Co-reporter:Jaya P. Shrestha, Coleman Baker, Yukie Kawasaki, Yagya P. Subedi, Nzuwah Nziko Vincent de Paul, Jon Y. Takemoto, Cheng-Wei Tom Chang
European Journal of Medicinal Chemistry 2017 Volume 126(Volume 126) pp:
Publication Date(Web):27 January 2017
DOI:10.1016/j.ejmech.2016.12.008
•These compounds were prepared in 2–3 steps with high overall yield.•Lead compounds are active against resistant fungi and bacteria.•Investigation reveals that dimeric CAAs can disrupt membrane integrity.•Computational study reveals designs for reviving the activity of QACs.•Lead compounds are selectively toxic to fungi and bacteria over human cells.A series of synthetic dimeric cationic anthraquinone analogs (CAAs) with potent antimicrobial activities against a broad range of fungi and bacteria were developed. These compounds were prepared in 2–3 steps with high overall yield and possess alkyl chain, azole, quinone, and quaternary ammonium complexes (QACs). In vitro biological evaluations reveal prominent inhibitory activities of lead compounds against several drug-susceptible and drug-resistant fungal and bacterial strains, including MRSA, VRE, Candida albicans and Aspergillus flavus. Mode of action investigation reveals that the synthesized dimeric CAA's can disrupt the membrane integrity of fungi. Computational studies reveal possible designs that can revive the activity of QACs against drug-resistant bacteria. Cytotoxicity assays in SKOV-3, a cancer cell line, show that the lead compounds are selectively toxic to fungi and bacteria over human cells.Download high-res image (78KB)Download full-size image
Co-reporter:Qian Zhang, Madher N. Alfindee, Jaya P. Shrestha, Vincent de Paul Nzuwah Nziko, Yukie Kawasaki, Xinrui Peng, Jon Y. Takemoto, and Cheng-Wei Tom Chang
The Journal of Organic Chemistry 2016 Volume 81(Issue 22) pp:10651-10663
Publication Date(Web):October 7, 2016
DOI:10.1021/acs.joc.6b01189
A concise and novel method for site-selective alkylation of 1,3,6′,3″-tetraazidokanamycin has been developed that leads to the divergent synthesis of three classes of kanamycin A derivatives. These new amphiphilic kanamycin derivatives bearing alkyl chains length of 4, 6, 7, 8, 9, 10, 12, 14, and 16 have been tested for their antibacterial and antifungal activities. The antibacterial effect of the synthesized kanamycin derivatives declines or disappears as compared to the original kanamycin A. Several compounds, especially those with octyl chain at O-4″ and/or O-6″ positions on the ring III of kanamycin A, show very strong activity as antifungal agents. In addition, these compounds display no toxicity toward mammalian cells. Finally, computational calculation has revealed possible factors that are responsible for the observed regioselectivity. The simplicity in chemical synthesis and the fungal specific property make the lead compounds ideal candidates for the development of novel antifungal agents.
Co-reporter:Qian Zhang, Xinrui Peng, Michelle Grilley, Jon Y. Takemoto and Cheng-Wei Tom Chang
Green Chemistry 2015 vol. 17(Issue 3) pp:1918-1925
Publication Date(Web):26 Dec 2014
DOI:10.1039/C4GC01659A
A library of fifteen commercially purchased and synthetic fluorogenic probes was employed for the investigation of biomass degradation using extracts of white-rot fungi. These probes were selected or designed to mimic the dominant linkages in celluloses, hemicelluloses, and lignin, the three most abundant polymers found in biomass. The results show that white-rot fungi display a high preference for cleaving mannose- and glucose-based probes, which mimic hemicelluloses. Low degrees of cleavages were noted for xylose- and cellobiose-based probes. No cleavages were observed for probes that mimic the linkages in lignin. Overall, these discoveries prove that it is possible to employ fungi for selective degradation or release of hemicelluloses from biomass.
Co-reporter:Jaya P. Shrestha, Yagya Prasad Subedi, Liaohai Chen and Cheng-Wei Tom Chang
MedChemComm 2015 vol. 6(Issue 11) pp:2012-2022
Publication Date(Web):25 Sep 2015
DOI:10.1039/C5MD00314H
Previously, we reported the synthesis and structure–activity relationship (SAR) study of a series of novel 4,9-dioxo-4,9-dihydro-1H-naphtho[2,3-d][1,2,3]triazol-3-ium salts, which had very potent anti-proliferative activities (low μM to nM GI50) against a broad range of cancer cells. These compounds, which can be viewed as cationic anthraquinone analogs (CAAs), are selective against cancer cells over bacteria or fungi as compared to the antibacterial CAAs that have also been reported by our group. Herein, we report a mode of action study of CAAs, which reveals that these compounds trigger apoptosis by generating extensive reactive oxygen species (ROS). The generation of extensive ROS causes oxidative stress, decrease in mitochondrial membrane potential, depletion of glutathione (GSH), and release of caspase-3, which ultimately kills cancer cells by programmed apoptosis. Furthermore, we have also shown that CAAs possess an 8-fold higher activity against the A549 cell line vs. the non-cancerous MRC-5 cell line.
