The anti-inflammatory activity of labdane diterpenoids has been attributed mainly to the inhibition of nuclear factor-κB (NF-κB) activity, the modulation of arachidonic acid (AA) metabolism and the reduction of nitric oxide (NO) production. This article provides extensive coverage of naturally occurring labdane diterpenes, discovered between 1981 and 2016, which have been verified as NF-κB, NO, or AA modulators. Herein, we also discuss the role of Michael acceptor, a common structural feature present in most of the active labdane diterpenes, and its association with NF-κB signaling inhibition. In the cases where a sufficient amount of data exists, structure-activity relationship (SAR) studies and clinical studies performed on the anti-inflammatory labdane diterpenoids are also discussed.This review summarises the labdane diterpenoids reported during 1981–2016 that have modulatory activities in the different pathways i.e. nuclear factor-kappaB (NF-κB), nitric oxide (NO), and arachidonic acid (AA) metabolite pathways, that lead to inflammation.Download high-res image (184KB)Download full-size image
Co-reporter:Liya Shi, Sridhar Santhanakrishnan, Yong Shung Cheah, Min Li, Christina Li Lin Chai, and Koon Gee Neoh
ACS Applied Materials & Interfaces 2016 Volume 8(Issue 48) pp:
Publication Date(Web):November 15, 2016
DOI:10.1021/acsami.6b07751
Dopamine (DA) protected by an o-nitrobenzyl functionality on its phenolic group was synthesized as a photolabile catecholamine derivative. This compound, o-nitrobenzyl dopamine (NBDA), was more stable than DA in basic solution at pH 8.5 and will not self-polymerize when protected from light. UV irradiation of a methanolic solution of NBDA at 365 nm for 40 min induced ca. 85% deprotection. Taking advantage of the stability of NBDA, a one-pot spray coating technique for modifying surfaces with polydopamine (PDA) was developed. Using ethylene glycol with Tris buffer (pH 8.5) as the solvent for this technique, stainless steel substrates can be coated with a robust PDA layer. Silver was deposited on the PDA-coated surface after treatment with silver nitrate solution, and >80% of the deposited silver remained on the surface after 1 week immersion in water. The NBDA-Ag surface was highly effective in inhibiting Staphylococcus aureus (S. aureus) biofilm formation.Keywords: antibacterial; photolabile; polydopamine; silver deposition; spray coating; UV-induced deprotection;
Co-reporter:Yong Shung Cheah, Sridhar Santhanakrishnan, Michael B. Sullivan, Koon Gee Neoh, Christina L.L. Chai
Tetrahedron 2016 Volume 72(Issue 41) pp:6543-6550
Publication Date(Web):13 October 2016
DOI:10.1016/j.tet.2016.08.068
The chemical reactivities of four catecholamines, N-acetyl dopamine (NADA) and its dehydro derivative (NAΔDA), N-acetyl 3,4-dihydroxy-phenylalanine methyl ester (NADOPAME) and its dehydro derivative (NAΔDOPAME), under oxidative nucleophilic trapping and polymerisation conditions were compared and contrasted. Despite their structural similarities, varying reactivities and regioselectivities for oxidative nucleophilic trapping with ethanethiol were observed. This has possible implications on the use of these natural building blocks and their derivatives in the design and synthesis of biomimetic materials.
Co-reporter:Dr. Mun Hong Ngai;Choon Leng So;Dr. Michael B. Sullivan;Dr. Han Kiat Ho;Dr. Christina L. L. Chai
ChemMedChem 2016 Volume 11( Issue 1) pp:72-80
Publication Date(Web):
DOI:10.1002/cmdc.201500475
Abstract