Co-reporter:Marina Fosso, Madher N. AlFindee, Qian Zhang, Vincent de Paul Nzuwah Nziko, Yukie Kawasaki, Sanjib K. Shrestha, Jeremiah Bearss, Rylee Gregory, Jon Y. Takemoto, and Cheng-Wei Tom Chang
The Journal of Organic Chemistry 2015 Volume 80(Issue 9) pp:4398-4411
Publication Date(Web):March 31, 2015
DOI:10.1021/acs.joc.5b00248
Novel fungicides are urgently needed. It was recently reported that the attachment of an octyl group at the O-4″ position of kanamycin B converts this antibacterial aminoglycoside into a novel antifungal agent. To elucidate the structure–activity relationship (SAR) for this phenomenon, a lead compound FG03 with a hydroxyl group replacing the 3″-NH2 group of kanamycin B was synthesized. FG03’s antifungal activity and synthetic scheme inspired the synthesis of a library of kanamycin B analogues alkylated at various hydroxyl groups. SAR studies of the library revealed that for antifungal activity the O-4″ position is the optimal site for attaching a linear alkyl chain and that the 3″-NH2 and 6″-OH groups of the kanamycin B parent molecule are not essential for antifungal activity. The discovery of lead compound, FG03, is an example of reviving clinically obsolete drugs like kanamycin by simple chemical modification and an alternative strategy for discovering novel antimicrobials.
Co-reporter:Jaya P. Shrestha, Marina Y. Fosso, Jeremiah Bearss, Cheng-Wei Tom Chang
European Journal of Medicinal Chemistry 2014 Volume 77() pp:96-102
Publication Date(Web):22 April 2014
DOI:10.1016/j.ejmech.2014.02.060
•The newly synthesized compounds show strong anticancer activities (nM GI50).•Structure–activity relationship for enhancing activities has been revealed.•Elements for tuning the biological activity has been identified.We have synthesized a series of novel 4,9-dioxo-4,9-dihydro-1H-naphtho[2,3-d][1,2,3]triazol-3-ium salts, which can be viewed as analogs of cationic anthraquinones. Unlike the similar analogs that we have reported previously, these compounds show relatively weak antibacterial activities but exert strong anticancer activities (low μM to nM GI50), in particular, against melanoma, colon cancer, non-small cell lung cancer and central nervous system (CNS) cancer. These compounds are structurally different from their predecessors by having the aromatic group, instead of alkyl chains, directly attached to the cationic anthraquinone scaffold. Further investigation in the structure–activity relationship (SAR) reveals the significant role of electron donating substituents on the aromatic ring in enhancing the anticancer activities via resonance effect. Steric hindrance of these groups is disadvantageous but is less influential than the resonance effect. The difference in the attached groups at N-1 position of the cationic anthraquinone analog is the main structural factor for the switching of biological activity from antibacterial to anticancer. The discovery of these compounds may lead to the development of novel cancer chemotherapeutics.Nine compounds were synthesized and evaluated for their anticancer and antibacterial activities. The results yield the structural motifs needed for tuning the activity from antibacterial to anticancer and vise versa.
Co-reporter:Qian Zhang, Jaya P. Shrestha, Cheng-Wei Tom Chang
Tetrahedron Letters 2014 Volume 55(Issue 10) pp:1839-1842
Publication Date(Web):5 March 2014
DOI:10.1016/j.tetlet.2014.01.129
Through a [2+3] cycloaddition reaction, a new environmentally friendly method was developed to enable the synthesis of bioactive 1-alkyl-1H-naphtho[2,3-d][1,2,3]triazole-4,9-diones and N-aryl-2-aminomethylene-1,3-indanediones using water as the solvent with good yields and minimum requirement of purification. This new green synthetic protocol is simple and suitable for scale-up synthesis.
Co-reporter:V. P. N. Nziko;Marina Y. Fosso
Medicinal Chemistry Research 2014 Volume 23( Issue 12) pp:5058-5062
Publication Date(Web):2014 December
DOI:10.1007/s00044-014-1069-y
A quantitative structure–activity relationship (QSAR) of a library of eighteen cationic anthraquinone analogs was done using the Hansch and Fujita models. This offers a clear understanding of the structural parameter that could be used to explain the antibacterial activity of this class of compounds. The physicochemical parameters taken into consideration here were the Mlog P, partial atomic charge, dipole moment, and the HOMO. These parameters correlate well (q2 > 0.8) with the observed antibacterial activity which provides valuable guidelines for new structural designs to further improve the antibacterial activity.