3-Substituted indolin-2-ones are an important class of compounds that display a wide range of biological activities. Sunitinib is an orally available multiple tyrosine kinase inhibitor that has been approved by the US Food and Drug Administration (FDA) for the treatment of renal cell cancer. Sunitinib and a related compound, semaxanib, exist as thermodynamically stable Z isomers, which photoisomerize to E isomers in solution. In this study, 17 3-substituted indolin-2-ones were synthesized, and the kinetics of their photoisomerization were studied by 1H NMR spectroscopy. The rate constants for photoisomerization ranged from 0.009 to 0.048 h−1. Selected compounds were tested for cytotoxicity in the TAMH liver cell line. E/Z mixtures of four compounds were also assessed for toxicity in the TAMH and HepG2 cell lines. In some cases, the stereochemically pure drug was more toxic than the E/Z mixtures, but a general statement cannot be made. Our studies show that each stereoisomer could contribute differently to toxicity, suggesting that stereochemical purity issues that could arise from isomerization cannot be ignored.
Co-reporter:Dr. Mun Hong Ngai;Choon Leng So;Dr. Michael B. Sullivan;Dr. Han Kiat Ho;Dr. Christina L. L. Chai
ChemMedChem 2016 Volume 11( Issue 1) pp:
Publication Date(Web):
DOI:10.1002/cmdc.201500583
Co-reporter:Kuan-Chieh Ching; Yiu-Wing Kam; Andres Merits; Lisa F. P. Ng
Journal of Medicinal Chemistry 2015 Volume 58(Issue 23) pp:9196-9213
Publication Date(Web):November 5, 2015
DOI:10.1021/acs.jmedchem.5b01047
Chikungunya virus (CHIKV) is a re-emerging vector-borne alphavirus and is transmitted to humans by Aedes mosquitoes. Despite the re-emergence of CHIKV as an epidemic threat, there is no approved effective antiviral treatment currently available for CHIKV. Herein, we report the synthesis and structure–activity relationship studies of a class of thieno[3,2-b]pyrroles and the discovery of a trisubstituted thieno[3,2-b]pyrrole 5-carboxamide 15c that exhibits potent inhibitory activity against in vitro CHIKV infection. Compound 15c displayed low micromolar activity (EC50 value of ca. 2 μM) and limited cytotoxic liability (CC50 > 100 μM) therefore furnishing a selectivity index of greater than 32. Notably, 15c not only controlled viral RNA production, but efficiently inhibited the expression of CHIKV nsP1, nsP3, capsid, and E2 proteins at a concentration as low as 2.5 μM. More importantly, 15c also demonstrated broad spectrum antiviral activity against other clinically important alphaviruses such as O’nyong–nyong virus and Sindbis virus.
Co-reporter:Paul B. Huleatt; Mui Ling Khoo; Yi Yuan Chua; Tiong Wei Tan; Rou Shen Liew; Balázs Balogh; Ruth Deme; Flóra Gölöncsér; Kalman Magyar; David P. Sheela; Han Kiat Ho; Beáta Sperlágh; Péter Mátyus
Journal of Medicinal Chemistry 2015 Volume 58(Issue 3) pp:1400-1419
Publication Date(Web):January 28, 2015
DOI:10.1021/jm501722s
To develop novel neuroprotective agents, a library of novel arylalkenylpropargylamines was synthesized and tested for inhibitory activities against monoamine oxidases. From this, a number of highly potent and selective monoamine oxidase B inhibitors were identified. Selected compounds were also tested for neuroprotection in in vitro studies with PC-12 cells treated with 6-OHDA and rotenone, respectively. It was observed that some of the compounds tested yielded a marked increase in survival in PC-12 cells treated with the neurotoxins. This indicates that these propargylamines are able to confer protection against the effects of the toxins and may also be considered as novel disease-modifying anti-Parkinsonian agents, which are much needed for the therapy of Parkinson’s disease.
Co-reporter:Shi Qing Tang, Yong Yang Irvin Lee, David Sheela Packiaraj, Han Kiat Ho, and Christina Li Lin Chai
Chemical Research in Toxicology 2015 Volume 28(Issue 10) pp:2019
Publication Date(Web):September 24, 2015
DOI:10.1021/acs.chemrestox.5b00247
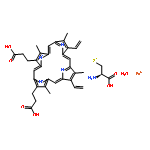
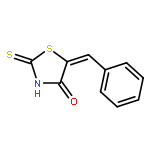
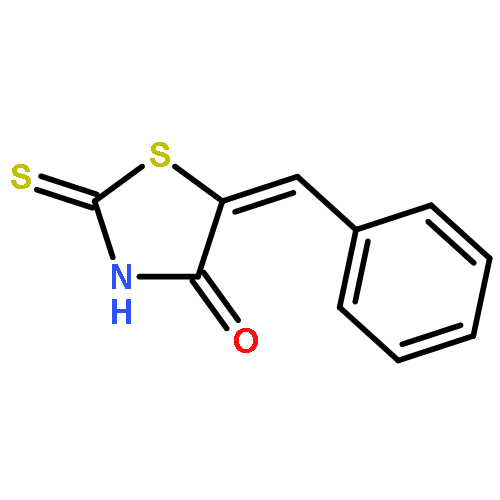