Co-reporter:Jaya P. Shrestha, Cheng-Wei Tom Chang
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 21) pp:5909-5911
Publication Date(Web):1 November 2013
DOI:10.1016/j.bmcl.2013.08.078
We have developed a new safe and easy route for the synthesis of 1,3-dimethyl-1,2,3-triazolium derivatives. We have reported the synthesis of 4,9-dioxo-1,3-dimethylnaphtho[2,3-d][1,2,3]triazol-3-ium chloride from methylation of 1-methyl-1H-naphtho[2,3-d][1,2,3]triazole-4,9-dione. The synthesis of 1-methyl-1H-naphtho[2,3-d][1,2,3]triazole-4,9-dione is inefficient as a significant amount of by-product is formed that is difficult to separate and also unsafe as it requires the use of hazardous methylazide as a starting material. It is, however, important to develop an improved method for the synthesis of 4,9-dioxo-1,3-dimethylnaphtho[2,3-d][1,2,3]triazol-3-ium salt due to its significant anticancer activities. Herein, we report a safe and convenient route for the synthesis of this compound, which lead to more detailed exploration of its profound anticancer activities. The improved method can be applicable for the synthesis of other 1,3-dimethyl-1,2,3-triazolium salts of interest without the use of potentially explosive methylazide. The compound synthesized in this new method shows significant anticancer activities against melanoma, colon cancer, non-small cell lung cancer and central nervous system (CNS) cancer with GI50 values ranging from low μM to nM.
Co-reporter:Venkatareddy Udumula, Young Wan Ham, Marina Y. Fosso, Ka Yee Chan, Ravi Rai, Jianjun Zhang, Jie Li, Cheng-Wei Tom Chang
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 6) pp:1671-1675
Publication Date(Web):15 March 2013
DOI:10.1016/j.bmcl.2013.01.073
Aminoglycoside represents a class of versatile and broad spectrum antibacterial agents. In an effort to revive the antibacterial activity against aminoglycoside resistant bacteria, our laboratory has developed two new classes of aminoglycoside, pyranmycin and amphiphilic neomycin (NEOF004). The former resembles the traditional aminoglycoside, neomycin. The latter, albeit derived from neomycin, appears to exert antibacterial action via a different mode of action. In order to discern that these aminoglycoside derivatives have distinct antibacterial mode of action, RNA-binding affinity and fluorogenic dye were employed. These studies, together with our previous investigation, confirm that pyranmycin exhibit the traditional antibacterial mode of action of aminoglycosides by binding toward the bacterial rRNA. On the other hand, the amphiphilic neomycin, NEOF004 disrupts the bacterial cell wall. In a broader perspective, it verifies that structurally modified neomycin can exert different antibacterial mode of action leading to the revival of activity against aminoglycoside resistant bacteria.Aminoglycoside represents a class of versatile and broad spectrum antibacterial agents. In an effort to revive the antibacterial activity against aminoglycoside resistant bacteria, our laboratory has developed two new classes of aminoglycoside, pyranmycin and amphiphilic neomycin (NEOF004). The former resembles the traditional aminoglycoside, neomycin. The latter, albeit derived from neomycin, appears to exert antibacterial action via a different mode of action. In order to discern that these aminoglycoside derivatives have distinct antibacterial mode of action, RNA-binding affinity and fluorogenic dye were employed. These studies, together with our previous investigation, confirm that pyranmycin exhibit the traditional antibacterial mode of action of aminoglycosides by binding toward the bacterial rRNA. On the other hand, the amphiphilic neomycin, NEOF004 disrupts the bacterial cell wall. In a broader perspective, it verifies that structurally modified neomycin can exert different antibacterial mode of action leading to the revival of activity against aminoglycoside resistant bacteria.