The thiazolidine and imidazolidine heterocyclic scaffolds, i.e., the rhodanines, 2,4-thiazolidinediones, 2-thiohydantoins, and hydantoins have been the subject of debate on their suitability as starting points in drug discovery. This attention arose from the wide variety of biological activities exhibited by these scaffolds and their frequent occurrence as hits in screening campaigns. Studies have been conducted to evaluate their value in drug discovery in terms of their biological activity, chemical reactivity, aggregation-based promiscuity, and electronic properties. However, the metabolic profiles and toxicities have not been systematically assessed. In this study, a series of five-membered multiheterocyclic (FMMH) compounds were selected for a systematic evaluation of their metabolic profiles and toxicities on TAMH cells, a metabolically competent rodent liver cell line and HepG2 cells, a model of human hepatocytes. Our studies showed that generally the rhodanines are the most toxic, followed by the thiazolidinediones, thiohydantoins, and hydantoins. However, not all compounds within the family of heterocycles were toxic. In terms of metabolic stability, 5-substituted rhodanines and 5-benzylidene thiohydantoins were found to have short half-lives in the presence of human liver microsomes (t1/2 < 30 min) suggesting that the presence of the endocyclic sulfur and thiocarbonyl group or a combination of C5 benzylidene substituent and thiocarbonyl group in these heterocycles could be recognition motifs for P450 metabolism. However, the stability of these compounds could be improved by installing hydrophilic functional groups. Therefore, the toxicities and metabolic profiles of FMMH derivatives will ultimately depend on the overall chemical entity, and a blanket statement on the effect of the FMMH scaffold on toxicity or metabolic stability cannot and should not be made.
Co-reporter:Dr. Eric K. W. Tam;Dr. Tuan Minh Nguyen;Cheryl Z. H. Lim;Puay Leng Lee;Zhimei Li;Xia Jiang;Dr. Sridhar Santhanakrishnan;Tiong Wei Tan;Yi Ling Goh;Sze Yue Wong;Haiyan Yang;Esther H. Q. Ong;Dr. Jeffrey Hill;Dr. Qiang Yu; Christina L. L. Chai
ChemMedChem 2015 Volume 10( Issue 1) pp:173-182
Publication Date(Web):
DOI:10.1002/cmdc.201402315
Abstract
3-Deazaneplanocin A (DzNep) is a potential epigenetic drug for the treatment of various cancers. DzNep has been reported to deplete histone methylations, including oncogenic EZH2 complex, giving rise to epigenetic modifications that reactivate many silenced tumor suppressors in cancer cells. Despite its promise as an anticancer drug, little is known about the structure–activity relationships of DzNep in the context of epigenetic modifications and apoptosis induction. In this study, a number of analogues of DzNep were examined for DzNep-like ability to induce synergistic apoptosis in cancer cells in combination with trichostatin A, a known histone deacetylase (HDAC) inhibitor. The structure–activity relationship data thus obtained provide valuable information on the structural requirements for biological activity. The studies identified three compounds that show similar activities to DzNep. Two of these compounds show good pharmacokinetics and safety profiles. Attempts to correlate the observed synergistic apoptotic activities with measured S-adenosylhomocysteine hydrolase (SAHH) inhibitory activities suggest that the apoptotic activity of DzNep might not be directly due to its inhibition of SAHH.
Co-reporter:Yun Xuan Tan, Sridhar Santhanakrishnan, Hai Yan Yang, Christina L. L. Chai, and Eric Kwok Wai Tam
The Journal of Organic Chemistry 2014 Volume 79(Issue 17) pp:8059-8066
Publication Date(Web):August 14, 2014
DOI:10.1021/jo501248e
The key cyclopentenyl intermediate 11b was synthesized in 4 steps from d-ribose in 41% overall yield via an efficient intramolecular Baylis–Hillman reaction. This novel key intermediate can be modified easily and transformed to neplanocin A (1a) and its 3′-epimer (1b).
Co-reporter:Jin Xu, Esther H.Q. Ong, Jeffrey Hill, Anqi Chen, Christina L.L. Chai
Bioorganic & Medicinal Chemistry 2014 22(23) pp: 6625-6637
Publication Date(Web):
DOI:10.1016/j.bmc.2014.10.006
Co-reporter:Dr. Eric K. W. Tam;Dr. Zhengqiu Li;Yi Ling Goh;Xiamin Cheng;Sze Yue Wong;Dr. Sridhar Santhanakrishnan;Dr. Christina L. L. Chai;Dr. Shao Q. Yao
Chemistry – An Asian Journal 2013 Volume 8( Issue 8) pp:1818-1828
Publication Date(Web):
DOI:10.1002/asia.201300303
Abstract
3-Deazaneplanocin A (DzNep), a global histone methylation inhibitor, has attracted significant interest in epigenetic research in recent years. The molecular mechanism of action and the cellular off-targets of DzNep, however, are still not well-understood. Our aim was to develop novel DzNep-derived small-molecule probes suitable to be used in live mammalian cells for identification of potential cellular targets of DzNep under physiologically relevant settings. In the current study, we have successfully designed, synthesized, and tested one such probe, called Dz-1. Dz-1 is a ‘clickable’ affinity-based probe (AfBP) derived from DzNep with minimal structural modifications. The probe was found to be highly cell-permeable, and possessed similar anti-apoptotic activities as DzNep in MCF-7 mammalian cells. Two additional control probes were made as negative labeling/pull-down probes in order to minimize false identification of background proteins due to unavoidable, intrinsic nonspecific photo-crosslinking reactions. All three probes were subsequently used for in-situ proteome profiling in live mammalian cells, followed by large-scale pull-down/LC-MS/MS analysis for identification of potential cellular protein targets that might interact with DzNep in native cellular environments. Our LC-MS/MS results revealed some highly enriched proteins that had not been reported as potential DzNep targets. These proteins might constitute unknown cellular off-targets of DzNep. Though further validation experiments are needed in order to unequivocally confirm these off-targets, our findings shed new light on the future use of DzNep as a validated chemical probe for epigenetic research and as a potential drug candidate for cancer therapy.
Co-reporter:Jin Xu;Dr. Anqi Chen;Dr. Joma Joy;Vanessa Joanne Xavier;Esther H. Q. Ong;Dr. Jeffrey Hill; Christina L. L. Chai
ChemMedChem 2013 Volume 8( Issue 9) pp:1483-1494
Publication Date(Web):
DOI:10.1002/cmdc.201300231
Abstract
Recent biological and computational advances in drug design have led to renewed interest in targeted covalent inhibition as an efficient and practical approach for the development of new drugs. As part of our continuing efforts in the exploration of the therapeutic potential of resorcylic acid lactones (RALs), we report herein the design, synthesis, and biological evaluation of conveniently accessible RAL enamide analogues as novel covalent inhibitors of MAP kinase interacting kinases (MNKs). In this study, we have successfully demonstrated that the covalent binding ability of RAL enamides can be tuned by attaching an electron-withdrawing motif, such as an acyl group, to enhance its reactivity toward the cysteine residues at the MNK1/2 binding sites. We have also shown that 1H NMR spectroscopy is a convenient and effective tool for screening the covalent binding activities of enamides using cysteamine as a mimic of the key cysteine residue in the enzyme, whereas mass spectrometric analysis confirms covalent modification of the kinases. Preliminary optimization of the initial hit led to the discovery of enamides with low micromolar activity in MNK assays. Cancer cell line assays have identified RAL enamides that inhibit the growth of cancer cells with similar potency to the natural product L-783,277.
Co-reporter:Song Wei Benjamin Tan, Christina L. L. Chai and Mark G. Moloney
Organic & Biomolecular Chemistry 2017 - vol. 15(Issue 8) pp:NaN1912-1912
Publication Date(Web):2017/01/31
DOI:10.1039/C6OB02828D
Mono and dihydroxypyrrolidinones are readily available by direct oxygenation of a pyroglutamate-derived bicyclic lactam with high diastereoselectivity, and these may be manipulated further in protected or unprotected form by Grignard addition to a pendant Weinreb amide to give acylhydroxypyrrolidinones, which are analogues of the natural product, pramanicin. Preliminary bioassay against S. aureus and E. coli indicated that some compounds exhibit selective Gram-negative antibacterial activity, and may offer promise for the development of novel systems suitable for antibacterial drug development.