Co-reporter:Marina Y. Fosso, Ka Yee Chan, Rylee Gregory, and Cheng-Wei Tom Chang
ACS Combinatorial Science 2012 Volume 14(Issue 3) pp:231
Publication Date(Web):February 10, 2012
DOI:10.1021/co2002075
We report the parallel synthesis of a series of novel 4,9-dioxo-4,9-dihydro-1H-naphtho[2,3-d][1,2,3]triazol-3-ium chloride salts, which are analogs to cationic anthraquinones. Three synthetic protocols were examined leading to a convenient and facile library synthesis of the cationic anthraquinone analogs that contain double alkyl chains of various lengths (C2–C12) at N-1 and N-3 positions. The antibacterial activities of these compounds were evaluated against Gram-positive bacterium Staphylococcus aureus and Gram-negative bacterium Escherichia coli. The antibacterial activities of these compounds were expected to be associated with the structural features of naphthoquinone, cation and lypophilic alkyl chain and, interestingly, they showed much higher levels of antibacterial activities against G+ than G– bacteria. In addition, when the total number of carbon atoms of the alkyl groups at both N-1 and N-3 positions lies between 9 and 18, the bactericidal activity against S. aureus increased with increasing alkyl chain length at both N-atoms with MIC ≤ 1 μg/mL.Keywords: anthraquinone analogs; antibacterial activity; Escherichia coli; Staphylococcus aureus
Co-reporter:Jianjun Zhang, Nathan Redman, Anthony Phillip Litke, Jia Zeng, Jixun Zhan, Ka Yee Chan, Cheng-Wei Tom Chang
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 1) pp:498-503
Publication Date(Web):1 January 2011
DOI:10.1016/j.bmc.2010.11.001
Reported previously by our group, one-pot cycloaddition using naphthoquinone, sodium azide and alkyl halides can lead to the formation of both 1-alkyl-1H- and 2-alkyl-2H-naphtho[2,3-d]triazole-4,9-diones. Herein, the effect of leaving group and additive in dictating the selectivity between the formation of 1-alkyl-1H- and 2-alkyl-2H-naphtho[2,3-d]triazole-4,9-diones has been further investigated. In the process of investigating the factors that control the selectivity and the biological activity associated with these two compounds, a novel class of antibacterial cationic anthraquinone analogs has been developed. Although these compounds are structurally similar, different antibacterial profiles are noted. One lead compound, 4e manifests high potency (MIC < 1 μg/mL) and selectivity against Gram positive (G+) pathogens including methicillin-resistant Staphylococcus aureus (MRSA) while exerting only modest activity against Gram negative (G−) bacteria. Other lead compounds (4f and 4g) exhibit broad antibacterial activity including MRSA and vancomycin-resistant Enterococcus faecalis (VRE) that is comparable to other commercially available cationic antiseptic chemicals. This unique difference in antibacterial profile may pave the way for the development of new therapeutic agents.In the process of investigating the factors that control the selectivity and the biological activity associated with 1-alkyl-1H and 2-alkyl-2H-naphtho[2,3-d]triazole-4,9-diones, a novel class of antibacterial cationic anthraquinone analogs has been developed. Although these compounds are structurally similar, different antibacterial profiles are noted. One lead compound, 4e manifests high potency (MIC < 1 μg/mL) and selectivity against Gram positive (G+) pathogens including methicillin-resistant Staphylococcus aureus (MRSA) while exerting only modest activity against Gram negative (G−) bacteria. Other lead compounds (4f and 4g) exhibit broad antibacterial activity including MRSA and vancomycin-resistant Enterococcus faecalis (VRE) that is comparable to other commercially available cationic antiseptic chemicals. This unique difference in antibacterial profile may pave the way for the development of new therapeutic agents.
Co-reporter:Ka Yee Chan, Jianjun Zhang, Cheng-Wei Tom Chang
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 21) pp:6353-6356
Publication Date(Web):1 November 2011
DOI:10.1016/j.bmcl.2011.08.107
Reported previously by our group, we have developed a novel class of antibacterial cationic anthraquinone analogs with superb potency (MIC <1 μg/mL) against Gram positive (G+) pathogens including Methicillin-resistant Staphylococcus aureus (MRSA). However, most of these compounds only manifest modest antibacterial activity against Gram negative (G−) bacteria. Further investigation on the antibacterial mode of action using fluorogenic dyes reveals that these compounds exert two different modes of action that account for the difference in their antibacterial profile. It was found that most of the compounds exert their antibacterial activity by disrupting the redox processes of bacteria. At high concentration, these compounds can also act as membrane disrupting agents. This information can help to design new therapeutics against various bacteria.Reported previously by our group, we have developed a novel class of antibacterial cationic anthraquinone analogs with superb potency (MIC <1 μg/mL) against Gram positive (G+) pathogens including Methicillin-resistant Staphylococcus aureus (MRSA). However, most of these compounds only manifest modest antibacterial activity against Gram negative (G−) bacteria. Further investigation on the antibacterial mode of action using fluorogenic dyes reveals that these compounds exert two different modes of action that account for the difference in their antibacterial profile. It was found that most of the compounds exert their antibacterial activity by disrupting the redox processes of bacteria. At high concentration, these compounds can also act as membrane disrupting agents. This information can help to design new therapeutics against various bacteria.