![4-Thiazolidinone, 5-[(3-hydroxyphenyl)methylene]-2-thioxo-, (5Z)-](http://img.cochemist.com/ccimg/911800/911714-30-2.png)
![4-Thiazolidinone, 5-[(3-hydroxyphenyl)methylene]-2-thioxo-, (5Z)-](http://img.cochemist.com/ccimg/911800/911714-30-2_b.png)


















![2H-Indol-2-one,3-[(3,5-dimethyl-1H-pyrrol-2-yl)methylene]-1,3-dihydro-, (3Z)-](http://img.cochemist.com/ccimg/194500/194413-58-6.png)
![2H-Indol-2-one,3-[(3,5-dimethyl-1H-pyrrol-2-yl)methylene]-1,3-dihydro-, (3Z)-](http://img.cochemist.com/ccimg/194500/194413-58-6_b.png)


![2,2'-spirobi[[1,4,2]oxathiazolo[2,3-a]pyridin-4-ium]](http://img.cochemist.com/ccimg/154600/154592-20-8.png)
![2,2'-spirobi[[1,4,2]oxathiazolo[2,3-a]pyridin-4-ium]](http://img.cochemist.com/ccimg/154600/154592-20-8_b.png)







![1-N-methyl-8-methoxy-9-hydroxy-1H-benzo[d,e][1,6]naphthyridin-4-ium monotrifluoroacetate](http://img.cochemist.com/ccimg/117200/117173-75-8.png)
![1-N-methyl-8-methoxy-9-hydroxy-1H-benzo[d,e][1,6]naphthyridin-4-ium monotrifluoroacetate](http://img.cochemist.com/ccimg/117200/117173-75-8_b.png)








![(1R,3aS,5aS)-1,3a,6-trimethyl-8-oxo-1,3a,4,5,5a,6,7,8-octahydrocyclopenta[c]pentalene-2-carboxylic acid](http://img.cochemist.com/ccimg/97800/97718-45-1.png)
![(1R,3aS,5aS)-1,3a,6-trimethyl-8-oxo-1,3a,4,5,5a,6,7,8-octahydrocyclopenta[c]pentalene-2-carboxylic acid](http://img.cochemist.com/ccimg/97800/97718-45-1_b.png)
![11,16,18,19-Tetraoxapentacyclo[12.2.2.16,9.01,15.010,12]nonadeca-6,8-diene-7-carboxylicacid, 12-methyl-4-(1-methylethenyl)-17-oxo-, methyl ester,(1R,4S,10R,12S,14R,15R)-](http://img.cochemist.com/ccimg/96000/95925-22-7.png)
![11,16,18,19-Tetraoxapentacyclo[12.2.2.16,9.01,15.010,12]nonadeca-6,8-diene-7-carboxylicacid, 12-methyl-4-(1-methylethenyl)-17-oxo-, methyl ester,(1R,4S,10R,12S,14R,15R)-](http://img.cochemist.com/ccimg/96000/95925-22-7_b.png)


![(1R,2R,4aS,5S,8S,8aS)-8-[(2R,5R)-5-chloro-2,6,6-trimethyltetrahydro-2H-pyran-2-yl]-1,5-diisocyano-2,5-dimethyldecahydronaphthalen-2-ol](http://img.cochemist.com/ccimg/91300/91294-83-6.png)
![(1R,2R,4aS,5S,8S,8aS)-8-[(2R,5R)-5-chloro-2,6,6-trimethyltetrahydro-2H-pyran-2-yl]-1,5-diisocyano-2,5-dimethyldecahydronaphthalen-2-ol](http://img.cochemist.com/ccimg/91300/91294-83-6_b.png)


![2-PROPANONE, 1-[4-(PENTYLOXY)PHENYL]-](http://img.cochemist.com/ccimg/84100/84023-39-2.png)
![2-PROPANONE, 1-[4-(PENTYLOXY)PHENYL]-](http://img.cochemist.com/ccimg/84100/84023-39-2_b.png)
![4-Thiazolidinone, 5-[(4-hydroxyphenyl)methylene]-2-thioxo-, (5Z)-](http://img.cochemist.com/ccimg/81200/81154-14-5.png)
![4-Thiazolidinone, 5-[(4-hydroxyphenyl)methylene]-2-thioxo-, (5Z)-](http://img.cochemist.com/ccimg/81200/81154-14-5_b.png)
![4-Thiazolidinone, 5-[(2-hydroxyphenyl)methylene]-2-thioxo-, (5Z)-](http://img.cochemist.com/ccimg/81200/81154-12-3.png)
![4-Thiazolidinone, 5-[(2-hydroxyphenyl)methylene]-2-thioxo-, (5Z)-](http://img.cochemist.com/ccimg/81200/81154-12-3_b.png)








![1-(4-Fluorobenzyl)-N-(1-(4-methoxyphenethyl)piperidin-4-yl)-1H-benzo[d]imidazol-2-amine](http://img.cochemist.com/ccimg/68900/68844-77-9.png)
![1-(4-Fluorobenzyl)-N-(1-(4-methoxyphenethyl)piperidin-4-yl)-1H-benzo[d]imidazol-2-amine](http://img.cochemist.com/ccimg/68900/68844-77-9_b.png)


![methyl 4-methyl-8-oxo-12-(prop-1-en-2-yl)-3,7,17-trioxatetracyclo[12.2.1.1~6,9~.0~2,4~]octadeca-1(16),9(18),14-triene-15-carboxylate](http://img.cochemist.com/ccimg/58800/58772-81-9.png)
![methyl 4-methyl-8-oxo-12-(prop-1-en-2-yl)-3,7,17-trioxatetracyclo[12.2.1.1~6,9~.0~2,4~]octadeca-1(16),9(18),14-triene-15-carboxylate](http://img.cochemist.com/ccimg/58800/58772-81-9_b.png)
![1,4-Benzenediol,2-[[(1R,2S,4aS,8aS)-1,2,3,4,4a,7,8,8a-octahydro-1,2,4a,5-tetramethyl-1-naphthalenyl]methyl]-](http://img.cochemist.com/ccimg/55400/55303-98-5.png)
![1,4-Benzenediol,2-[[(1R,2S,4aS,8aS)-1,2,3,4,4a,7,8,8a-octahydro-1,2,4a,5-tetramethyl-1-naphthalenyl]methyl]-](http://img.cochemist.com/ccimg/55400/55303-98-5_b.png)
![Pyridine, 5-[(3,7-dimethyl-6-octenyl)oxy]-2-methyl-](http://img.cochemist.com/ccimg/54000/53953-30-3.png)
![Pyridine, 5-[(3,7-dimethyl-6-octenyl)oxy]-2-methyl-](http://img.cochemist.com/ccimg/54000/53953-30-3_b.png)