Co-reporter:Jianjun Zhang, Anthony Litke, Katherine Keller, Ravi Rai, Cheng-Wei Tom Chang
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 4) pp:1396-1405
Publication Date(Web):15 February 2010
DOI:10.1016/j.bmc.2010.01.027
Using allylic azide rearrangement, a convenient method has been developed for the synthesis of 2′,3′-dideoxyaminoglycosides that are, otherwise, difficult to be prepared. The antibacterial activity of these novel aminoglycosides also confirms the indispensable role of 2′-NH2 group for both neomycin and kanamycin classes of aminoglycosides. A novel structural motif containing the hexylaminocarbonyl groups at O-5 and/or O-6 of 2′,3′-dideoxyneamine could lead to the production of new aminoglycosides against resistant bacteria.Using allylic azide rearrangement, a convenient method has been developed for the synthesis of 2′,3′-dideoxyaminoglycosides that are, otherwise, difficult to be prepared. The antibacterial activity of these novel aminoglycosides also confirms the indispensable role of 2′-NH2 group for both neomycin and kanamycin classes of aminoglycosides. A novel structural motif containing the hexylaminocarbonyl groups at O-5 and/or O-6 of 2′,3′-dideoxyneamine could lead to the production of new aminoglycosides against resistant bacteria.
Co-reporter:Christabel T. Tanifum, Jianjun Zhang, Cheng-Wei T. Chang
Tetrahedron Letters 2010 Volume 51(Issue 33) pp:4323-4327
Publication Date(Web):18 August 2010
DOI:10.1016/j.tetlet.2010.06.027
Co-reporter:Cheng-Wei Tom Chang
Chemistry & Biology 2009 Volume 16(Issue 6) pp:579-580
Publication Date(Web):26 June 2009
DOI:10.1016/j.chembiol.2009.06.001
An innovative approach for manipulating glycosyltransferase-catalyzed glycosylation has now been developed (Truman et al.). Created using a domain-swapping strategy, these chimeric glycotransferases have predictable substrate specificity and may lead to the breakthrough developments in the preparation of carbohydrate-containing molecules of biological interest.
Co-reporter:Jianjun Zhang, Katherine Keller, Jon Y Takemoto, Mekki Bensaci, Anthony Litke, Przemyslaw Greg Czyryca and Cheng-Wei Tom Chang
The Journal of Antibiotics 2009 62(10) pp:539-544
Publication Date(Web):July 24, 2009
DOI:10.1038/ja.2009.66
A library of 5″-modified neomycin derivatives were synthesized for an antibacterial structure–activity optimization strategy. Two leads exhibited prominent activity against both methicillin-resistant Staphylococcus aureus (MRSA) and vancomycin-resistant enterococci (VRE). Antibacterial activities were measured when combined with other clinically used antibiotics. Significant synergistic activities were observed, which may lead to the development of novel therapeutic practices in the battle against infectious bacteria.
Co-reporter:Jianjun Zhang and Cheng-Wei Tom Chang
The Journal of Organic Chemistry 2009 Volume 74(Issue 11) pp:4414-4417
Publication Date(Web):May 5, 2009
DOI:10.1021/jo9004926
A one-pot three-component [2+3] cycloaddition for the synthesis of 1-alkyl 1H-naphtho[2,3-d][1,2,3]triazole-4,9-dione and 2-alkyl 2H-naphtho[2,3-d][1,2,3]triazole-4,9-dione has been developed. By taking the advantage of difference in basicity, both products can be obtained in good purity. The unique heterocyclic scaffolds of these two products could exert interesting chemical and biological properties. The synthetic protocol is concise and suitable for scale-up synthesis of the desired products.
Co-reporter:Jianjun Zhang ; Fang-I. Chiang ; Long Wu ; Przemyslaw Greg Czyryca ; Ding Li
Journal of Medicinal Chemistry 2008 Volume 51(Issue 23) pp:7563-7573
Publication Date(Web):November 14, 2008
DOI:10.1021/jm800997s
A facile synthetic protocol for the production of neomycin B derivatives with various modifications at the 5′′ position has been developed. The structural activity relationship (SAR) against aminoglycoside resistant bacteria equipped with various aminoglycoside-modifying enzymes (AMEs) was investigated. Enzymatic and molecular modeling studies reveal that the superb substrate promiscuity of AMEs allows the resistant bacteria to cope with diverse structural modifications despite the observation that several derivatives show enhanced antibacterial activity compared to the parent neomycin. Surprisingly, when testing synthetic neomycin derivatives against other human pathogens, two leads exhibit prominent activity against both methicillin-resistant Staphylococcus aureus (MRSA) and vancomycin-resistant enterococci (VRE) that are known to exert a high level of resistance against clinically used aminoglycosides. These findings can be extremely useful in developing new aminoglycoside antibiotics against resistant bacteria. Our result also suggests that new biological and antimicrobial activities can be obtained by chemical modifications of old drugs.