![2-((Thiocyanatomethyl)thio)benzo[d]thiazole](http://img.cochemist.com/ccimg/21600/21564-17-0.png)
![2-((Thiocyanatomethyl)thio)benzo[d]thiazole](http://img.cochemist.com/ccimg/21600/21564-17-0_b.png)
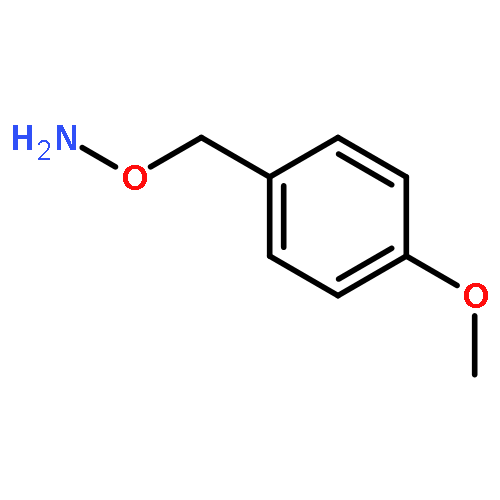


![Benzamide, N-[2-(benzoyloxy)ethyl]-](http://img.cochemist.com/ccimg/16200/16180-99-7.png)
![Benzamide, N-[2-(benzoyloxy)ethyl]-](http://img.cochemist.com/ccimg/16200/16180-99-7_b.png)
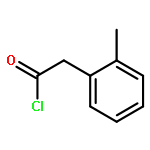
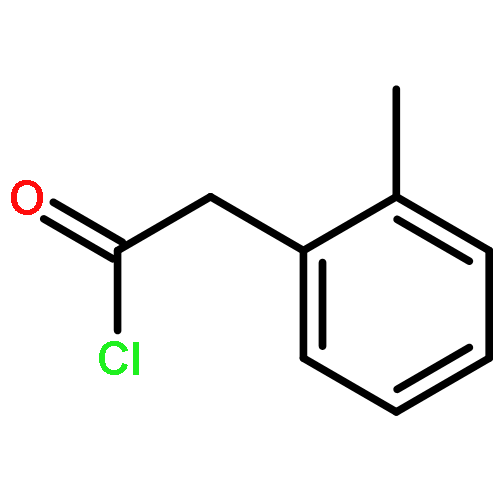
![7H-Furo[3',2':4,5]furo[2,3-c]xanthen-7-one,3a,12c-dihydro-8-hydroxy-6-methoxy-, (3aR,12cS)-](http://img.cochemist.com/ccimg/10100/10048-13-2.png)
![7H-Furo[3',2':4,5]furo[2,3-c]xanthen-7-one,3a,12c-dihydro-8-hydroxy-6-methoxy-, (3aR,12cS)-](http://img.cochemist.com/ccimg/10100/10048-13-2_b.png)

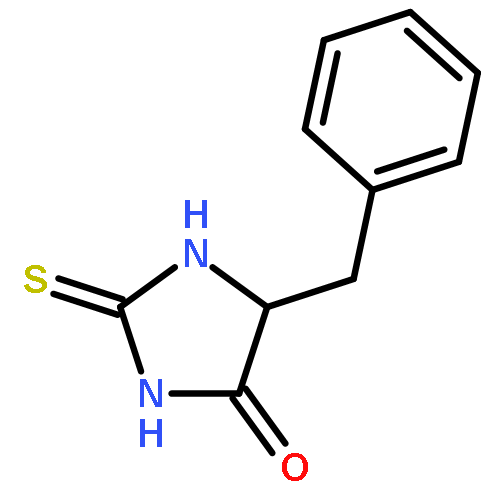



![1H-Isoindole-1,3(2H)-dione, 2-[(4-methoxyphenyl)methoxy]-](http://img.cochemist.com/ccimg/2100/2014-61-1.png)
![1H-Isoindole-1,3(2H)-dione, 2-[(4-methoxyphenyl)methoxy]-](http://img.cochemist.com/ccimg/2100/2014-61-1_b.png)