Co-reporter:Jianjun Zhang, Massoud Garrossian, Dale Gardner, Arash Garrossian, Young-Tae Chang, Yun Kyung Kim, Cheng-Wei Tom Chang
Bioorganic & Medicinal Chemistry Letters 2008 Volume 18(Issue 4) pp:1359-1363
Publication Date(Web):15 February 2008
DOI:10.1016/j.bmcl.2008.01.017
A diversity-oriented synthesis has been developed for facile construction of a library of carbohydrate–cyclopamine conjugates. The synthetic protocol is suitable for generating cyclopamine derivatives with various structural motifs for exploring the desired activity. From this initial library, we have observed one derivative that exhibits improved activity against lung cancer cell as compared to cyclopamine.A diversity-oriented synthesis has been developed for facile construction of a library of carbohydrate–cyclopamine conjugates. The synthetic protocol is suitable for generating cyclopamine derivatives with various structural motifs for exploring the desired activity. From this initial library, we have observed one derivative that exhibits improved activity against lung cancer cell as compared to cyclopamine.

![1,4,6,7(6aH,10H)-Naphthacenetetrone,8-acetyl-10-[[5-[[2,6-dideoxy-4-O-(2,4-dimethyl-1-oxo-2-heptenyl)-3-C-methyl-a-L-xylo-hexopyranosyl]oxy]tetrahydro-6-methyl-2H-pyran-2-yl]oxy]-10a,11-dihydro-5,6a,9,10a-tetrahydroxy-2-methoxy-12-methyl-,[2S-[2a(6aR*,10S*,10aS*),5b(2Z,4S*),6a]]- (9CI)](http://img.cochemist.com/ccimg/146700/146663-67-4.png)
![1,4,6,7(6aH,10H)-Naphthacenetetrone,8-acetyl-10-[[5-[[2,6-dideoxy-4-O-(2,4-dimethyl-1-oxo-2-heptenyl)-3-C-methyl-a-L-xylo-hexopyranosyl]oxy]tetrahydro-6-methyl-2H-pyran-2-yl]oxy]-10a,11-dihydro-5,6a,9,10a-tetrahydroxy-2-methoxy-12-methyl-,[2S-[2a(6aR*,10S*,10aS*),5b(2Z,4S*),6a]]- (9CI)](http://img.cochemist.com/ccimg/146700/146663-67-4_b.png)
![(2z)-2-[(3r,4s,5s,8s,9s,10s,11r,13r,14s,16s)-16-acetyloxy-3,11-dihydroxy-4,8,10,14-tetramethyl-2,3,4,5,6,7,9,11,12,13,15,16-dodecahydro-1h-cyclopenta[a]phenanthren-17-ylidene]-6-methylhept-5-enoic Acid](http://img.cochemist.com/ccimg/7000/6990-06-3.png)
![(2z)-2-[(3r,4s,5s,8s,9s,10s,11r,13r,14s,16s)-16-acetyloxy-3,11-dihydroxy-4,8,10,14-tetramethyl-2,3,4,5,6,7,9,11,12,13,15,16-dodecahydro-1h-cyclopenta[a]phenanthren-17-ylidene]-6-methylhept-5-enoic Acid](http://img.cochemist.com/ccimg/7000/6990-06-3_b.png)










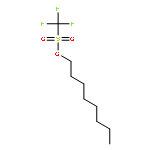

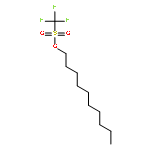
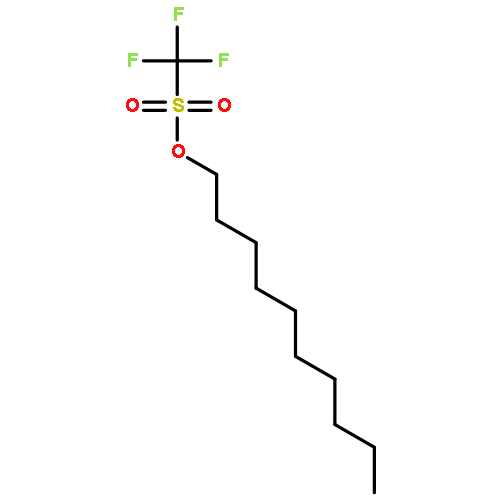
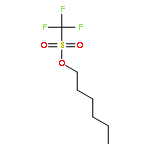
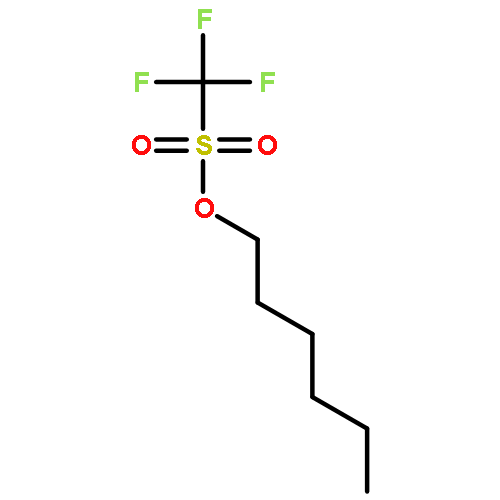
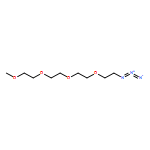
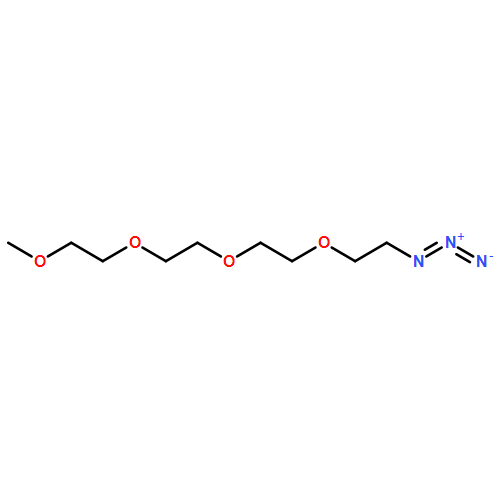

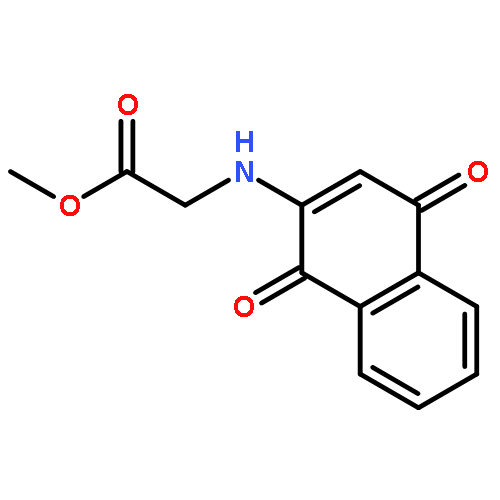




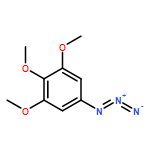
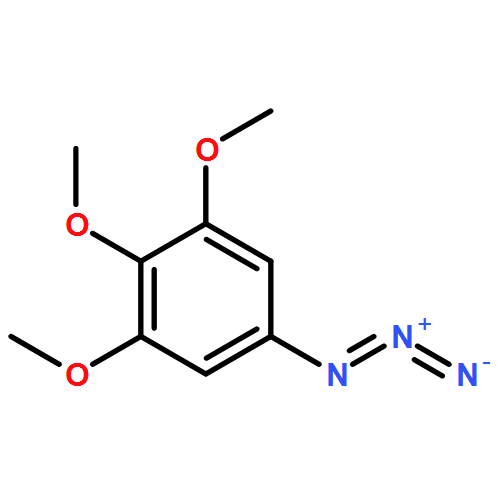
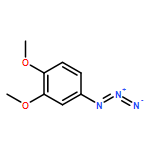
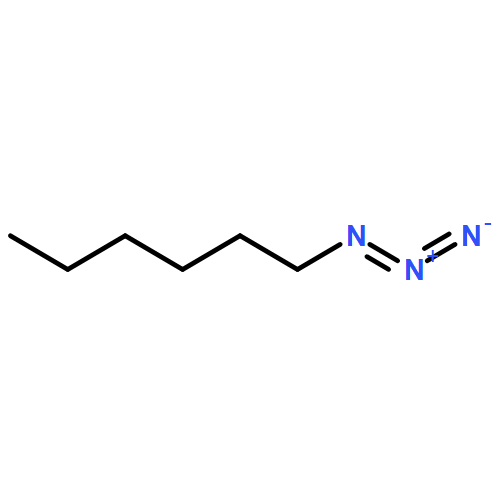
![β-D-Glucopyranoside, phenyl 2,3-bis-O-(phenylmethyl)-4,6-O-[(R)-phenylmethylene]-1-thio-](/data/chemimg/1372400/87470-70-0.png)
![β-D-Glucopyranoside, phenyl 2,3-bis-O-(phenylmethyl)-4,6-O-[(R)-phenylmethylene]-1-thio-](/data/chemimg/1372400/87470-70-0_b.png)












![2-[(phenylmethyl)amino]-1,4-Naphthalenedione](http://img.cochemist.com/ccimg/24200/24137-17-5.png)
![2-[(phenylmethyl)amino]-1,4-Naphthalenedione](http://img.cochemist.com/ccimg/24200/24137-17-5_b.png)





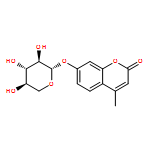
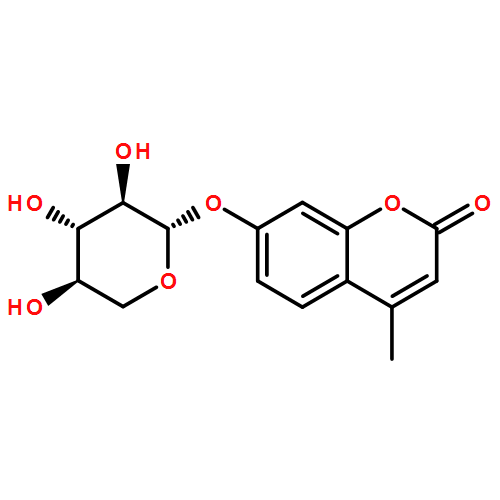




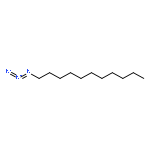
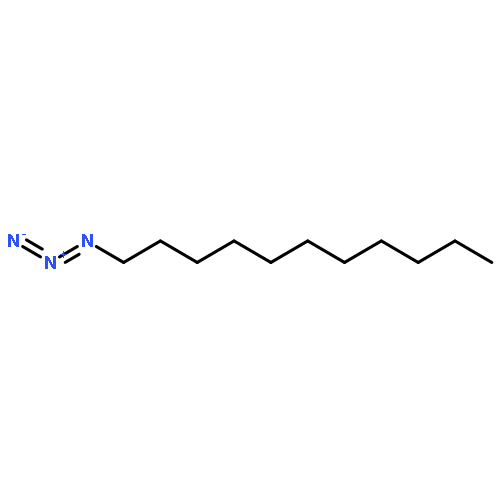

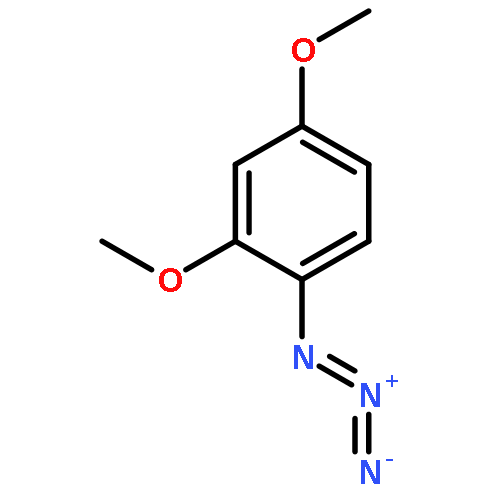



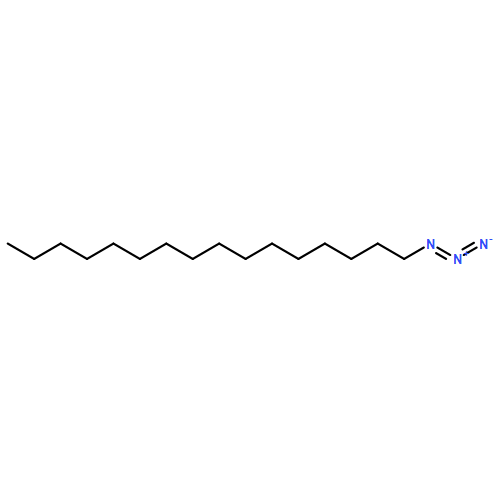


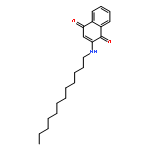

![1-phenyl-1H-naphtho[2,3-d][1,2,3]triazole-4,9-dione](http://img.cochemist.com/ccimg/5500/5466-47-7.png)
![1-phenyl-1H-naphtho[2,3-d][1,2,3]triazole-4,9-dione](http://img.cochemist.com/ccimg/5500/5466-47-7_b.png)
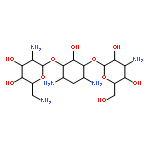


![1H-Naphtho[2,3-d]triazole-4,9-dione, 1-(phenylmethyl)-](/data/chemimg/3722500/79707-04-3.png)
![1H-Naphtho[2,3-d]triazole-4,9-dione, 1-(phenylmethyl)-](/data/chemimg/3722500/79707-04-3_b.png)